


The genus Cephalotaxus produces a variety of important natural products with complex structures and diverse biological activities. One of the most significant of these is homoharringtonine, which was approved by the US food and drug administration (FDA) for the treatment of chronic myeloid leukemia. Equally significant are the cephalotane diterpenoids, another major class of secondary metabolites isolated from the genus Cephalotaxus. These compounds have garnered considerable attention due to their remarkable anti-cancer activity and unique structural features.
Structurally, cephalotane diterpenoids are characterized by a 7/6/5/6-fused tetracyclic carbon skeleton and a bridged lactone (Figure 1). They can be classified into five categories based on variations in the A-ring: prototype diterpenoids, 17-nor-cephalotane diterpenoids, A-ring-seco nordi-terpenoids, benzenoid cephalotane-type norditerpenoids, and their dimers.[1,2] Prototype diterpenoids possess an intact C20 carbon skeleton with a 7/6/5/6-fused tetracy-clic carbon framework. 17-Nor-cephalotane diterpenoids (C19) also feature a 7/6/5/6-fused tetracyclic carbon framework and are further divided into two subcategories: those with a tropone moiety, known as Cephalotaxus troponoids, and those without, referred to as tropone-free 17-nor-cephalotane diterpenoids. Benzenoid cephalotane-type norditerpenoids (C19 or C18) contain a benzene ring and exhibit a 6/6/5/6-fused tetracyclic carbon framework. A-ring-seco norditerpenoids contain both a six-membered lactone ring and a five-membered ring instead of benzene rings. Norditerpenoid dimers are formed by the coupling of two monomer troponoids, which form a new eight-mem-bered ring. These benzenoid derivatives may originate from Cephalotaxus troponoids via a ring-contraction process.
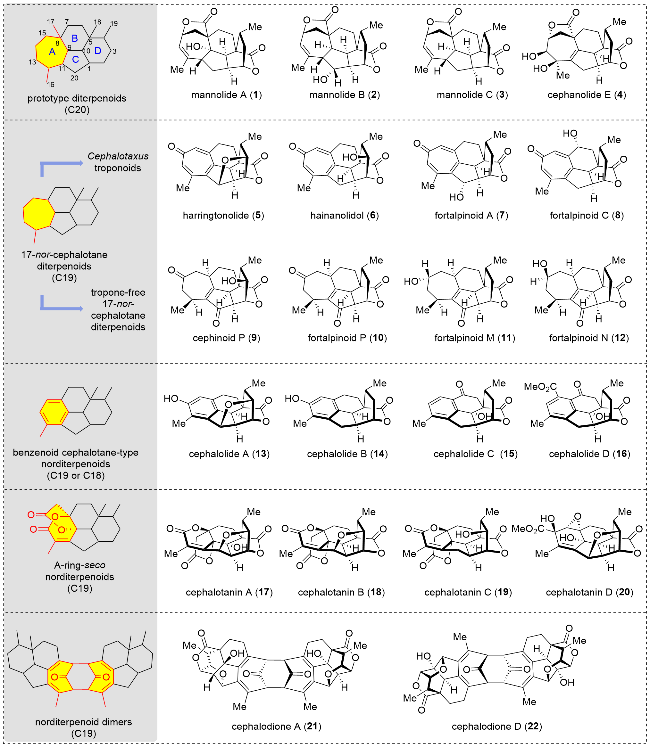
Cephalotane diterpenoids have demonstrated diverse biological activities, including inhibition of plant growth,[3] antiviral,[4] anti-inflammatory,[5] antitumor,[6-11] and NF-kB inhibitory activities.[12] Among these, the most important bioactivity is antitumor. For example, (+)-harringtonolide has shown significant activity against human KB cells with an IC50 of 43 nmol/L.[13] Their intricate structures and promising bioactivities have attracted significant attention from the chemical community. In 2012, Nay and colleagues[13] introduced early investigations of Cephalotaxus troponoids, covering their structural characteristics and synthetic studies. Chen and co-workers[14] provided a comprehensive overview of the total synthesis of benzenoid cephalotane-type norditerpenoids. In 2021, Hua and colleagues[1] described the structural features and bioactivities of over 70 cephalotane diterpenoids. Recently, Yue and colleagues[2] offered a detailed review of more than 100 cephalotane diterpenoids, encompassing their structural features, bioactivities, and progress in biosynthesis and chemical synthesis. To date, ten research groups have achieved the total synthesis of 24 cephalotane diterpenoids, and one research group has reported a semi-synthetic approach.[15-25] This review aims to introduce the latest progress in the synthesis of cephalotane diterpenoids, highlighting the importance of innovative synthetic strategies in the efficient synthesis of complex natural products and their potential significance in advancing drug discovery.
1 Biosynthetic studies of cephalotane di-terpenoids
Ever since the discovery of the first member, (+)-harringtonolide, in 1978, the biosynthetic pathway of cephalotane diterpenoids has been long debated. Pioneers such as Nay,[13] Yue,[8] and Cai[5] have proposed different biosynthesis proposals based on their speculations of the inter-relationships among various cephalotane diterpenoids (Scheme 1). Nay et al.[13] proposed that the cephalotane diterpenoid core shares a common origin with abietanes, both deriving from pimaradiene (Scheme 1a). His primary concern was the co-isolated C-ring-aromatic abietane diterpenoids, which might share this common origin with Cephalotaxus diterpenoids. Nay proposed that the oxidation of pimaradiene triggers a cascade of Wagner-Meerwein rearrangements. This process leads to the migrations of the C-17, C-19, and C-20 methyl groups, ultimately resulting in the formation of the aromatic C-ring. The five-membered ring closes through the oxidation of the C-20 methyl group, while the benzene ring expands into a seven-membered ring via oxidation. Finally, the troponoid norditerpenoids are formed through decarboxylation and pseudo-aromatization in the later stages.
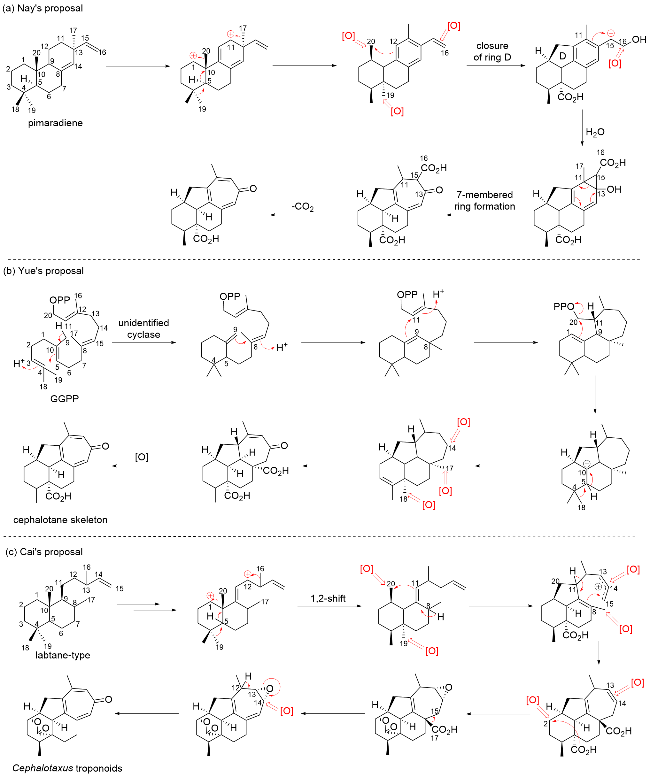
Yue, in contrast, proposed that the cephalotane core results from a stereospecific carbocation-driven cascade cyclization of geranylgeranyl pyrophosphate (GGPP), followed by a sequence of oxidation, decarboxylation, and aromatization (Scheme 1b). In 2016, Yue and colleagues[8] made a significant discovery by identifying cephalotane diterpenoids with an intact C20 skeleton from Cephalotaxus mannii, concluding that cephalotanes are the true precursors of Cephalotaxus troponoids. Building on this finding, Yue's group[8] proposed a de novo biosynthetic pathway (Scheme 1b), hypothesizing the existence of undiscovered cyclases responsible for catalyzing the stereospecific carbocation-driven cascade cyclization of GGPP, forming the cephalot-3-ene skeleton. A series of oxidative processes, including decarboxylation at C-8 and A-ring aromatization, along with further biosynthetic modifications, eventually led to the formation of Cephalotaxus troponoids.
In 2018, Cai and colleagues[5] proposed a biogenetic hypothesis suggesting that Cephalotaxus troponoids originate from labdane-type diterpenoids (Scheme 1c). According to this hypothesis, labdane diterpenoids undergo a series of 1,2-shifts, which include the migration of three methyl groups. Following this, new bonds are formed between C-20 and C-11, as well as C-8 and C-15, resulting in the formation of the tetracyclic cephalotane skeleton. Subsequent decarboxylation and aromatization reactions then lead to the evolution of the troponoid structure. Cai's hypothesis parallels Nay's proposal closely, but it begins with labdane diterpenoids rather than pimaradiene.
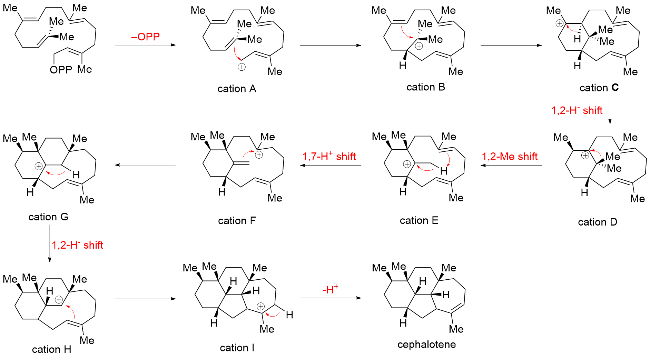
While these three proposals are plausible and inspiring, the key intermediates responsible for the skeleton reorganization remain undisclosed. However, a breakthrough was made in 2023 by Dai[26] and Wang,[27] who independently reported the discovery of the diterpene synthase that catalyzes the direct cyclization of GGPP into a distinctive cephalot-12-ene skeleton, which is believed to be a putative precursor to other cephalotane-type diterpenoid biosynthesis (Scheme 2). A plausible biosynthetic mechanism en route to tetracyclic ring formation was proposed based on extensive isotopic labeling experiments, density functional theory (DFT) calculations, molecular docking, and molecular dynamics simulation combined with site-directed mutagenesis. Although the enzymes responsible for the late-stage modifications have yet to be identified, the research by Dai and Wang marks a significant breakthrough in clarifying the complete biosynthetic pathway of cephalotane diterpenoids, providing strong support for Yue's proposed biosynthetic proposal.
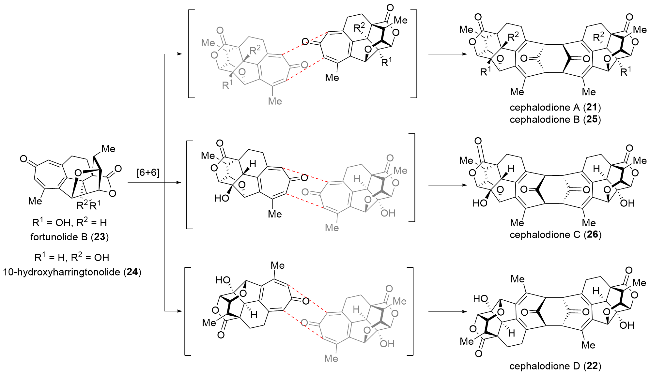
While dimeric natural products formed through Diels-Alder or [2+2]-cycloaddition reactions are quite common, dimeric compounds arising from a [6+6]-cycloaddition had not been previously confirmed, either chemically or biosynthetically. Following the identification of C19-nordi-terpenoid dimers, Yue[21] suggested that these dimers result from homo-or cross-dimerization of related monomers (Scheme 3). The mechanism involves an exo-selective [6+6]-cycloaddition reaction, which, according to the Woodward-Hoffmann rules, is thermodynamically forbidden but allowed under photochemical conditions.
2 Total synthesis of cephalotane diterpenoids via ring expansions
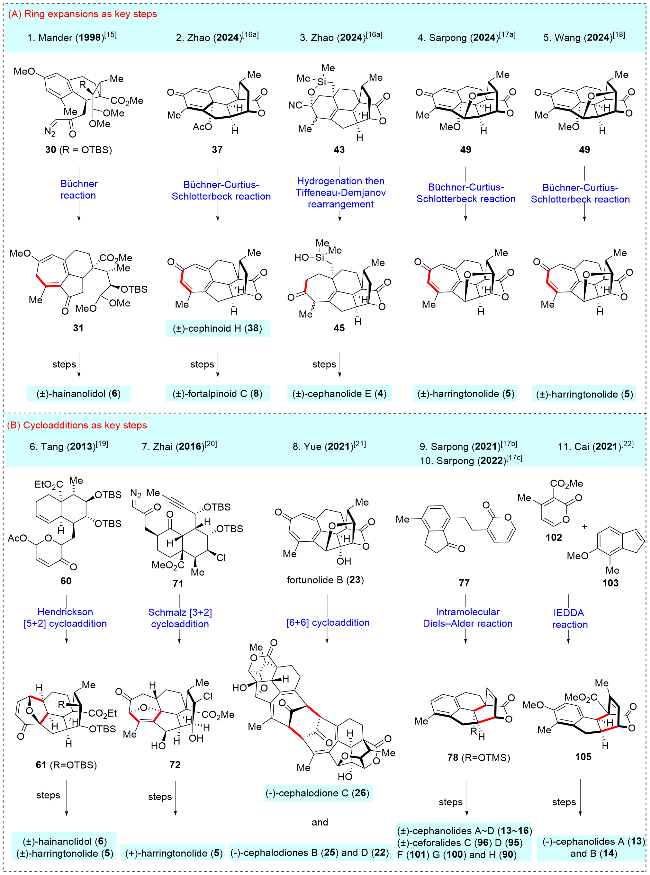
In 1998, Mander and colleagues[15] made a landmark contribution to this field by reporting the first total synthesis of (±)-hainanolidol (6) (Scheme 4). The synthesis featured the use of the Büchner reaction, a novel approach for constructing the poly-substituted cycloheptatriene skeleton, which was quite inspiring for later synthesis. The process commenced with the preparation of the tricyclic compound 28, derived from benzoic acid (27) through an elaborate six-step sequence. This was followed by converting compound 28 into dimethyl acetal 29 via oxidative cleavage using Pb(OAc)4, a Wittig reaction, and oxidation with 3-chloroperoxybenzoic acid (m-CPBA). Product 29 then underwent a series of reactions: tert-butyldimethylsilyl (TBS) protection of the secondary alcohol, selective hydrolysis of the methyl ester, and condensation with diazomethane to yield diazoketone 30. Compound 30 was subsequently transformed into cycloheptatriene 31 using the Büchner reaction catalyzed by Rh2(mandelate)4. Following this, a three-step transformation-deprotection of the acetal, intramolecular aldol reaction, and transannular lactonization yielded product 32, which established the core skeleton of (±)-hainanolidol (6). Finally, the synthesis was completed through a three-step conversion involving desilylation, reduction of the ketone, and acid-mediated elimination, resulting in the successful total synthesis of (±)-hainano-lidol (6). Albeit racemic, Mander's 20-step synthesis of (±)-hainanolidol (6) opened the gateway to the chemical synthesis of other cephalotane diterpenoids.
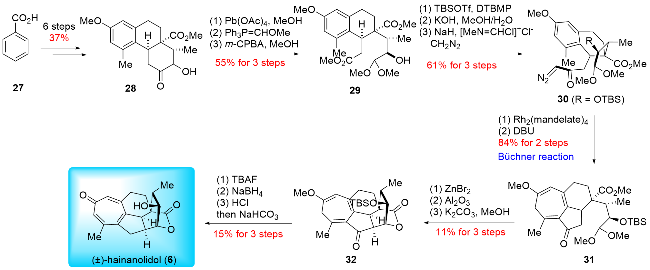
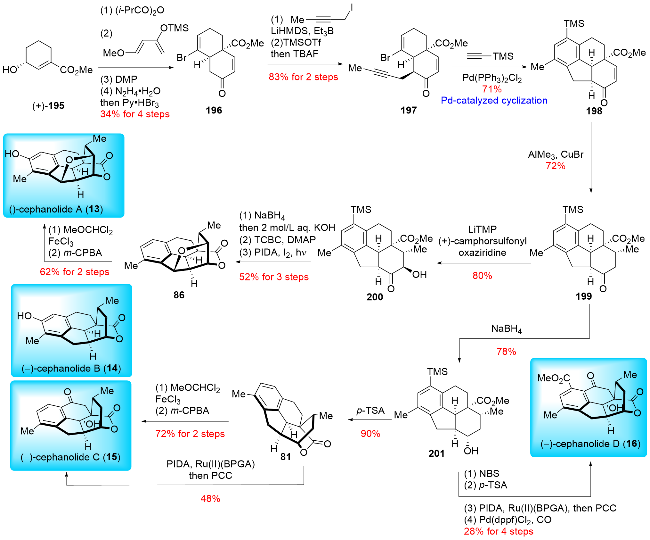
In 2024, Zhao and co-workers[16a] reported the total synthesis of (±)-cephinoid H (38) and (±)-fortalpinoid C (8) (Scheme 5). They applied the ring expansion strategy to construct the key cycloheptatriene skeleton through Büchner-Curtius-Schlotterbeck (BCS) reaction. The synthesis began with their previously synthesized (±)-cephanolide B (14),[16b] which underwent oxidative dearomatization to yield dienone 37. This compound was then subjected to the Büchner-Curtius-Schlotterbeck reaction, producing (±)-cephinoid H (38) and its isomer 39 in a ratio of 1∶3.5. To enhance the regio-selectivity in the ring expansion, (±)-cephanolide B (14) was first chlorinated to give compound 33, which was subsequently converted into a chlorine-substituted dienone 34 using the same procedure. Ring expansion of 34 proceeded smoothly, yielding the desired product 35 and its isomer 36 in a ratio of 3∶1. Pd-catalyzed dehalogenation of 35 then afforded (±)-cephinoid H (38). SeO₂-mediated oxidation of (±)-cephinoid H (38), followed by ketone reduction, resulted in compound 40. Finally, compound 40 underwent a Mitsunobu reaction and hydrolysis to complete the synthesis of (±)-fortalpinoid C (8). Zhao and co-workers demonstrated a remarkable advancement in synthetic chemistry, achieving transformations beyond what is achievable through biosynthesis. They were the first to apply the Büchner-Curtius-Schlotter-beck (BCS) reaction to construct the tropone ring. This innovative approach highlighted the strength of synthetic chemistry, enabling the conversion of benzenoid cephalotane-type norditerpenoids into 17-nor-cephalotane diter-penoids via carbon insertion, contrary to the natural biosynthetic pathways. This breakthrough underscores the potential of synthetic methods in overcoming limitations found in nature.
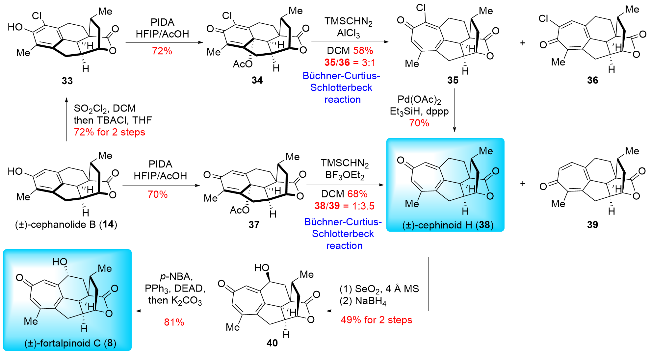
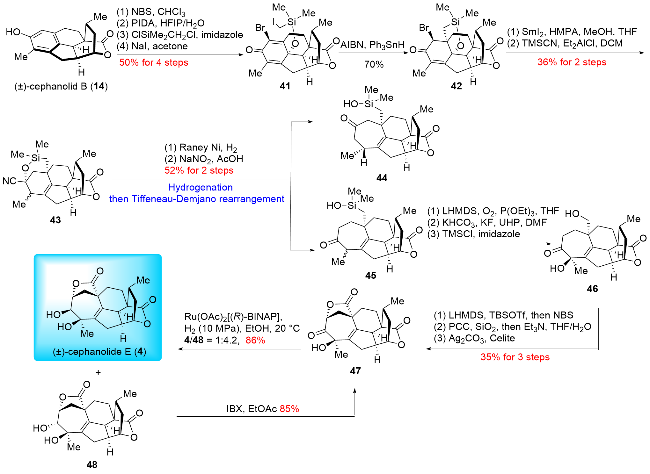
In 2024, Zhao and co-workers[16a] also accomplished the synthesis of (±)-cephanolide E, utilizing key steps such as Ueno-Stork radical cyclization and Tiffeneau-Demjanov rearrangement (Scheme 6). The synthesis started from their previously synthesized (±)-cephanolide B (14),[16b] which was converted into bromine-substituted dienone 41 through a four-step operation. The Ueno-Stork-type radical cyclization was then performed, yielding the desired cyclization product 42. Subsequent cleavage of the C—O bond in 42 was followed by nucleophilic addition of a cyanide group, producing compound 43. The cyanide group in 43 was reduced to a primary amine, which was then oxidized and subjected to a Tiffeneau-Demjanov rearrangement to afford ring expansion products 44 and 45. Compound 45 underwent a three-step transformation involving α-hy-droxylation, Fleming oxidation, and trimethylsilyl (TMS) protection to yield 46. α-Bromination of 46 provided the bromide intermediate. Oxidation of the primary alcohol followed by hydrolysis produced a hemiacetal, which was then oxidized with pyridinium chlorochromate (PCC) to form lactone 47. Finally, Noyori hydrogenation of 47 was performed to afford (±)-cephanolide E (4) and its epimer 48. Compound 48 could be converted back to its precursor 47 via oxidation with 2-iodoxybenzoic acid (IBX). Thus, Zhao and co-workers employed the Tiffeneau-Demjanov rearrangement to construct the seven-membered ring on tropone-free 17-nor-cephalotane diterpenoids. Notably, in this instance, they opted for the Tiffeneau-Demjanov rearrangement instead of the Büchner-Curtius-Schlotterbeck (BCS) reaction to achieve the one-carbon insertion strategy, further demonstrating their innovative approach to complex ring construction.
In 2024, Sarpong and co-workers[17a] reported their synthesis of (±)-harringtonolide (5) (Scheme 7), utilizing an innovative late-stage benzenoid-to-troponoid skeletal modification strategy, which contrasted with previous synthetic approaches. Their synthesis began with the oxidative dearo-matization of (±)-cephanolide A (13), a compound obtained through their previous synthetic work.[17b,17c] This process led to the formation of dienone 49. After extensive optimization, the crucial benzenoid-to-troponoid ring expansion was achieved by treating 49 with TMSCHN2 in the presence of AlCl3, enabling the total synthesis of (±)-har-ringtonolide (5) in 12 steps (linear longest sequence, LLS). Notably, harringtonolide (5) serves as a biosynthetic precursor to cephanolide A (13), and the Sarpong group's re-verse-biomimetic approach to synthesizing (±)-harringto-nolide (5) represents a significant milestone, as this compound is otherwise inaccessible via natural biosynthesis. Interestingly, both Sarpong and Zhao independently and simultaneously utilized the Büchner-Curtius-Schlotterbeck (BCS) reaction to construct the tropone rings in troponoids, further demonstrating the synthetic utility of this transformation.
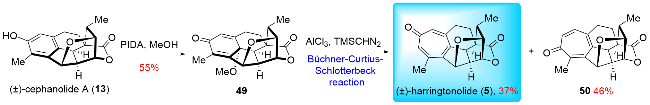
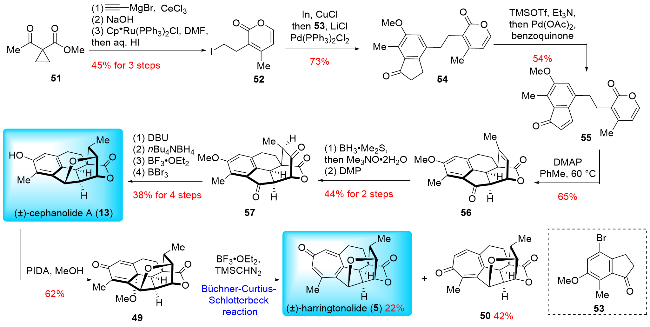
In 2024, Wang and co-workers[18] reported a concise synthesis of (±)-cephanolide A (13) and (±)-harringtono-lide (5) (Scheme 8). Key transformations in their synthesis included a Pd-catalyzed Csp²-Csp³ cross-coupling, an electron-deficient intramolecular Diels-Alder reaction, and a late-stage Büchner-Curtius-Schlotterbeck (BCS) reaction. Their synthesis began with the preparation of α-pyrone 52 and indanone 53. The α-pyrone 52 was synthesized in three steps from compound 51, involving nucleophilic addition with acetylene magnesium bromide, hydrolysis of the methyl ester, and Ru-catalyzed regioselective cyclization. Next, the Pd-catalyzed C(sp²)—C(sp³) cross-coupling between α-pyrone 52 and indanone 53 yielded pyrone 54. This was followed by Saegusa oxidation and an electron-defi-cient intramolecular Diels-Alder reaction to produce pentacyclic product 56. Subsequent diastereoselective hydroboration-oxidation and Dess-Martin oxidation were employed to convert the olefin to ketone, resulting in product 57. After inverting the configuration of the methyl group with 1,8-diazabicyclo[5.4.0]undecane-7-ene (DBU), both ketone groups were reduced using n-Bu₄NBH₄ to afford a diol, which was then subjected to BF₃•OEt₂-mediated etherification to form the tetrahydrofuran ring. Demethylation with BBr₃ successfully yielded (±)-cephanolide A (13). Oxidative dearomatization of (±)-cephanolide A (13) with (diacetoxyiodo)benzene (PIDA) in MeOH produced the p-quinol derivative 49, which underwent the Büchner-Curtius-Schlotterbeck (BCS) reaction to form the tropone ring, completing the synthesis of (±)-harringtonolide (5) in 16 steps (LLS). Notably, while Zhao, Sarpong, and Wang all employed analogous strategies for tropone ring construction, their methodologies diverged in key aspects, particularly in the choice of substrates and reaction conditions used for assembling the core skeleton and executing the ring expansion.
3 Total synthesis of cephalotane diterpenoids via cycloaddition reactions
In 2013, Tang and colleagues[19] reported a significant advance in natural product synthesis with their racemic total synthesis of both (±)-hainanolidol (6) and (±)-harring-tonolide (5) (Scheme 9). Their innovative synthetic route incorporated several key transformations: an Achmatowicz rearrangement, an intramolecular oxidopyrylium-based [5+2] cycloaddition, and a DBU-promoted Kornblum-DeLaMare rearrangement. The synthesis began with the preparation of furanylketone 59, derived from the bicyclic compound 58 through a 16-step process. Compound 59 was then converted into dihydropyranone 60 via a three-step transformation that included reduction of the ketone, VO-(acac)2-catalyzed epoxidation, Achmatowicz rearrangement, and esterification. Under DBU conditions, the intramolecular oxidopyrylium-based [5+2] cycloaddition smoothly produced the pentacyclic product 61. Subsequently, compound 61 underwent several transformations: addition of a methyl Grignard reagent, selective desilylation, and transannular lactonization to yield lactone 62. The introduction of a phenylthiol group was achieved through a BF3•OEt2-mediated SN1 displacement, followed by opening of the oxygen bridge using lithium diisopropylamide/ hexa-methylphosphoramide (LDA/HMPA) to afford diene 63. After triethylsilyl (TES) protection of the secondary alcohol, the phenylthiol group was oxidized to sulfoxide with magnesium monoperoxyphthalate hexahydrate (MMPP), and a reductive cleavage of the C—S bond with SmI2 produced product 64. A [4+2] cycloaddition between the diene and singlet oxygen generated peroxide 65. DBU-promoted Kornblum-DeLaMare rearrangement formed the ketone and tertiary alcohol, which underwent acid-mediated elimination to yield (±)-hainanolidol (6). Finally, treatment of (±)-hainanolidol with Pb(OAc)4 resulted in the formation of the tetrahydrofuran skeleton, thereby completing the synthesis of (±)-harringtonolide (5) in 27 steps (LLS). Tang and co-workers were the pioneers in applying the oxidopyrylium-based [5+2] cycloaddition to the construction of the poly-substituted cycloheptatriene skeleton, representing a significant advancement in the synthesis of cephalotane diterpenoid skeletons via a cycloaddition strategy.

In 2016, Zhai and co-workers[20a] achieved the first asymmetric total synthesis of (+)-harringtonolide (5). Their synthetic route was distinguished by using an intramolecular Diels-Alder reaction and a rhodium-catalyzed intramolecular [3+2] cycloaddition (Scheme 10). The synthesis began with the asymmetric reduction of compound 66, yielding chiral secondary alcohol 67. This was followed by a four-step sequence: condensation with sorbic acid, an intramolecular Diels-Alder reaction, deconjugation of the olefin, and reduction of the lactone, leading to the formation of hemiacetals 68. Subsequently, hemiacetals 68 underwent nucleophilic addition with 1-propynylmagne-sium bromide, followed by selective protection of the propargyl alcohol. The remaining secondary alcohol was oxidized to ketone, producing epimers 69 and 70. Compound 70 was further transformed through a five-step process to yield the key precursor 71. Utilizing Rh(OAc)4 as a catalyst, the intramolecular [3+2] cycloaddition efficiently formed the pentacyclic skeleton, which was then desilylated to produce compound 72. Compound 69 could also be converted to 72 in 9 steps with an overall yield of 2.7%. The synthesis continued with an SN2 displacement under NaH conditions to form the tetrahydrofuran ring 73. To invert the configuration of the hydroxy group, a redox operation was performed, followed by lactonization under basic conditions to yield lactone 74. Finally, the carbonyl group was transformed into an enol ether, and a Me2AlCl-mediated cleavage of the C—O bond/elimination cascade reaction generated the tropone ring, completing the total synthesis of (+)-harringtonolide (5) in 21 steps (LLS). In contrast to Tang's [5+2] cycloaddition, Zhai and co-workers made a groundbreaking contribution by employing a Rh-catalyzed [3+2] cycloaddition to the construction of the key poly-substituted cycloheptatriene skeleton.
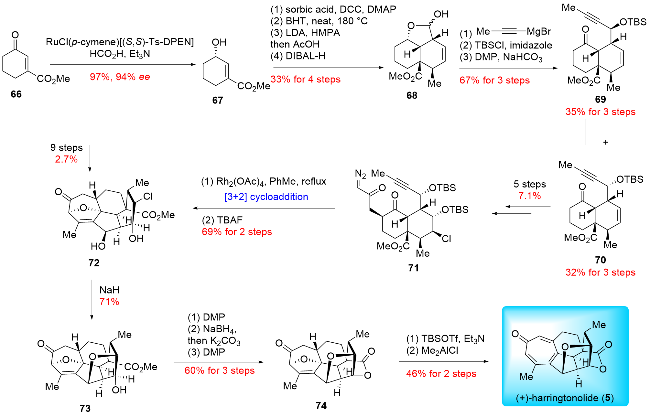
In 2021, inspired by their proposed biosynthetic pathway, Yue and co-workers[21] successfully achieved the biomimetic transformation of the co-isolated monomer, fortuno-lide B (23), into cephalodiones B~D. This transformation was accomplished via an ultraviolet-enabled [6+6] cycloaddition (Scheme 11). Upon treatment with a mercury lamp under argon, the monomer underwent dimerization, yielding three distinct dimers: (-)-cephalodiones B~D. Remarkably, Yue achieved the semi-synthesis of these three products in a single step. This [6+6] cycloaddition is unprecedented in organic synthesis, and they successfully achieved the biomimetic reaction using a high-pressure mercury lamp. This work not only confirmed the proposed biosynthetic pathway for dimers but also proved that these dimers are not artifacts of the isolation or purification processes.
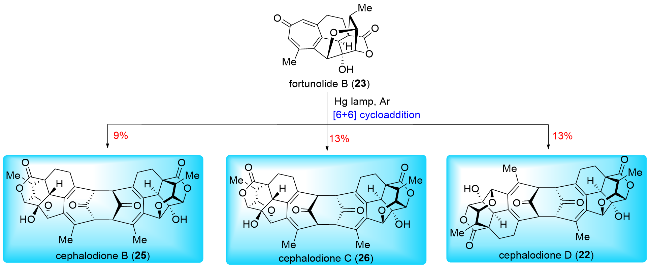
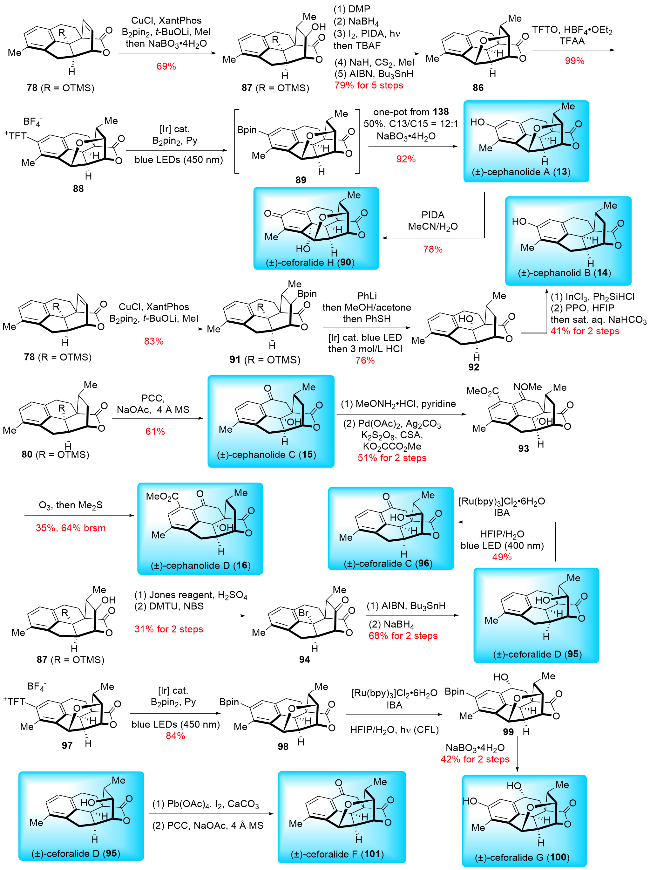
In 2021, Sarpong and co-workers[17b] reported their synthesis of (±)-cephanolides A~D (Scheme 12), employing C(sp2)—C(sp3) cross-coupling, an intramolecular inverse-demand Diels-Alder reaction, and late-stage oxidations. Their synthesis began with the preparation of indanone derivative 77 from 7-hydroxy-4-methylindanone through a triflation and C(sp2)—C(sp3) Suzuki cross-coupling sequence. The ketone moiety of 77 was then converted to a silyl enol ether, which underwent an intramolecular inverse-demand Diels-Alder reaction to form the basic framework of benzenoid cephalotane norditerpenoids. This was followed by Mukaiyama hydration to yield ketone, and a modified olefination with Ti(Oi-Pr)2Cl2 and Nysted reagent provided exo-methylene 79. Hydrogenation of 79 produced 80, which was then subjected to desilylation and ionic deoxygenation to afford 81. Direct oxygenation of 81 produced (±)-cephanolide B (14) and its isomer 82 in a 1.3∶1 ratio with a combined yield of 74%. Alternatively, compound 80 underwent benzylic oxidation with PCC, followed by desilylation to yield (±)-cephanolide C (15). Similarly, benzylic oxidation of 80 followed by condensation and acetylation to form acetyl oxime 83. Oxime-directed ortho C—H acetoxylation was performed, followed by hydrolysis and oxidative removal of the oxime to give 84. (±)-Cepha-nolide D (16) was obtained from 84 through triflation and Pd-catalyzed methoxy carbonylation. For cephanolide A (13), compound 79 underwent a five-step transformation, including allylic oxidation, hydrogenation, epimerization, ketone reduction, and Suárez C—H oxidation to produce 85. Barton-McCombie deoxygenation of 85 gave 86, which was further oxidized by cyclopropane malonyl peroxide to afford (±)-cephanolide A (13) and its isomer in a 6∶1 ratio with a combined yield of 39%.
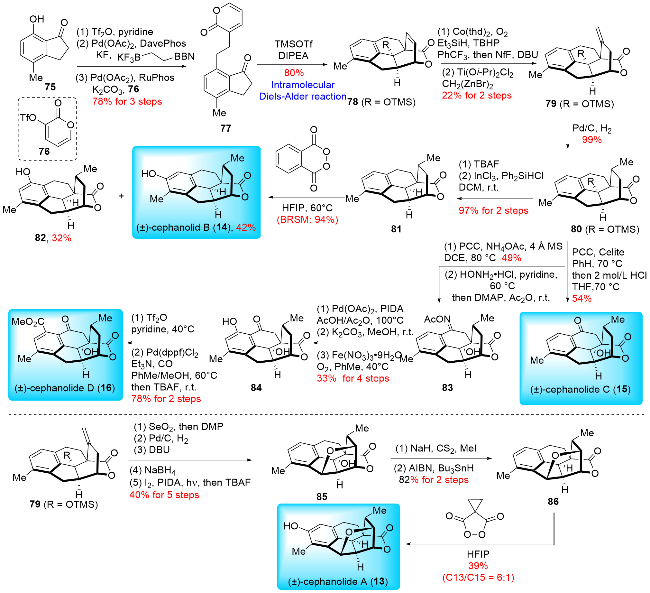
In 2022, Sarpong and co-workers[17c] reported an improved total synthesis of benzenoid cephalotane-type norditerpenoids (Scheme 13). Their synthesis commenced with an optimized Cu-catalyzed methylboration of 78, followed by an oxidation to give 87. Compound 87 underwent a five-step functional group manipulation to produce 86. A C—H thianthrenation of 86 then formed thianthrenium salt 88, which was converted to boronic ester 89. Subsequent oxidation of the Bpin moiety in 89 resulted in (±)-cephano-lide A (13). (±)-Cephanolide A (13) was further subjected to oxidative dearomatization to give (±)-ceforalide H (90). The Cu-catalyzed methylboration of 78 was also performed to produce boronic ester 91. Treatment of 91 with PhLi generated the corresponding borate, which was then reduced through protodeboronation and desilylation to yield 92. Compound 92 was transformed into (±)-cephanolide B (14) through a two-step process involving InCl3-catalyzed ionic deoxygenation and phthaloyl peroxide-mediated oxygenation. (±)-Cephanolide C (15) was synthesized by PCC-mediated benzylic oxidation of 80. A methyl ester group was introduced to give 93 via methyloxime-directed C—H methoxycarbonylation. Cleavage of the methyloxime in 93 under ozonolysis conditions yielded (±)-cephanolide D (16). Compound 87 was oxidized with Jones reagent and brominated to form bromide 94, which underwent reductive debromination and carbonyl group reduction to produce (±)-ceforalide D (95). (±)-Ceforalide C (96) was obtained from (±)-ceforalide D (95) through selective benzylic oxidation under photoinduced conditions. To access (±)-ceforalide G (100), thianthrenium salt 97 was borylated to give boronic ester 98. Photoredox oxygenation of 98 introduced a hydroxyl group at the desired position, and oxidation of the Bpin moiety provided (±)-ceforalide G (100). (±)-Ceforalide D (95) could also be converted to (±)-ceforalide F (101) through a two-step process involving Suárez C—H oxidation and PCC-mediated benzylic oxidation. Unlike Tang and Zhai's approach, which constructed the A ring via [5+2] and [3+2] cycloaddition reactions, respectively, Sarpong synthesized the B and D rings in a single step using a Diels-Alder reaction. This strategy demonstrated high efficiency, as an intramolecular Diels-Alder reaction was employed to form three rings in one step. The longest linear sequences (LLS) for the obtained products ranged from 6 to 13 steps, with total yields varying from 1.5% to 14%, calculated from known precursors. Although racemic, Sarpong's synthesis remains the most efficient method to date for producing cephalotane diterpenoids.
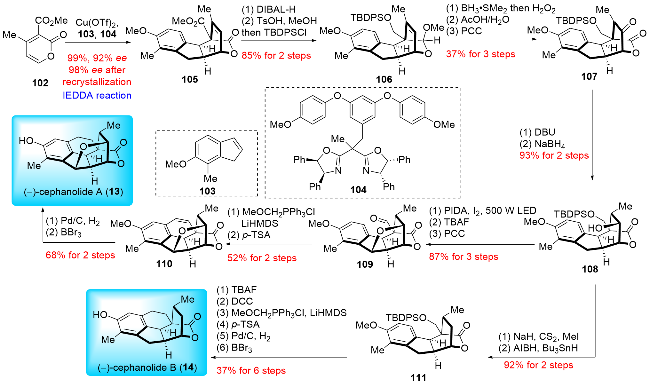
In 2021, Cai and co-workers[22] reported the total synthesis of (-)-cephanolides A and B, utilizing a Cu-cataly-zed asymmetric inverse-electron-demand Diels-Alder (IEDDA) reaction as a key step (Scheme 14). Their synthesis commenced with the Cu-catalyzed asymmetric inverse-electron-demand Diels-Alder (IEDDA) reaction between 4-methyl-2-pyrone 102 and indene 103, yielding the desired product 105 with 99% yield and 92% ee on a gram scale. Following this, the lactone and ester groups were reduced using diisobutylaluminium hydride (DIBAL-H), and the resulting compound was subjected to acetalization and silylation to give 106. This was followed by hydroboration-oxidation, hydrolysis, and PCC oxidation to produce 107. The configuration of the methyl group was then inverted, and the ketone was reduced with NaBH4 to obtain alcohol 108. Suárez C—H oxidation was performed to construct the tetrahydrofuran ring, which was then followed by desilylation and oxidation to give aldehyde 109. Wittig reaction generated methylenol ether, which underwent an intramolecular Friedel-Crafts reaction to afford product 110. Finally, (-)-cephanolide A (13) was synthesized through a two-step process involving hydrogenation and demethylation. To synthesize (-)-cephanolide B (14), compound 106 was also used. The hydroxyl group of 106 was reduced by Barton-McCombie deoxygenation to yield product 111, which was then converted to (-)-cephanolide B (14) using the same steps as those described for the synthesis of (-)-cephanolide A (13). Similar to Sarpong's strategy, Cai and co-workers also constructed the D ring using a Diels-Alder reaction. However, Cai advanced the approach by developing a Cu-catalyzed asymmetric intermolecular inverse-electron-demand Diels-Alder reaction (IEDDA). This reaction enabled the efficient, one-step construction of an asymmetric poly-substituted [2.2.2] lactone ring. Cai's synthesis demonstrated a 14-step LLS with a total yield of 8.8% for (-)-cephanolide A and a 15-step LLS with a 9.7% total yield for (-)-cephanolide B, calculated from known precursors. To date, this represents the most efficient asymmetric synthesis for these compounds.
4 Total synthesis of cephalotane diterpenoids via Pauson-Khand reaction
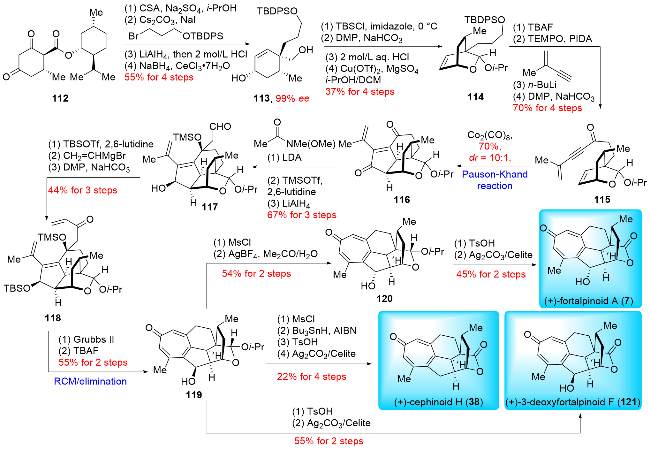
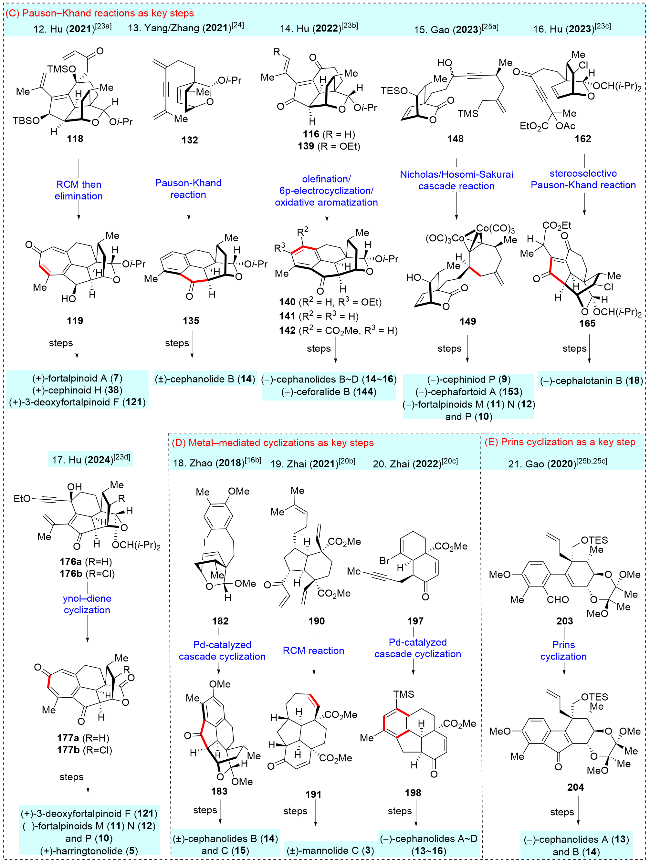
Given the presence of a fully substituted cyclopentane, the Pauson-Khand reaction could be a viable method for constructing the B ring. In 2021, Hu and co-workers reported an innovative synthesis of (+)-fortalpinoid A (7), (+)-cephinoid H (38), and (+)-3-deoxyfortalpinoid F (121), featuring a diastereoselective Pauson-Khand reaction and a ring-closing metathesis (RCM) reaction as key steps (Scheme 15).[23a] Their synthesis began with chiral compound 112, previously reported by Mayer's group.[29] Compound 112 underwent a four-step transformation involving enol etherification, alkylation, and reduction of both ester and carbonyl groups, resulting in enantiopure diol 113 (>99% ee). Subsequently, the allylic alcohol was selectively protected with TBS, followed by Dess-Martin oxidation to produce acetal 114. After desilylation, the aldehyde was converted into isopropyl acetal in the presence of Cu(OTf)2. Further functional group manipulations included desilylation, 2,2,6,6-tetramethyl-1-piperinedinyl-oxy (TEMPO)-mediated oxidation, nucleophilic addition with alkynyl lithium reagent, and Dess-Martin oxidation to generate key intermediate 115. The diastereoselective Pauson-Khand reaction of 115 with Co2(CO)8 efficiently yielded the tetracyclic product 116. To introduce the Weinreb amide moiety, nucleophilic addition was performed, followed by protection of the tertiary alcohol with TMS and reduction with LiAlH4 to produce aldehyde 117. After TBS protection of the secondary alcohol, the aldehyde underwent addition with vinylmagnesium bromide and Dess-Martin oxidation to form compound 118. The RCM reaction successfully constructed the seven-membered ring, which was then subjected to desilylation and elimination cascade to form the tropone skeleton, yielding 119. Further, hydrolysis of the acetal moiety and oxidation with Fetizon's reagent completed the synthesis of (+)-3-deoxyfortal-pinoid F (121). To synthesize (+)-cephinoid H (38), the hydroxyl group of 119 was removed by chlorination and dechlorination, followed by the same lactone-forming procedure. Finally, inversion of the hydroxyl group configuration in compound 119, followed by the same lactone-forming process, accomplished the synthesis of (+)-fortal-pinoid A (7) in 25 steps (LLS). Hu and co-workers strategically employed the Pauson-Khand reaction to construct the B/C ring system and utilized ring-closing metathesis (RCM) to form the poly-substituted cycloheptatriene A ring. This approach highlights the critical capability of these reactions in synthesizing highly-strained and poly-substi-tuted rings within complex natural products.
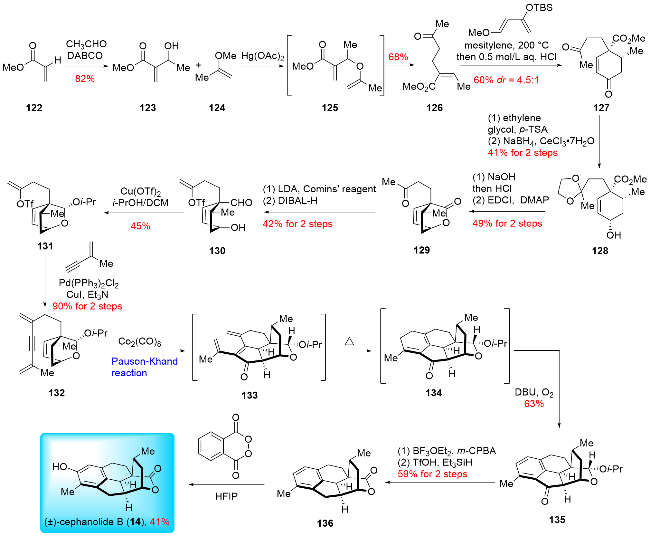
In 2021, Yang and Zhang,[24a] along with their co-wor-kers, reported the total synthesis of (±)-cephanolide B, utilizing an intramolecular Pauson-Khand reaction, 6π-electrocyclization, and oxidative aromatization cascade as key steps (Scheme 16). Their synthesis began with the pre-paration of cyclohexene ketone 127 from methyl acrylate 122 through a three-step process. This included a Morita-Baylis-Hillman reaction, etherification/[3,3]sigmatropic rearrangement cascade reaction, and an intermolecular Diels-Alder reaction. Following this, regioselective ketalization was performed, and the resulting product was reduced using Luche reduction to yield allylic alcohol 128. The hydrolysis of the methyl ester was carried out, followed by intramolecular lactonization to produce product 129. Treatment of methyl ketone with LDA and Comins’ reagent afforded a triflate, which was then reduced with DIBAL-H to yield aldehyde 130. Aldehyde 130 was converted to isopropyl acetal 131 in the presence of Cu(OTf)2. The key intermediate 132 was formed via a Sonogashira reaction between isopropyl acetal 131 and 2-methylbut-1-en-3-yne. A tandem reaction involving Pauson-Khand reaction, 6π-electrocyclization, and oxidative aromatization led to the formation of the desired product 135. The isopropyl acetal of 135 was then oxidized to lactone, and the ketone moiety was removed under reductive conditions to give 136. Finally, a hydroxyl group was introduced on the benzene ring using phthaloyl peroxide, culminating in the synthesis of (±)-cephanolide B (14). Yang and co-workers[24b] are undoubtedly expert in the Pauson-Khand reaction, as demonstrated by their continuous application of this method in complex natural product synthesis. Their expertise was pivotal in the synthesis of (±)-cephanolide B, showcasing the Pauson-Khand reaction's effectiveness in constructing poly-substituted cyclopentanones. This example highlights the reaction's potential for synthesizing more challenging natural products.
In 2022, Hu and co-workers[23b] reported the asymmetric and divergent total synthesis of (-)-cephanolides B~D and (-)-ceforalide B (Scheme 17). Their synthesis hinged on several key transformations, including a diastereoselective Pauson-Khand reaction, an olefination/6π-electro-cyclization/oxidative aromatization cascade reaction and a late-stage oxidation. Their synthesis commenced with the chiral compound (-)-137. A nucleophilic addition of 137 with alkynyl lithium reagent was first occurred to give secondary alcohol which went through an oxidation with IBX to afford ketone 138. The Pauson-Khand reaction occurred to afford pentacyclic product 139. Then Wittig olefination was performed to generate terminal olefine followed by 6π-electrocyclization/oxidative aromatization cas-cade reaction to form benzenic ring giving 140. (-)-Cepha-nolide B (14) was obtained through a three-step functional group manipulation including oxidation of methyl acetal, reduction of ketone and demethylation. Next compound 137 was converted to pentacyclic product 116 in a three-step transformation. And olefination/6π-electrocyclization/oxi-dative aromatization cascade reaction of 116 processed smoothly to afford product 141 which was subjected to a three-step conversion involving oxidation of methyl acetal, reduction of ketone and site-selective benzylic oxidation to afford (-)-cephanolide C (15). Furthermore, compound 116 underwent an Horner-Wadsworth-Emmons (HWE) olefination followed by 6π-electro-cyclization/oxidative aromatization cascade reaction to give product 142 which was subjected to acetal oxidation and ketone reduction to provide 143. Finally, by using the optimized benzylic oxidative condition, (-)-ceforalide B (144) and (-)-cephano-lide D (16) were obtained, respectively.
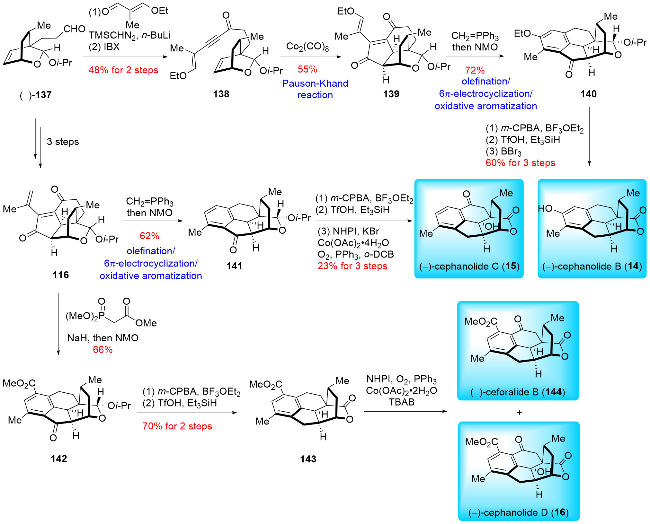
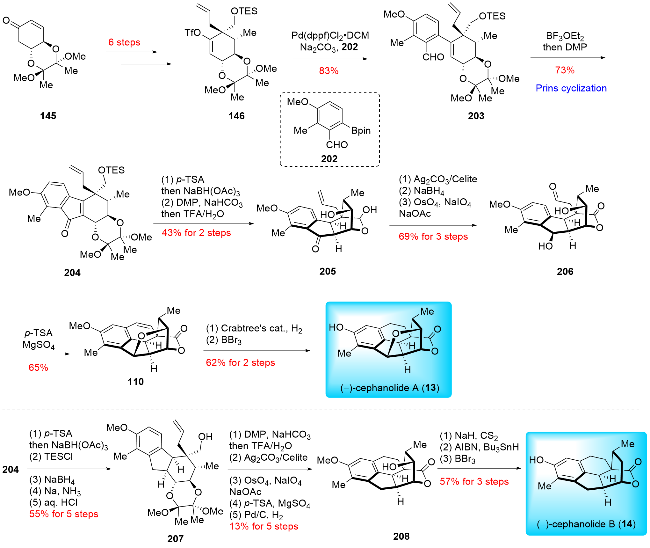
In 2023, Gao and co-workers[25a] reported the asymmetric synthesis of (-)-cephinoid P, (-)-cephafortoid A, and (-)-fortalpinoids M, N, P (Scheme 18). Their synthetic route featured a Nicholas/Hosomi-Sakurai cascade reaction and an intramolecular Pauson-Khand reaction. The synthesis commenced with chiral compound 145, derived from (-)-quinic acid. This compound underwent a six-step transformation, including 1,4-addition, esterification, Pd-catalyzed decarboxylative allylation, hydroxymethylation, TES protection, and triflation, resulting in 146. Compound 146 was then subjected to a further six-step process involving Pd-catalyzed hydrogenolysis, Dess-Martin oxidation, Pinnick oxidation, condensation with 1-(3-dimethyl-aminopropyl)-3-ethyl carbodiimide hydrochloride (EDCI), and anti-Markovnikov Wacker oxidation to yield aldehyde 147. An alkynyl lithium reagent was added nucleophilically to aldehyde 147, producing propargylic alcohol 148. This compound was treated with Co₂(CO)₈ to form a dicobalt hexacarbonyl complex, which was subsequently subjected to TMSOTf-promoted Hosomi-Sakurai cyclization, yielding product 149. The lactone of 149 was then converted to an isopropyl acetal group over two steps, followed by oxidation of the secondary alcohol to generate product 150. The Pauson-Khand reaction was carried out smoothly in the presence of tetramethylthiourea (TMTU), producing product 152. A four-step transformation, including selective reduction of the carbonyl group, oxidative cleavage of the terminal olefin, deprotection of the acetal group, and oxidation of the hemiacetal, completed the synthesis of (-)-cephinoid P (9). (-)-Cephinoid P (9) was then reduced with NaBH₄ to yield (-)-cephafortoid A (153) and 14-epi-cephafortoid A (154) in a 1∶1.5 ratio. Further reduction of (-)-cephinoid P (9) using Barton-McCombie deoxygenation provided (-)-fortalpinoid P (10), which was subsequently reduced by NaBH₄ to produce (-)-fortalpinoids M (11) and N (12) in a 1.5∶1 ratio.
Gao's synthesis is both adventurous and straightforward. The preinstalled dicobalt hexacarbonyl complex serves dual purposes: it facilitates the Hosomi-Sakurai reaction and acts as an ideal precursor for alkyne in the subsequent Pauson-Khand reaction. By leveraging these two key reactions, Gao efficiently assembled the 7/6/5/6-fused tetracyclic carbon framework, achieving the first synthesis of tropone-free 17-nor-cephalotane diterpenoids of this type.
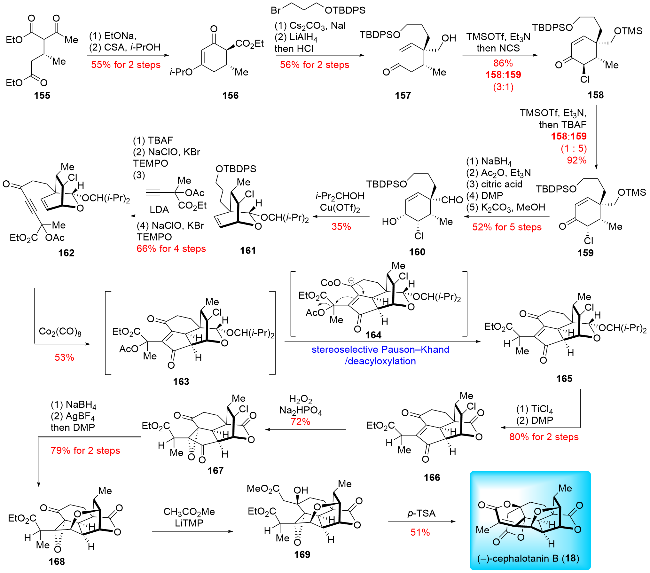
In 2023, Hu and co-workers[23c] reported the total synthesis of (-)-cephalotanin B (Scheme 19). The key steps of their synthetic route included a divergent asymmetric Michael addition reaction, a Pauson-Khand/deacyloxylation cascade reaction, and an epoxide-opening/elimination/dual-lactonization cascade reaction. Their synthesis commenced with the asymmetric Michael addition to prepare the chiral compound 155, which was obtained in 95% yield with 98% ee under optimized conditions. Compound 155 then underwent an intramolecular Dieckmann condensation and enol etherification to give 156. Subsequent alkylation and reductive processes formed cyclohexanone 157, which underwent α-chlorination to provide a pair of diastereomers (dr=3∶1). The major product 158 was further epimerized to the desired 159. After a five-step functional group manipulation, aldehyde 160 was obtained and subjected to acetalization with 2,4-dimethyl-3-pentanol to afford 161. Compound 161 was then transformed into 162 through a four-step sequence. The Pauson-Khand/deacyloxylation cascade reaction was carried out in the presence of Co2(CO)8, providing the tetracyclic product 165. The lactone 166 was derived from 165 through acetal deprotection and oxidation. Epoxidation of 166 yielded 167, which was then subjected to ketone reduction and AgBF4-promoted etherification to form the tetrahydrofuran ring, followed by Dess-Martin oxidation to furnish 168. Compound 168 underwent nucleophilic addition with the lithium enolate of methyl acetate, providing 169. Finally, a pyridinium p-toluenesulfonate (PPTS)-promoted epoxide-opening/elimination/dual lactonization cascade reaction was performed, completing the synthesis of (-)-cephalotanin B (18).
In 2024, Hu and co-workers[23d] reported the divergent total synthesis of (+)-3-deoxyfortalpinoid F, (+)-harring-tonolide and (-)-fortalpinoids M/N/P (Scheme 20). Their synthetic strategy featured a Pauson-Khand reaction and a novel ynol-diene cyclization. The synthesis began with the preparation of compounds 170 and cyclohexanone 156. Compound 171 was obtained from these starting materials through a two-step process involving alkylation and reduction. The primary alcohol of 171 was oxidized to an alde-hyde, followed by a reductive process to yield 173a. This compound underwent acetalization with 2,4-dimethyl-3-pentanol in the presence of Cu(OTf)₂, followed by desilylation and TEMPO-mediated oxidation to produce 174a. A Pauson-Khand reaction then furnished the tetracyclic product 175a, which was further reacted with an alkynyl lithium reagent to give 176a. The tropone skeleton was constructed via ynol-diene cyclization of 176a, using Ti(Oi-Pr)Cl₃ as a catalyst, and the resulting hemiacetal was oxidized with TEMPO to afford product 177a. Reduction of 177a with NaBH₄ yielded (+)-3-deoxyfortalpinoid F (112), while hydrogenation of the tropone moiety in 177a provided (-)-fortalpinoid P (10), which was further reduced to give (-)-fortalpinoids M (11) and N (12).
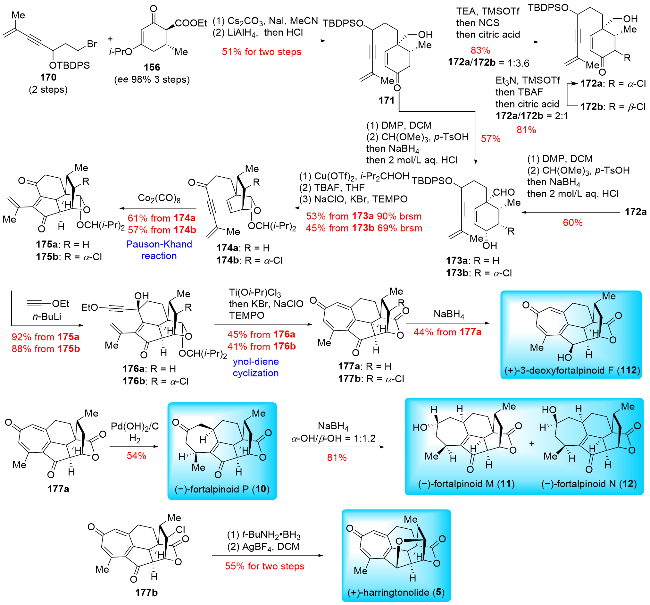
For the synthesis of (+)-harringtonolide (5), compound 171 was first subjected to α-chlorination and epimerization, resulting in 172a. This intermediate then underwent a series of similar transformations as previously described, leading to the formation of product 177b. Finally, 177b was reduced with t-BuNH₂•BH₃ and underwent etherification, catalyzed by AgBF₄, to complete the synthesis of (+)-harringtonolide (5). Hu and co-workers have synthesized the largest number of cephalotane diterpenoids to date, totaling 13 members, using their general and practical strategy. The Pauson-Khand reaction played a crucial role in constructing the shared cyclopentanone B ring, while other cyclization reactions, such as ring-closing metathesis (RCM), 6π-electro-cyclization, and ynol-diene cyclization, were employed to build the benzene or tropone A ring. The successful synthesis of these 13 cephalotane diterpenoids clearly demonstrates the effectiveness of their approach.
5 Total synthesis of cephalotane diterpenoids via metal-mediated cyclozation
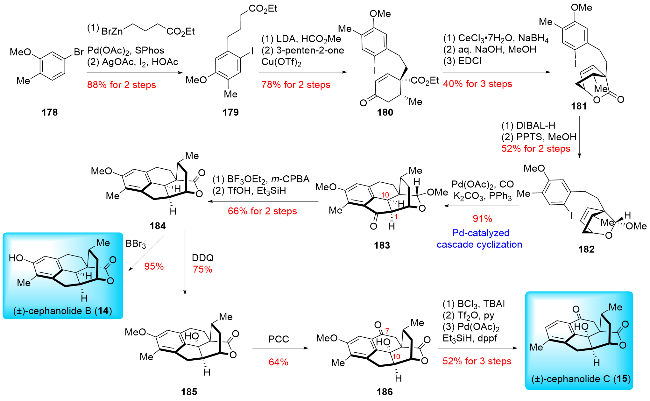
In 2018, Zhao and co-workers[16b] reported the first total synthesis of (±)-cephanolides B and C using a Pd-cata-lyzed cascade cyclization (Scheme 21). The synthesis began with the preparation of precursor 182. Initially, 5-bromo-2-methylanisole (178) and alkyl zinc bromide were coupled via a Negishi reaction, followed by aromatic iodination to yield product 179. Ester 179 was then subjected to a series of reactions, including formylation and Robinson annulation, resulting in an inseparable diastereomeric mixture of 180. The Luche reduction of mixture 180 afforded separable allylic alcohols, which were then hydrolyzed and condensed to form the key precursor 181. The Pd-catalyzed carbonylative annulation reaction initially produced an undesired C1/C10 diastereomer. Optimization revealed that the desired diastereomer 183 was obtained by transforming the lactone into a sterically hindered methyl acetal 182. Methyl acetal 182 was subsequently oxidized to lactone under m-CPBA/BF3•OEt2 conditions, followed by ketone reduction to afford product 183. Demethylation of 184 with BBr3 yielded (±)-cephanolide B (14) in high yield. To synthesize (±)-cephanolide C (15), compound 184 was subjected to a two-step oxidation with 2,3-dichloro-5,6-dicyano-1,4-ben-zoquinone (DDQ) and PCC, introducing a hydroxy group at C10 and a carbonyl group at C7 in product 186. (±)-Cep-hanolide C (15) was then synthesized through a three-step transformation involving demethylation, triflation, and Pd-catalyzed reduction. In contrast to previous syntheses of the cyclopentanone (B ring) via the Pauson-Khand reaction, Zhao developed an unprecedented Pd-catalyzed carbonylative annulation reaction. This breakthrough expanded the strategies for synthesizing fully substituted cyclopentanones and laid the foundation for the synthesis of other cep-halotane diterpenoid types.
In 2021, Zhai and co-workers[20b] reported the first asy-mmetric total synthesis of (+)-mannolide C (3), employing Ru-complex catalyzed double ring-closing metathesis (RCM) reactions as a key step (Scheme 22). The synthesis began with the preparation of racemic alcohol 188 through a four-step sequence from the known Diels-Alder adduct 187. The racemic alcohol 188 was then converted to the chiral enone 189 via a lipase-mediated biosynthetic resolution followed by Dess-Martin oxidation. After seven additional steps, the tetraene 190 was obtained. Using the Hoveyda-Grubbs 2nd generation catalyst, double RCM reactions were conducted to produce the tetracyclic product 191. Subsequent inversion of configuration by DBU and Ni-catalyzed methylation yielded the Michael adduct, which was further processed through reduction and lactonization to afford the pentacyclic product 192. An allylic oxidation followed by stereoselective α-methylation produced product 193. Treatment of 193 with 4-methylbenzenesul-fonhydrazide generated the tosylhydrazone, which was then reduced with catecholborane to yield product 194. Finally, a two-step functional manipulation involving diastereoselective dihydroxylation and dehydration completed the synthesis of (+)-mannolide C (3). Zhai's work strategically applied the ring-closing metathesis (RCM) reaction to simultaneously construct both the seven-membered ring and the six-membered ring in a single step, highlighting its critical capability in forming highly-strained rings during the synthesis of complex natural products.
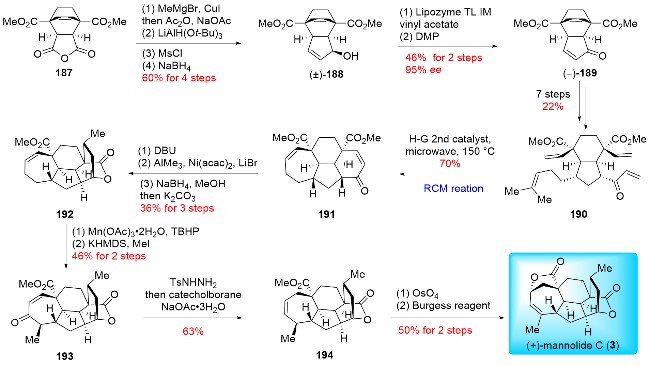
In 2022, Zhai and co-workers reported the asymmetric and divergent total syntheses of (-)-cephanolides A~D (Scheme 23).[20c] Their synthetic route featured a diastereoselective intermolecular Diels-Alder reaction, a Pd-cata-lyzed formal bimolecular [2+2+2] cycloaddition and a late-stage oxidative diversification. Their synthesis began with the chiral alcohol (+)-195, which underwent acylation with isobutyric anhydride, followed by an intermolecular Diels-Alder reaction with Danishefsky's diene. Subsequent Dess-Martin oxidation and formation of vinyl bromide afforded the bicyclic product 196. Monoalkylation and epimerization provided product 197. Next, the Pd-catalyzed formal bimolecular [2+2+2] cycloaddition constructed the tetracyclic skeleton 198. Enone 198 underwent a stereoselective Michael addition, introducing a methyl group to furnish product 199, which served as a common precursor for the synthesis of (-)-cephanolides A-D.
For the synthesis of (-)-cephanolide A (13), a stereoselective α-hydroxylation of 199 was performed, yielding product 200. The lactone and tetrahydrofuran rings were then formed through a four-step sequence: reduction, hydrolyzation, condensation, and Suárez C—H oxidation, resulting in product 86. Finally, regioselective hydroxylation on the benzene ring of 86 was achieved through Friedel-Crafts acylation and Baeyer-Villiger oxidation, completing the synthesis of (-)-cephanolide A (13). For the synthesis of (-)-cephanolides B~D, compound 199 was reduced to form alcohol 201, which underwent intra-molecular lactonization to give product 81. Friedel-Crafts acylation followed by Baeyer-Villiger oxidation afforded (-)-cephanolide B (14). Compound 81 then underwent a Ru-catalyzed chemoselective and site-selective C—H oxidation developed by Uchida, followed by PCC oxidation, to provide (-)-cephanolide C (15). Finally, compound 201 was subjected to bromination, Ru-catalyzed C—H oxidation, PCC oxidation, and Pd-catalyzed methoxy carbonylation, yielding (-)-cephanolide D (16). In contrast to previous syntheses of the poly-substituted benzene ring (A ring) from benzene-containing substrates, Zhai developed a de novo strategy for benzene ring (A ring) synthesis using an unprecedented Pd-catalyzed [2+2+2] annulation reaction. This breakthrough expanded the approaches for synthesizing poly-substituted benzene rings and laid the groundwork for the synthesis of other cephalotane diter-penoid types via late-stage benzene oxidation.
6 Total synthesis of cephalotane diterpenoids via Prins reaction
Before the widespread use of the Pauson-Khand reaction to construct fully substituted cyclopentanes (B ring), Gao and co-workers developed an alternative approach using the Prins reaction. This strategy is both convergent and straight-forward. In 2020, Gao and co-workers[25b] reported the first asymmetric total synthesis of (-)-cephanolide A, utilizing Suzuki-Miyaura cross-coupling and Prins cyclization as key steps (Scheme 24). Their synthesis began with the preparation of triflate 146 from compound 145 through a six-step process. The Pd-catalyzed Suzuki-Miyaura cross-coupling then formed coupling product 203. This was followed by a BF3•OEt2-mediated intramolecular Prins cyclization and Dess-Martin oxidation to yield tetracyclic compound 204. Desilylation and reduction of the double bond led to the formation of hydroxyl group, which was then oxidized. Subsequent deprotection of the cyclic acetal produced hemiacetal 205. Treatment of 205 with Fetizon's reagent provided the lactone, followed by ketone reduction and oxidative cleavage of the terminal olefin to afford product 206. Under p-toluenesulfonic acid (p-TSA) conditions, a cation-mediated etherification and intramolecular Friedel-Crafts reaction formed C—O and C—C bonds, respective-ly, yielding product 110. Final hydrogenation and demethylation completed the synthesis of (-)-cephanolide A (13). These synthetic strategies were also applied to the synthesis of (-)-cephanolide B (14).[25c] Compound 204 underwent a five-step functional group manipulation, including reduction of the tetra-substituted olefin and carbonyl group to produce compound 207. Following a similar procedure, compound 207 was converted to pentacyclic product 208. Finally, Barton-McCombie deoxygenation and demethylation with BBr3 afforded (-)-cephanolide B (14).
7 Conclusion
In this review, we have discussed recent progress in the total syntheses of 24 cephalotane diterpenoids achieved by 10 different research groups. As summarized in Scheme 25, various inspiring strategies, including ring expansion, cycloaddition, Pauson-Khand reaction, metal-mediated cyclization, and prins cyclization, have been developed to construct the rigid 7/6/5/6 or 6/6/5/6-fused tetracyclic architecture characteristic of cephalotane diterpenoids. These total syntheses undoubted demonstrate the efficacy of key reactions in building highly rigid ring systems. More importantly, the synthetic efforts by Mander,[28] Huang,[29] Camp,[30] Li,[31] Nay,[32] Xie,[33] Hu,[34] and Zhao,[35] along with those reviewed, could serve as valuable tools for structural modification, potentially aiding in the discovery of cephalotane-related drugs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It is foreseeable that more research on the synthesis of cephalotane diterpenoids will be reported in the future. Furthermore, it is noteworthy that although over twenty strategies have been developed for the synthesis of cephalotane diterpenoids, a bioinspired approach has yet to be realized. For instance, synthesizing benzenoid cephalotane-type norditerpenoids from Cephalotaxus troponoids remains an unexplored avenue. With ongoing advancements in decoding biosynthetic pathways and developing synthetic methodologies, emulating the biosynthesis of cephalotane diterpenoids chemically is likely to become feasible in the future.
(Lu, Y.)