


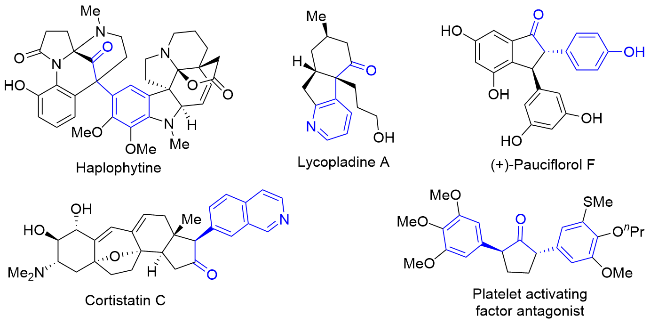
手性α-芳基酮广泛存在于天然产物、药物分子、农药分子、香料和材料等活性和功能分子中. 杀虫剂Haplophytine、生物碱Lycopladine A、抗抑郁药Cortistatin C、天然产物(+)-Pauciflorol F和血小板活化因子拮抗剂等均含有手性α-芳基酮结构片段(图1)[1]. 同时, 手性α-芳基酮也是有机合成和制药工业中合成其它手性片段和骨架的重要合成前体. 因此, 手性α-芳基酮类化合物的合成已经成为一个重要的研究领域, 受到了化学工作者的广泛关注. 传统合成方法从手性原料出发, 需要多步反应来实现. 因此, 发展催化合成手性α-芳基酮类化合物的方法成为有机合成领域的研究热点之一. 近年来, Colacot课题组[2a], Rossi课题组[2b], Licini课题组[2c]以及其他课题组[2d-2g]综述了手性α-芳基酮的部分合成方法, 但是目前缺乏手性α-芳基酮催化合成的全面概述. 因此, 该综述总结了近20年来手性α-芳基酮类化合物的催化合成研究进展, 详细讨论了每种方法的反应机理、优点和局限性及其应用, 并进一步展望了手性α-芳基酮的催化合成新方法、新策略和新方向.
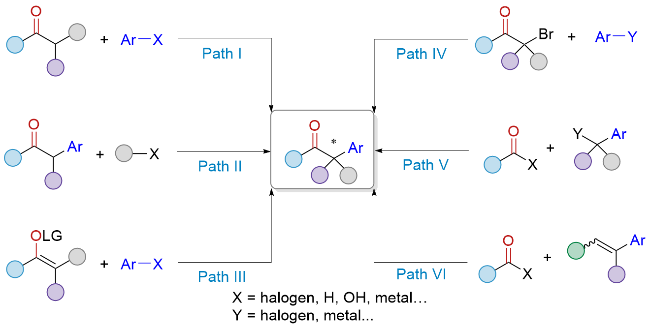
根据反应的成键方式和反应前体, 目前构建手性α-芳基酮的催化合成方法可以分为以下几类: (1)酮α位C—H键不对称芳基化反应(Path I); (2)外消旋α-芳基酮的不对称官能团化反应(Path II); (3)烯醇不对称芳基化反应(Path III); (4) α-溴代酮与芳基金属试剂的不对称交叉偶联反应(Path IV); (5)酰基亲电试剂的不对称苄基化反应(Path V); (6)芳基烯烃的不对称氢酰基化反应(Path VI)等(Scheme 1).
1 酮α位C—H键不对称芳基化反应
使用芳基化试剂对酮α位C-H键进行不对称α-芳基化反应是构建手性α-芳基酮类化合物的重要策略. 其中, 芳基卤代物、芳基三氟甲磺酸盐、芳炔以及苯醌类化合物均可以作为芳基化试剂. 值得注意的是, 催化策略的应用和手性配体的开发在酮α位的不对称芳基化反应中具有关键作用. 接下来, 我们将该策略分为酮α位C—H键分子间与分子内不对称芳基化反应进行详细阐述.
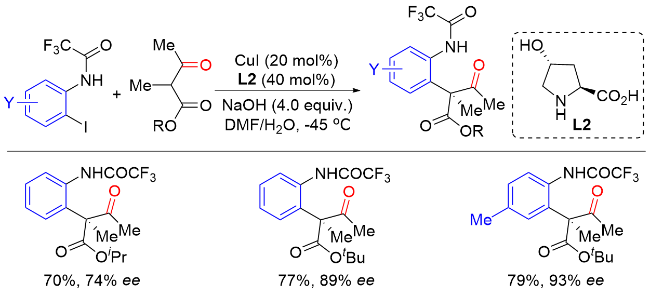
1.1 酮α位C—H键分子间不对称芳基化反应
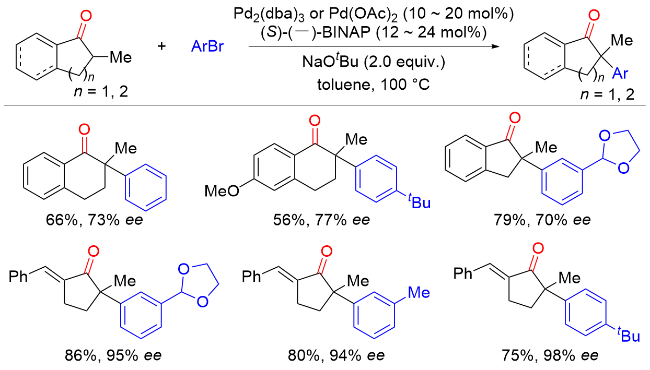
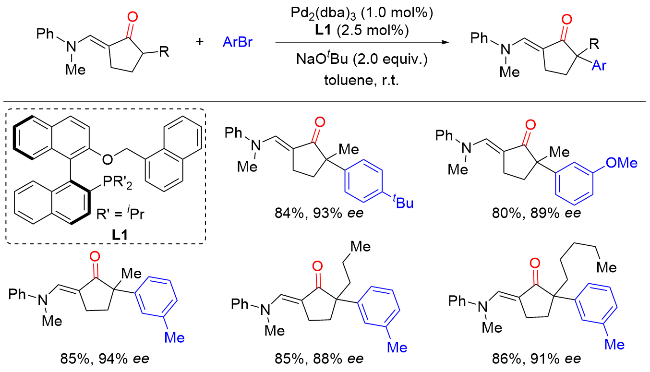
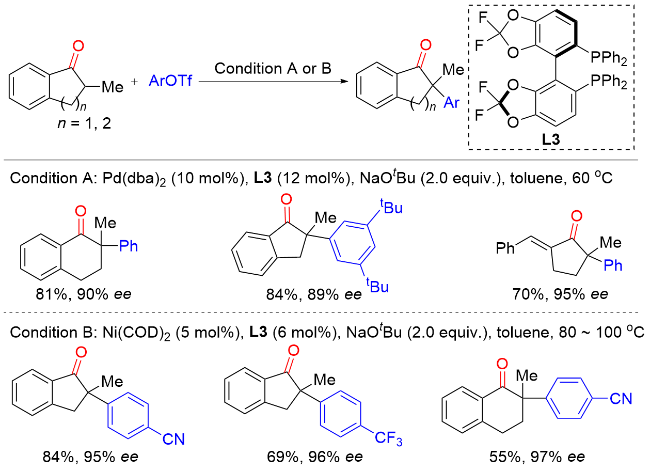
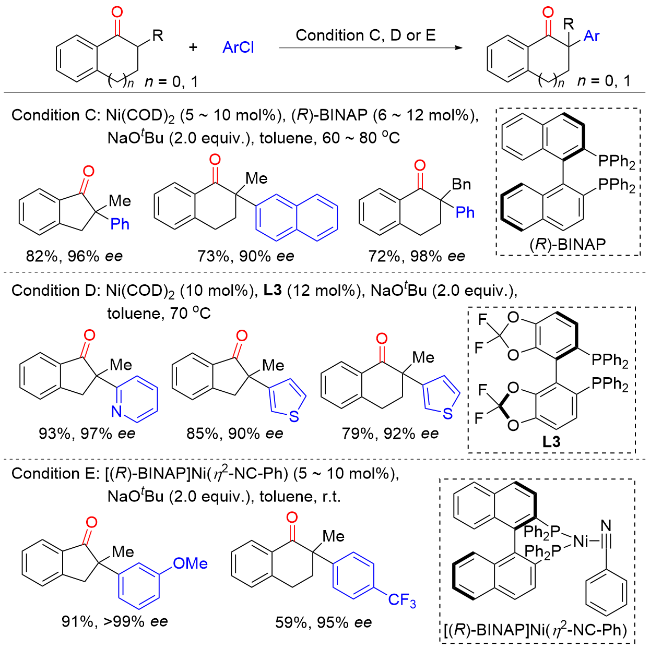
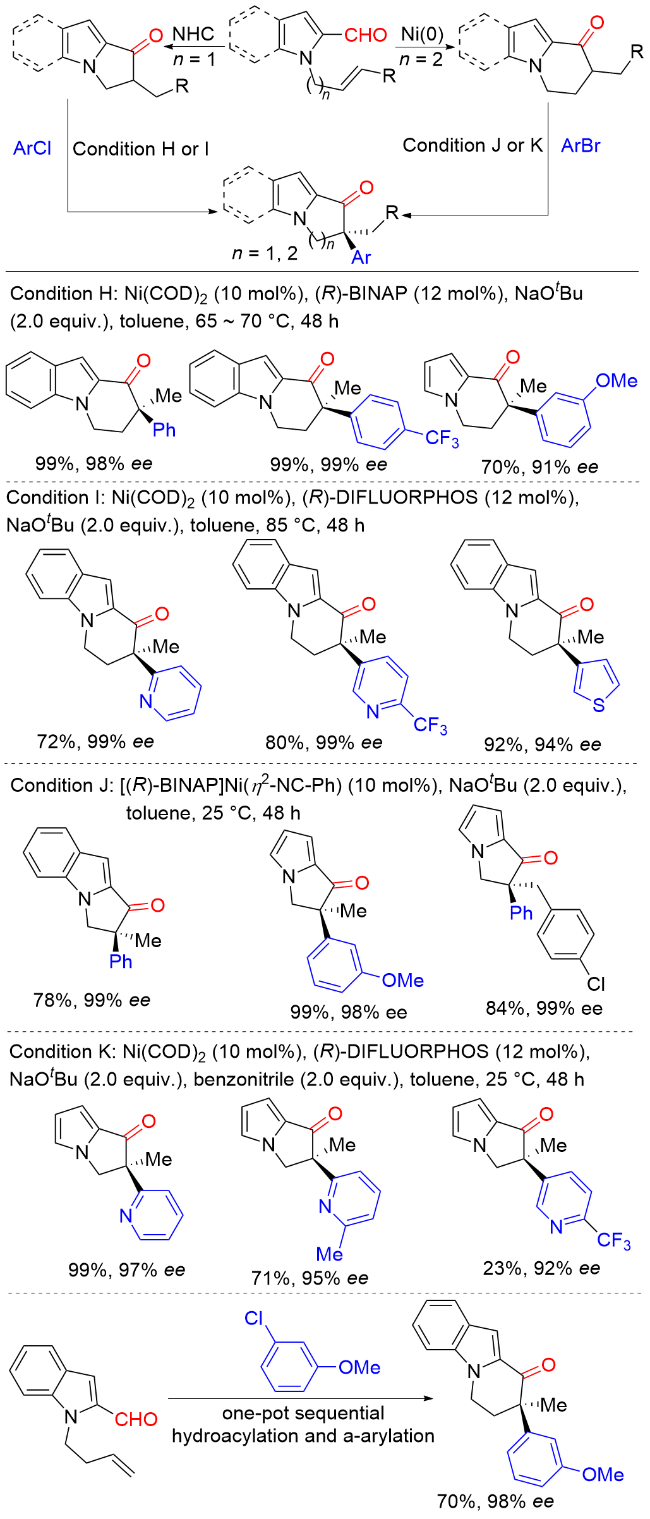
2011年, Hartwig课题组[7]报道了Ni(COD)2/(R)-(+)-联萘二苯基膦[(R)-BINAP]催化环状酮与芳基氯化物以及Ni(COD)2/5,5'-双(二苯基磷)-四氟-二-1,3-苯二氧杂环[(R)-DIFLUORPHOS]催化环状酮与杂环芳基氯化物的不对称α-芳基化反应(Scheme 6). 该反应条件温和, 底物范围广, 以良好的收率和优异的对映选择性实现了一系列含季碳手性中心的α-芳基酮类化合物的合成. 当使用预制备的[(R)-BINAP]Ni(η2-NC-Ph)络合物作为催化剂时, 该反应在室温条件下即可高效进行. 与卤代芳烃通常的反应性相反, 溴代芳烃在该反应中的产率和对映选择性均低于相应的氯代芳烃, 这很可能是因为溴代芳烃的反应活性更高, 通过Ni(0)或Ni(II)物种的分解形成了选择性较低的催化剂.
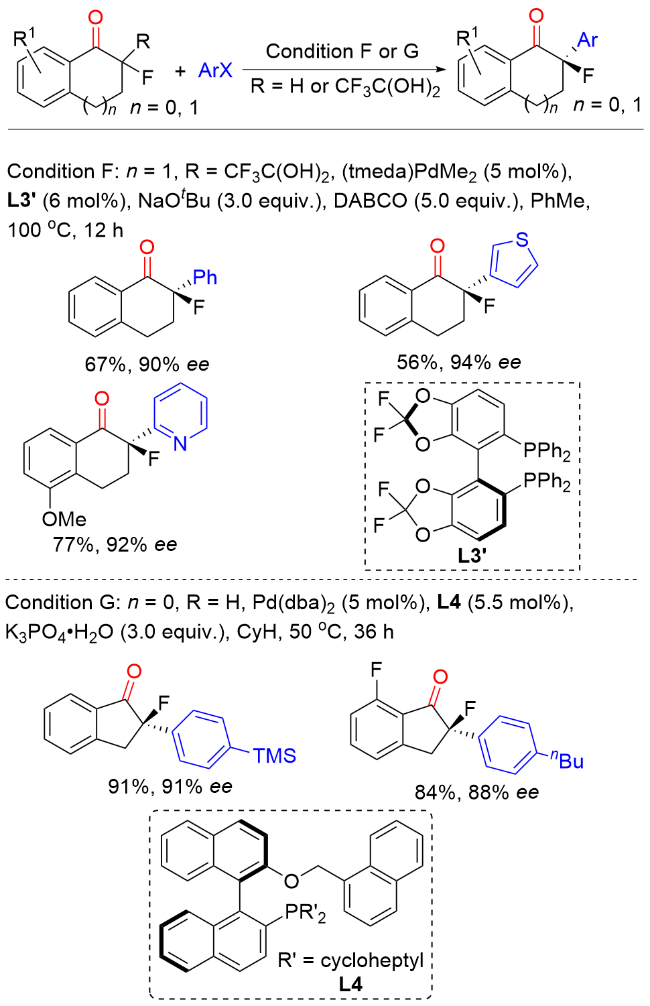
2016年, Stanley课题组[9]报道了一种烯烃氢酰化反应生成环状酮和镍催化环状酮不对称α-芳基化反应的串联策略, 合成了一系列含季碳手性中心的α-芳基环状酮类化合物(Scheme 8). 在该反应中, 烯烃的催化氢酰化反应可以通过改变条件调控生成五元或者六元环状酮. 镍催化分子内氢酰化反应通过选择性调控以良好的收率生成α-取代的六元含氮杂环酮类化合物. 氮杂环卡宾(NHC)催化的分子内氢酰基化反应同样可以选择性进行, 以较高的产率生成α-取代的五元杂环酮, 随后通过镍催化实现五元和六元含氮杂环酮与芳基卤代物的不对称α-芳基化反应构建酮α-季碳手性中心. 此外, 该反应也可以在一锅进行, 以良好的产率和优异的对映选择性得到目标产物.
1.2 酮α位C—H键分子内不对称芳基化反应
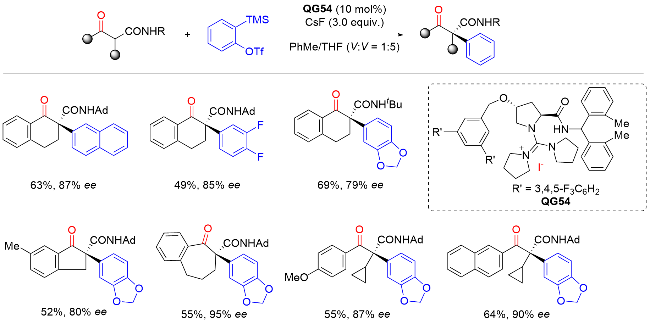
2016年, 贾义霞课题组[14]利用醋酸钯/L-脯氨酸协同催化的去对称化策略, 实现了4-取代环己酮类化合物的分子内不对称α-芳基化反应, 以良好的收率和优异的对映选择性地合成了一系列具有两个手性中心的桥环酮类化合物(Scheme 13). 受产物中桥环刚性结构影响, 酮羰基α-叔碳手性中心在碱性条件下不会发生消旋化, 从而实现了构建叔碳手性中心的不对称羰基α-芳基化反应. 机理研究表明, L-脯氨酸作为催化剂与酮生成烯胺物种, 芳基卤代物与零价钯发生氧化加成生成芳基钯中间体; 该中间体随后对烯胺发生Heck反应, 实现对映选择性羰基α-芳基化. 值得指出的是该反应获得的产物骨架是Morphan类生物碱的重要结构单元.
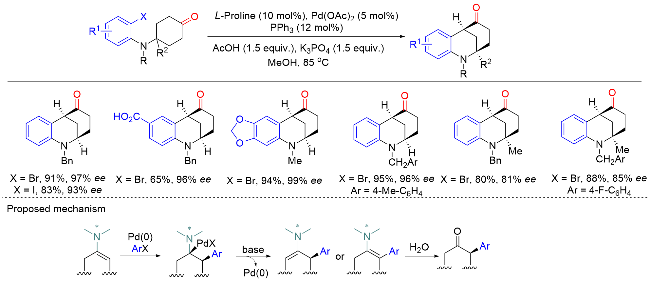
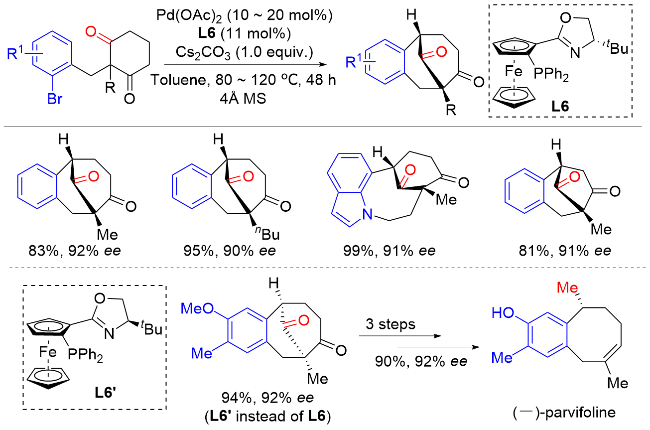
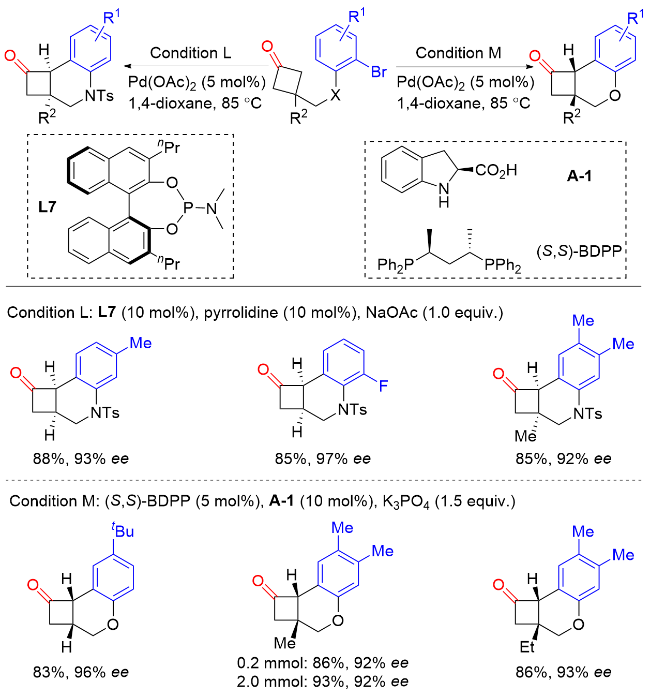
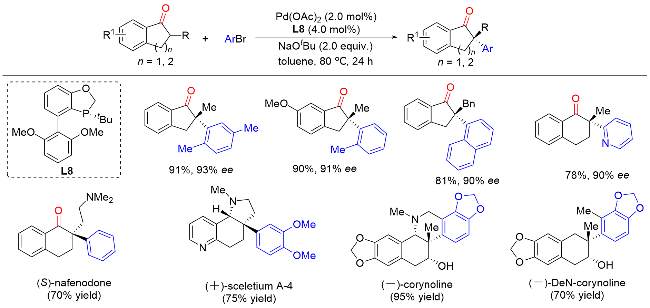
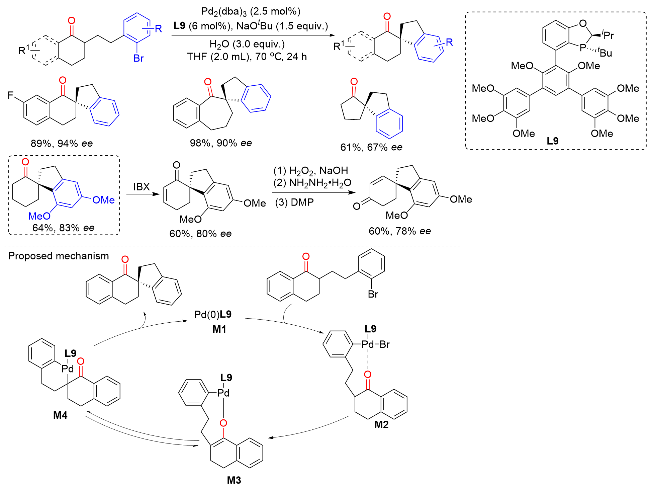
在此基础上, 汤文军课题组[18]进一步开发了一种空间位阻更大且富电子的膦中心手性单膦联芳基配体L9. 使用该配体可以实现环状酮的分子内不对称α-芳基化反应, 从而构建含有α-季碳手性中心的螺环酮类化合物(Scheme 17). 该反应具有优异的对映选择性和良好的底物普适性, 并实现了药物分子(-)-cannabispiren-ones A和B的全合成, 展现了该方法在天然产物全合成和药物开发方面的潜力. 此外, 水的加入可以显著提高反应的对映选择性. 机理研究表明, 零价钯物种M1与芳基溴化物发生氧化加成生成二价钯物种M2. 在碱的作用下, 该中间体通过去质子化发生分子内配体交换生成钯的烯醇中间体M3. M3通过共振生成α-钯酮中间体M4, 该类中间体经还原消除生成最终产物, 零价钯物种M1完成催化循环.
2 外消旋α-芳基酮的不对称官能团化反应
通过对外消旋α-芳基酮的α-碳氢键进行不对称官能团化, 是手性α-芳基酮的另一种催化合成策略. 根据不同的成键类型, 可以分为以下几类.
2.1 外消旋α-芳基酮α位不对称C—C键构建
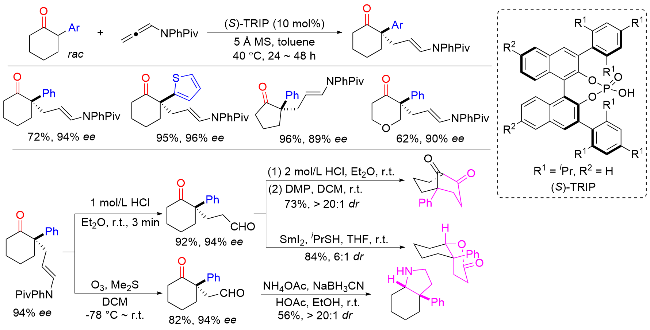
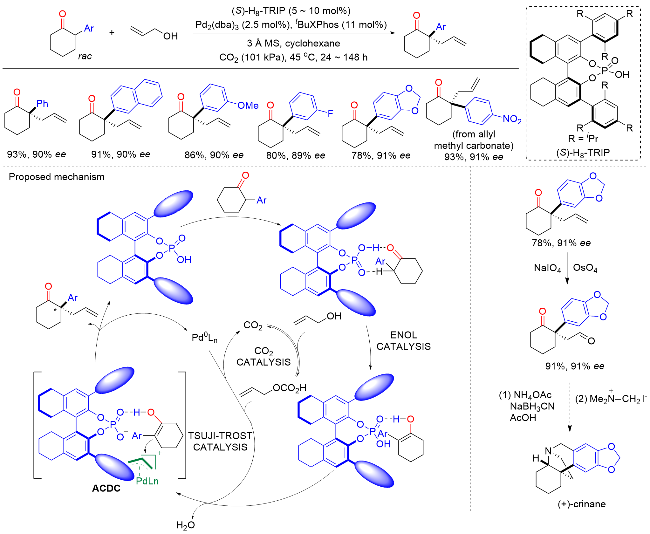
2016年, List课题组[20]开发了一种钯催化α-芳基环状酮与烯丙醇的不对称α-烯丙基化反应, 以优异的对映选择性合成了一系列含有季碳手性中心的α-芳基环状酮产物(Scheme 19). 该方法对各种芳基都具有良好的耐受性, 但底物范围仅限于环状酮. 此外, 该反应收率高, 原子经济性好, 水是唯一的副产物. 反应机理表明, 手性磷酸首先与酮相互作用, 生成烯醇中间体. 同时, 钯催化烯丙醇与二氧化碳反应形成相应的碳酸酯. 随后, 钯与碳酸酯发生氧化加成得到烯丙基钯亲电试剂, 同时释放水和二氧化碳. 最后, 烯丙基钯亲电试剂经过π-烯丙基阳离子的连续亲核进攻、还原消除以及手性磷酸和钯催化剂再生的过程, 得到含季碳手性中心的α-芳基环状酮产物. 此外, 作者以市售的2-苯基环已酮为初始原料, 四步合成了药物分子(+)-crinane, 证明了该方法的实用性.
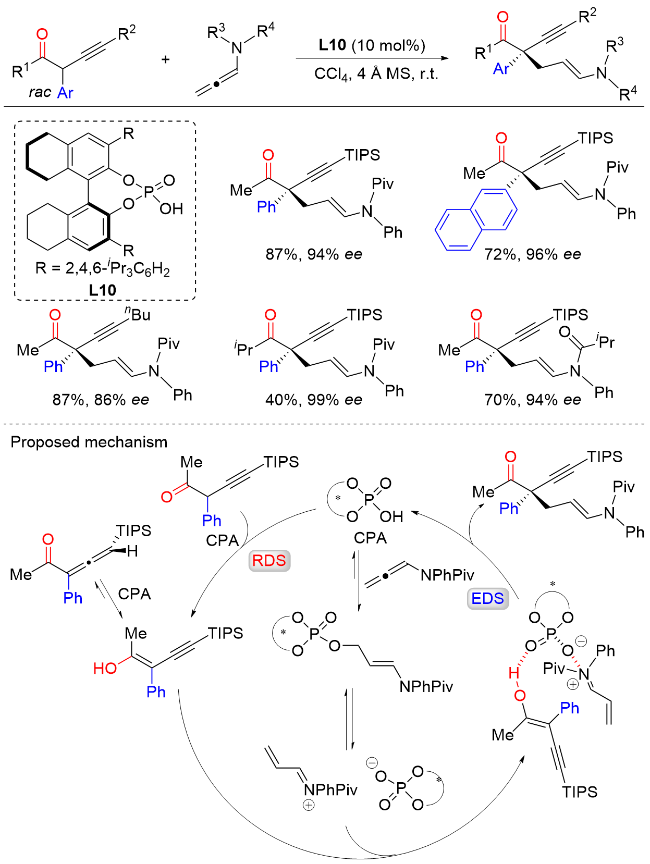
2021年, 杨晓瑜课题组[21]报道了手性磷酸催化外消旋α-炔基-α-芳基酮与N-酰基联烯胺的不对称加成反应, 以优异的区域选择性和较高的对映选择性实现了一系列含有α-季碳手性中心的α-芳基酮类化合物的高效构建(Scheme 20). 该方法对于酮α-位各种取代的芳基、炔基以及烷基都具有较好的适用性, 同时也能兼容各种N-取代芳基以及N-取代酰基. 机理研究表明, 在该反应中, 手性磷酸催化剂具有双重作用, 一是活化α-炔基酮底物生成烯醇中间体, 该过程是反应的决速步(RDS); 二是与N-酰基联烯胺发生加成反应, 生成共价中间体, 随后再发生消除反应生成α,β-不饱和亚胺正离子中间体. 该正离子中间体在手性阴离子诱导下与烯醇中间体发生不对称共轭加成反应, 从而实现产物中非环状手性季碳中心的高效构建, 该步骤是反应的对映选择性决定步骤(EDS). 鉴于反应产物中含有丰富的官能团, 包括酮羰基、炔基和烯酰胺等, 作者也通过实验证明可以利用这些官能团实现一系列具有手性季碳中心的多样性手性骨架的不对称合成.
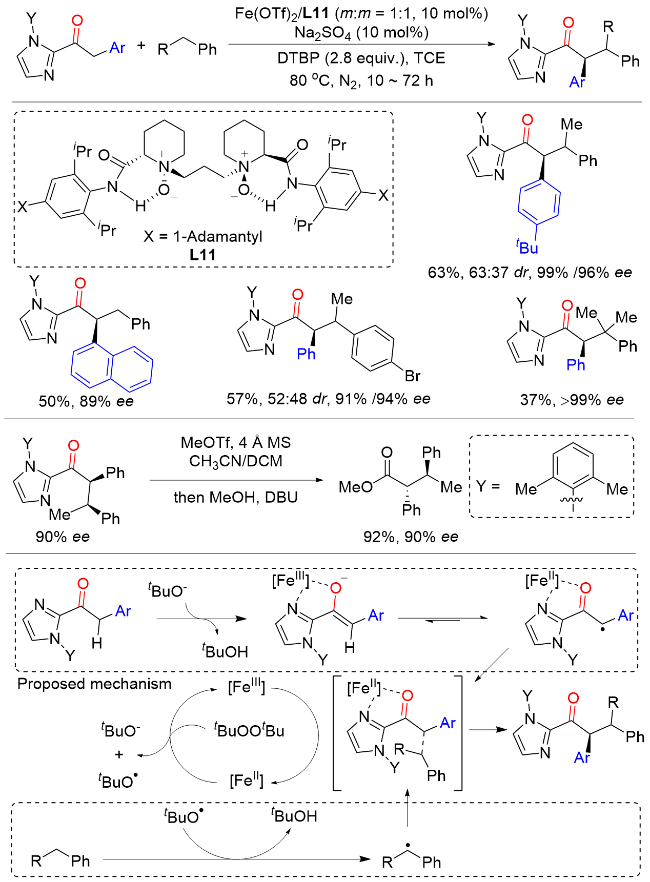
2024年, 冯小明课题组[22]利用独具特色的手性双氮氧配体和廉价易得的三氟甲磺酸亚铁作为催化剂, 二叔丁基过氧化物作为氧化剂, 实现了2-酰基咪唑与非活化烷烃的高对映选择性交叉脱氢偶联反应, 为非环状酮的不对称烷基化提供了一条具有原子经济性和步骤经济性的策略(Scheme 21). 该策略底物普适性较为广泛, 不但适用于各类甲苯衍生物, 对于简单烯烃和惰性的简单环烷烃也能取得良好的结果. 此外, 在保持手性控制的情况下, 能以高收率脱去咪唑基团, 得到含有α手性中心的非环状酯. 遗憾的是, 该反应对于非对映选择性的控制较差, 且底物仅限于咪唑酮类化合物. 机理研究表明, 首先手性铁络合物与二叔丁基过氧化物反应生成Fe(III)物质以及叔丁氧基阴离子和叔丁氧基自由基. 随后, 2-酰基咪唑在叔丁氧基阴离子的作用下进行去质子化和烯醇化, 与Fe(III)物质配位后, 通过配体到金属电荷转移(LMCT)的过程形成自由基. 同时, 叔丁氧基自由基与烷基作用, 苄基C—H键断裂得到自由基. 最后, 通过不对称自由基-自由基偶联得到目标产物.
2.2 外消旋α-芳基酮α位不对称C—N键构建
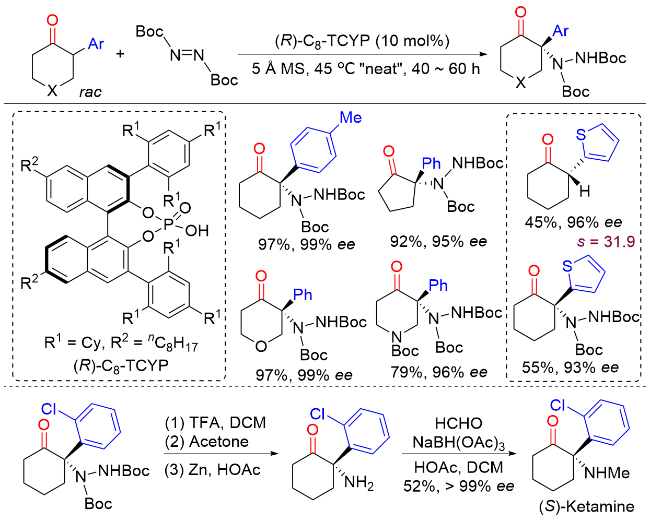
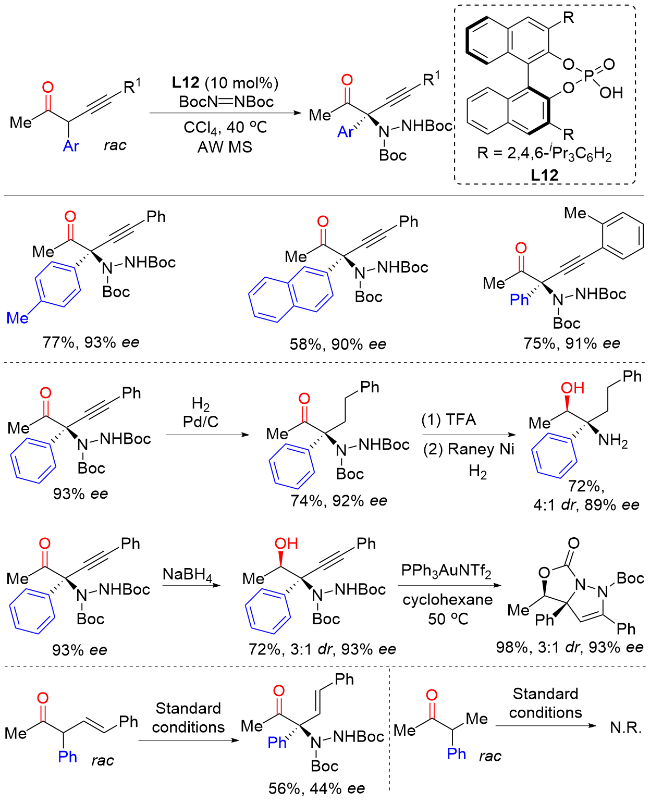
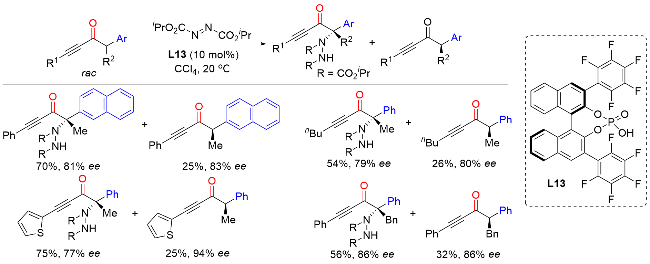
2.3 外消旋α-芳基酮α位不对称C—O键构建
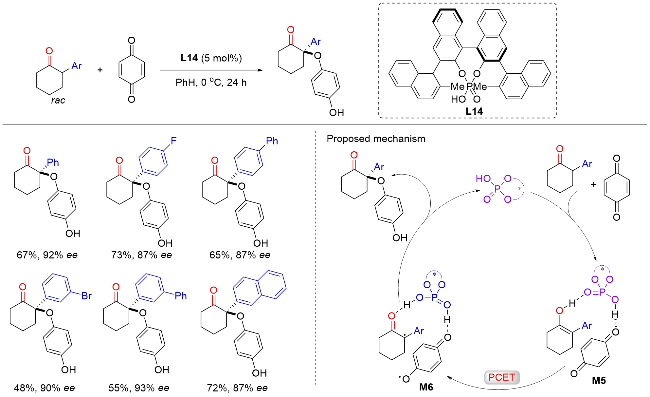
2018年, List课题组[26]报道了手性磷酸催化外消旋α-芳基环状酮与1,4-苯醌的不对称α-芳氧基化反应, 以良好的产率和优异的对映选择性实现了一系列手性α-芳基-α-芳氧基环状酮类化合物的构建(Scheme 25). 该反应可以兼容在芳基间位或者对位含有给电子或吸电子基团的α-芳基酮, 但遗憾的是, 该反应底物范围仅限于六元环状酮以及1,4-苯醌. 初步的机理实验表明, 该反应是通过质子偶合电子转移(PCET)和自由基重组进行的. 催化循环由磷酸催化酮烯醇化引发, 随后, 醌衍生物通过氢键配位得到中间体M5. 中间体M5经过PCET途径生成双自由基配合物M6. 最后通过自由基重组, 形成目标产物以及再生手性磷酸催化剂, 完成催化循环.
3 烯醇的不对称芳基化反应
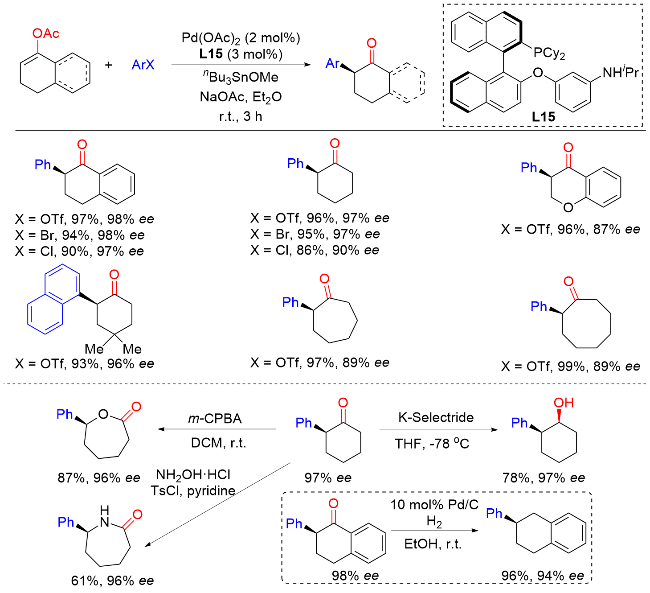
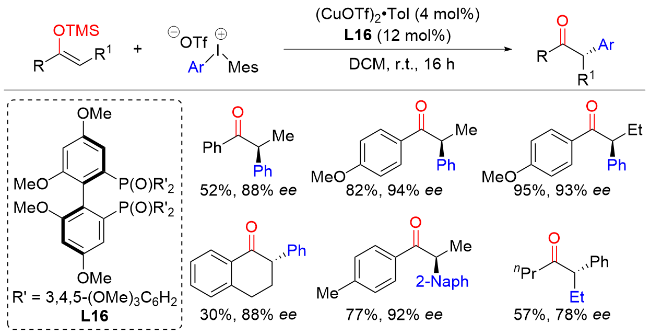
4 α-溴代酮与芳基金属试剂的不对称交叉偶联反应
近年来, 利用过渡金属催化外消旋α-卤代酮化合物与不同芳基金属试剂的不对称交叉偶联反应实现手性α-芳基酮类化合物的构建已成为研究的热点.
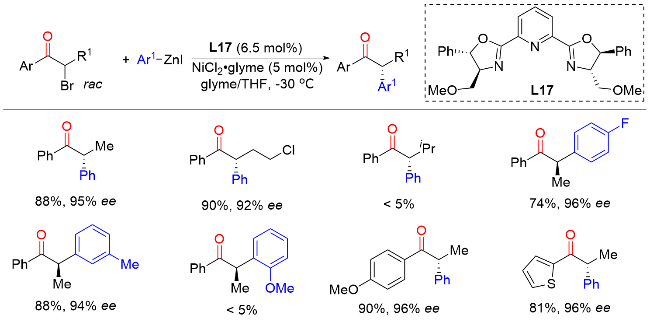
2010年, Fu课题组[30]报道了镍催化烷基亲电试剂的不对称Kumada反应, 即外消旋α-溴代酮与芳基格氏试剂的不对称偶联反应, 以较高的产率和优异的对映选择性直接构建了一系列手性α-芳基酮类化合物(Scheme 29). 该方法对于简单烷烃、卤化物、叠氮、酯或杂环等底物均有良好的耐受性. 此外, 手性α-芳基酮产物可以转化为相应的手性醇和手性胺, 表明了该反应的实用性. 2019年, 该课题组[31]还探索了反应机理, 为自由基链途径提供了进一步的支持. 机理研究表明, 首先NiI中间体从亲电试剂中攫取一个卤原子, 生成烷基自由基和NiII中间体. NiII中间体与亲核试剂反应生成芳基NiII中间体. 随后, 烷基自由基与芳基NiII中间体发生自由加成反应得到NiIII中间体. 最后, NiIII中间体还原消除得到目标产物, 重新生成NiI中间体完成催化循环.
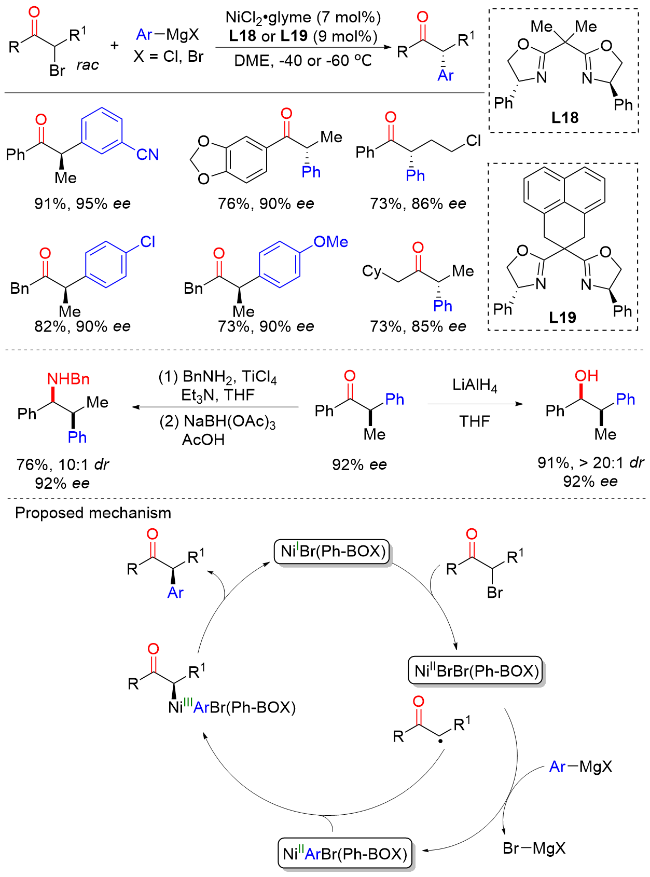
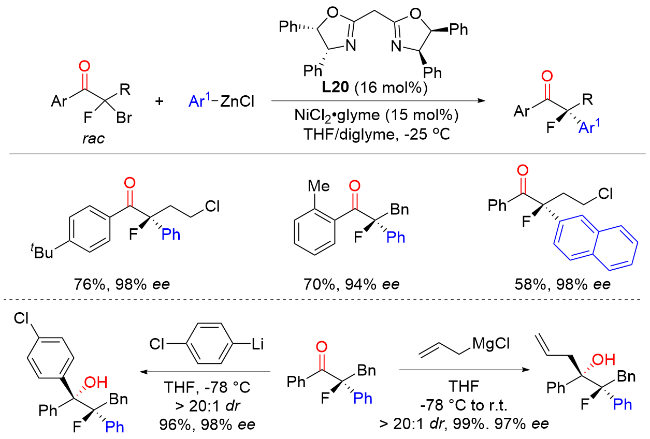
2023年, 陆展课题组[33]报道了钴催化外消旋α-溴代酮与芳基锌试剂的不对称交叉偶联反应, 以较高的产率和优异的对映选择性实现了一系列含有α-叔立体中心的手性α-芳基酮类化合物的构建(Scheme 31). 在该反应中, 具有空间位阻的2-氟苯基、β-仲烷基以及叔烷基链的α-溴酮都可以很好地兼容. 作者发现, 调整手性不对称N,N,N-三齿配体的电子效应对于提高该转化的反应性和选择性至关重要. 对照实验和动力学研究表明, 反应涉及自由基中间体, 还原消除是速率决定步骤. 机理研究表明, 钴(II)前催化剂在体系中被锌试剂还原为CoI物种M7, 然后与外消旋α-溴代酮进行单电子氧化加成生成自由基M8和CoII中间体M9. 该CoII中间体M9与芳基锌试剂进行配体交换生成芳基CoII物种M10. 随后, 芳基CoII物种M10被自由基M8捕获发生自由基加成反应得到CoIII中间体M11. 最后, 还原消除生成最终产物并再生CoI物种M7, 完成催化循环.
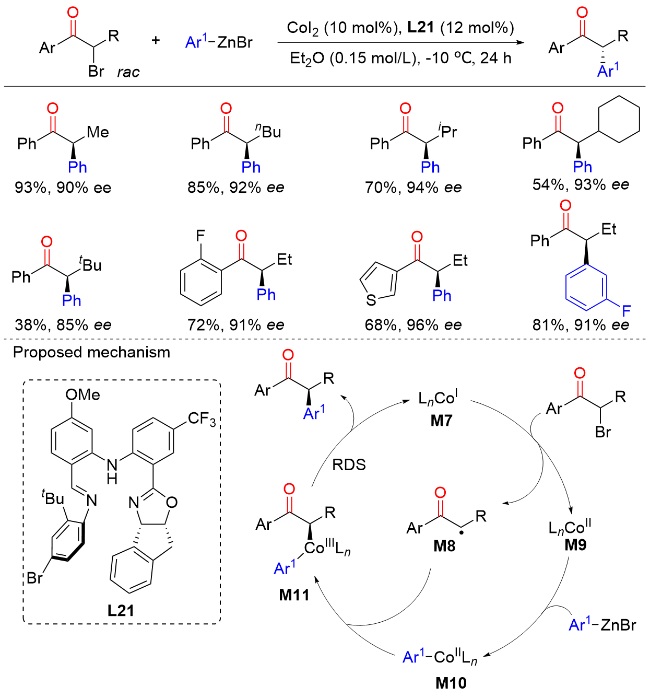
5 酰基亲电试剂的不对称苄基化反应
此外, 使用不同类型的酰基亲电试剂(如酰氯、醛、硫酯、羧酸和酸酐等)进行不对称的苄基化反应是高效构建手性α-芳基酮类化合物的另一种策略.
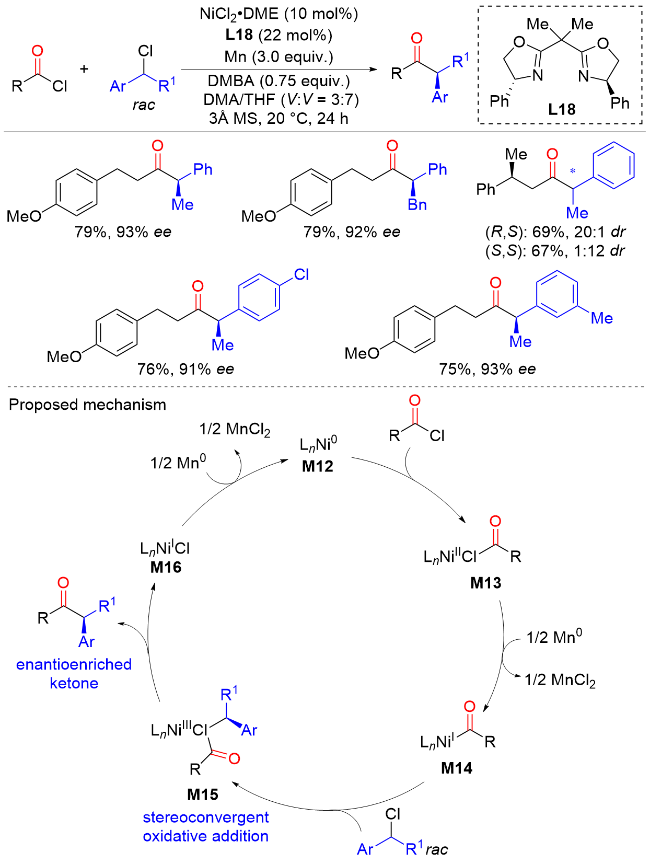
2013年, Reisman课题组[34]报道了以锰粉为还原剂, 镍与双(噁唑啉)配体催化外消旋苄基氯化物与酰氯的不对称交叉偶联反应, 实现了一系列手性α-芳基-α-烷基酮的快速构建(Scheme 32). 该反应具有优异的产率和对映选择性, 且不需要化学计量手性助剂或预生成有机金属试剂, 具有良好的底物兼容性和官能团耐受性. 但遗憾的是, 含有邻位取代基的苄基氯反应效果较差, 得到的酮产物产率和ee值都很低. 机理研究表明, Ni0物种M12与酰氯进行氧化加成可生成NiII酰基中间体M13, 该中间体被Mn还原生成NiI酰基化合物M14. 随后, 与外消旋的苄基氯化物进行氧化加成反应, 生成NiIII中间体M15. 这一步的机制类似于镍催化的仲烷基卤化物和有机金属试剂之间的立体会聚式交叉偶联反应. 随后, NiIII中间体M15还原消除生产手性α-芳基-α-烷基酮产物与NiI络合物M16. 最后, NiI络合物M16被Mn还原生成Ni0物种M12完成催化循环.
2019年, Melchiorre课题组[37]报道了可见光介导镍催化4-烷基二氢吡啶和对称酸酐的不对称酰基化偶联反应(Scheme 35). 该反应不需要外源光催化剂, 由4-烷基二氢吡啶化合物直接引发, 以良好的产率和优异的对映选择性实现了一系列手性α-芳基酮的合成. 其中, 4-烷基二氢吡啶作为该反应的自由基源和还原剂, 促进手性催化镍络合物的转化是该反应中极为重要的部分. 机理研究表明4-烷基二氢吡啶在光照作用下到达激发态M17, 激发态M17通过两次单电子转移过程将NiⅡ还原为Ni0中间体M18, 同时产生二级自由基M19. Ni0中间体M18对酸酐进行氧化加成产生NiⅡ-酰基复合物M20, 随后自由基M19与M20相互作用产生NiⅢ中间体M21. 最后M21经还原消除产生手性偶联产物与NiⅠ中间体M22, 最后再经单电子转移过程再生Ni0催化剂.
2021年, 霍浩华课题组[38]报道了镍和光氧化还原双重催化烷基芳烃与原位活化羧酸的不对称苄位C—H键酰基化反应, 以良好的产率和优异的对映选择性实现了一系列手性α-芳基酮的合成(Scheme 36). 该反应具有广泛的底物范围和出色的官能团耐受性, 底物包括卤代物、醚、腈、酯、烯烃以及各种杂芳烃等, 同时也可以应用于药物相关分子的后期修饰. 机理研究表明, 催化反应是通过Ni0催化剂M23与原位活化的羧酸氧化加成生成NiII中间体M24而引发的. 随后捕获由溴自由基介导的氢原子转移(HAT)过程产生的前手性苄基自由基, 提供NiIII配合物M25, 配合物M25经过还原消除生成所需产物和NiI中间体M26. 最后, NiI中间体M26和还原光催化剂之间的单电子转移(SET)使Ni0催化剂M23和基态光催化剂再生, 从而完成两个催化循环. 其中, 还原消除是立体化学的决定步骤.
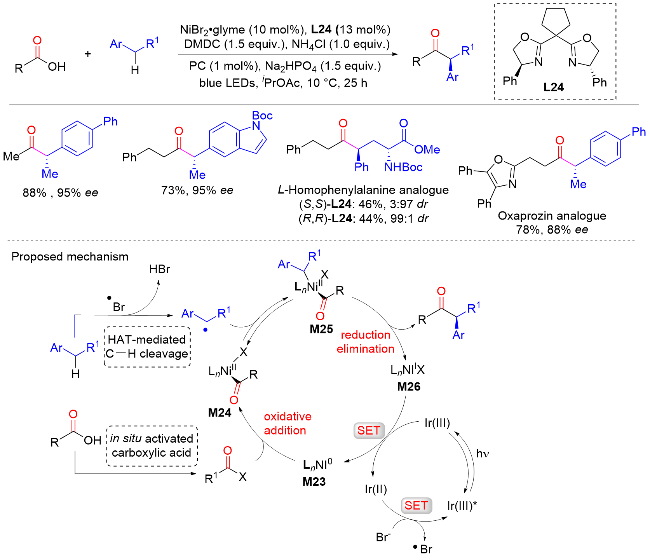
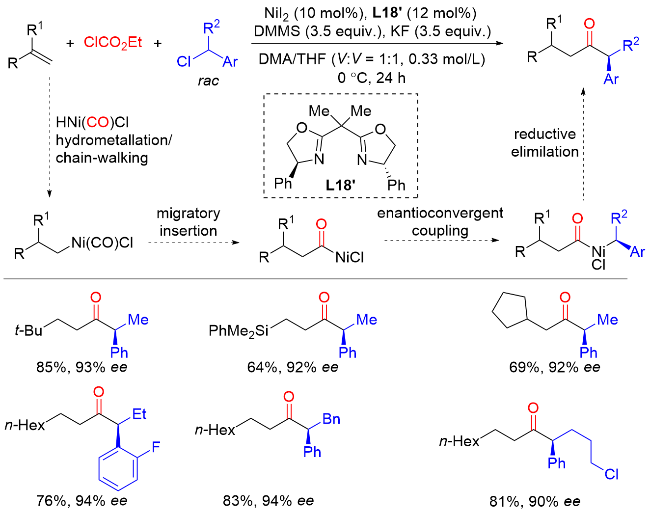
2021年, 朱少林课题组[39]在镍催化下, 以手性双噁唑啉作为配体, 硅烷作为还原剂, 氯甲酸酯作为安全的CO源, 外消旋仲苄基作为烷基化试剂, 实现了未活化烯烃还原氢羰基化反应, 合成了一系列功能化的手性α-芳基酮类化合物(Scheme 37). 该方法可以兼容多种有用的官能团, 包括酯、酰胺、缩醛、醚、卤素、硼酸酯、氰、酮和硅烷等, 并且可应用于一些药物分子的后期修饰. 机理研究表明, 原位生成的CO可以被X— Ni(II)—H捕获, 然后进行氢金属化和随后的CO插入, 生成酰基镍(II)配合物, 避免产生过量的CO使催化剂失活. 最后与外消旋仲烷基亲电试剂(如苄基氯)进行对映选择性的交叉偶联反应.
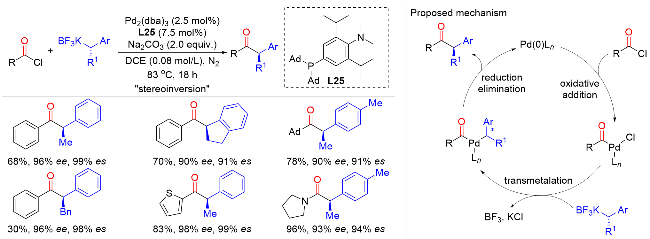
2023年, Lee课题组[40]报道了钯与膦配体催化酰氯或氨基甲酰氯与苄基三氟硼酸盐的立体定向suzuki-miyaura交叉偶联反应, 以较高的收率和优异的对映选择性制备了一系列有价值的手性α-芳基酮类化合物(Scheme 38). 该反应具有良好的底物普适性和官能团兼容性, 适用于各种不同类型的三氟硼酸酯、芳基酰氯、杂芳基酰氯以及烷基酰氯. 作者通过密度泛函理论计算提出了以下的反应机理: 首先, 酰氯底物与配位后的Pd(0)Ln发生氧化加成, 进一步与苄基三氟硼酸盐底物发生转金属化, 最后通过还原消除重生钯催化剂. 其中, 使用具有较大空间位阻和富电子的膦配是该反应成功的关键. 此外, 作者在过渡态中发现了一种独特的金 属−卤素相互作用, 有效地降低了反应势能, 促进转金属化反应的发生.
6 芳基烯烃的不对称氢酰基化反应
烯烃是一类广泛易得的基础化工原料. 近年来, 烯烃的催化不对称氢酰基化反应得到了快速发展. 其中, 芳基烯烃的不对称酰基化反应成为高效构建手性α-芳基酮类化合物的热门方法.
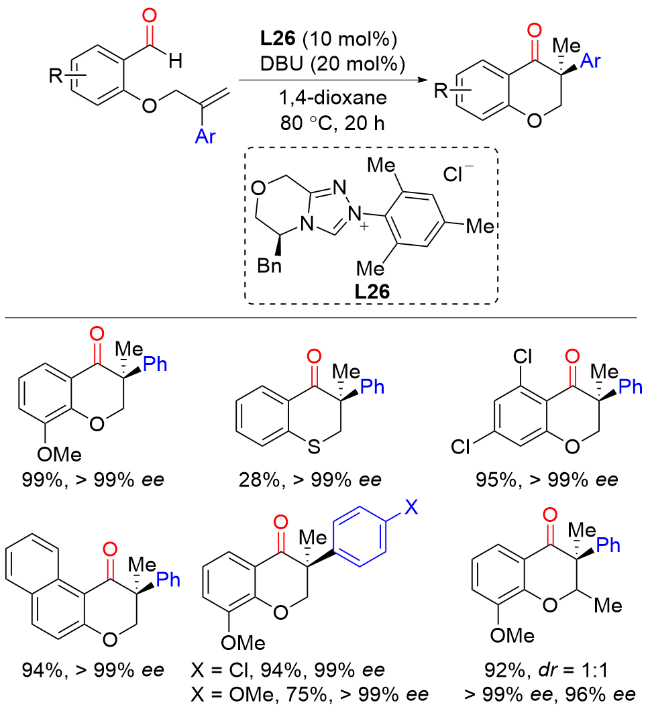
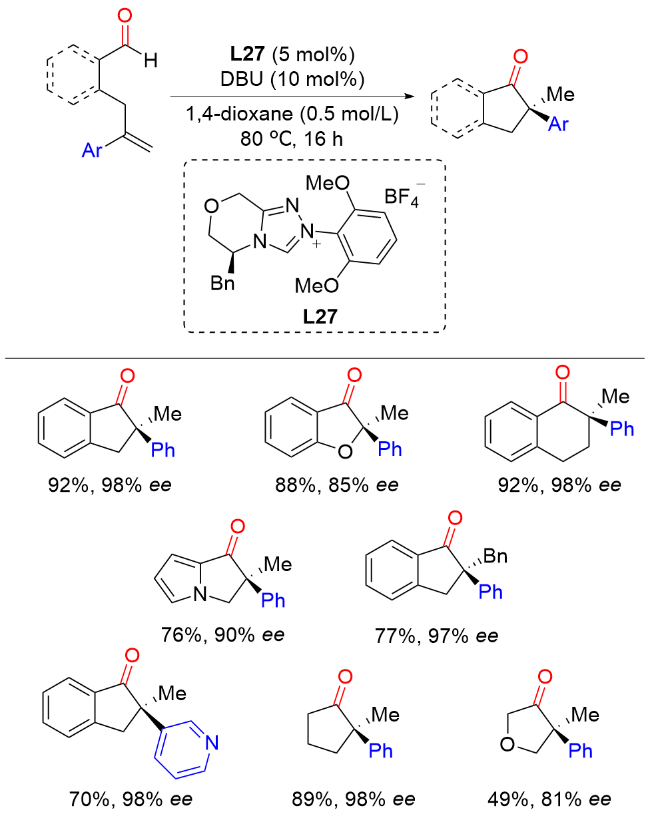
2016年, Buchwald课题组[44]报道了铜催化芳基烯烃与芳基羧酸酐的不对称还原偶联反应, 以较好的产率和优异的对映选择性实现了一系列手性酮或醇类化合物的构建(Scheme 42). 值得注意的是, 该反应以相对快速的对映选择性氢酰化和较慢的非对映选择性酮还原的双催化循环完成. 作者提出了可能的催化循环: 首先通过L*CuH物种M27与芳基烯烃进行不对称马氏氢金属化生成手性苄基铜中间体M28, 然后与酰基亲电试剂进行亲电取代反应, 产生手性酮产物M29和L*CuX物种M30. L*CuX物种M30需与硅烷反应再生L*CuH物种M27. 在适当条件下, L*CuH物种M27可在还原循环中对手性酮产物M29进行高度非对映选择性还原, 生成手性硅基醚产物. 理想情况下, 这两种催化过程可以在同一反应容器中与共同的亲电试剂进行, 从而根据所采用的反应条件实现手性酮或醇产物的快速合成.
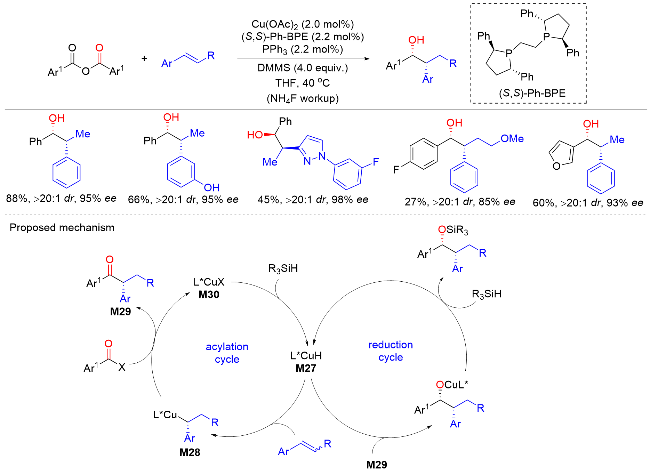
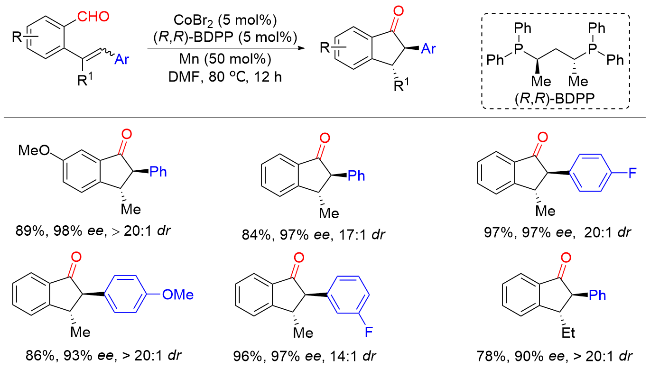
2021年, 戴辉雄课题组[47]报道了以硫酯为酰基亲电试剂, tBuNC为氨基甲酰基试剂, 钯催化烯烃的不对称酰基氨基甲酰基化反应. 以良好的产率和优异的对映选择性合成了一系列含α-季碳手性中心的环状酮类化合物(Scheme 45). 该方法条件温和, 底物范围广, 可以很好地构建含有五元环和六元环的α-芳基酮. 机理研究表明, Pd0物种M31与反应物进行氧化加成反应生成酰基PdII中间体M32. 然后酰基PdII中间体M32在烯烃中的分子内迁移插入与C(sp3)-Pd物种形成α-手性季环状酮M33, 其随后通过1,1-迁移插入tBuNC形成中间体M34. 中间体M34水解得到中间体M35, 最后发生还原消除并进一步互变异构以生成最终产物.
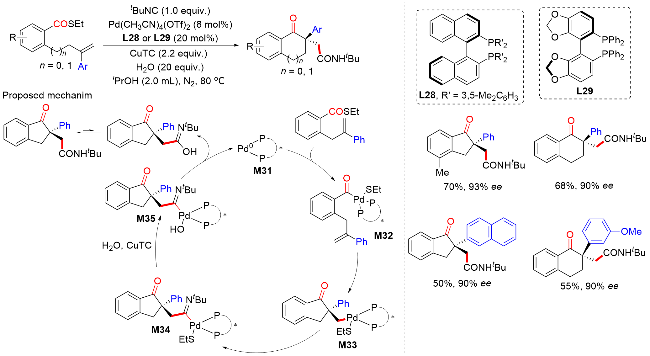
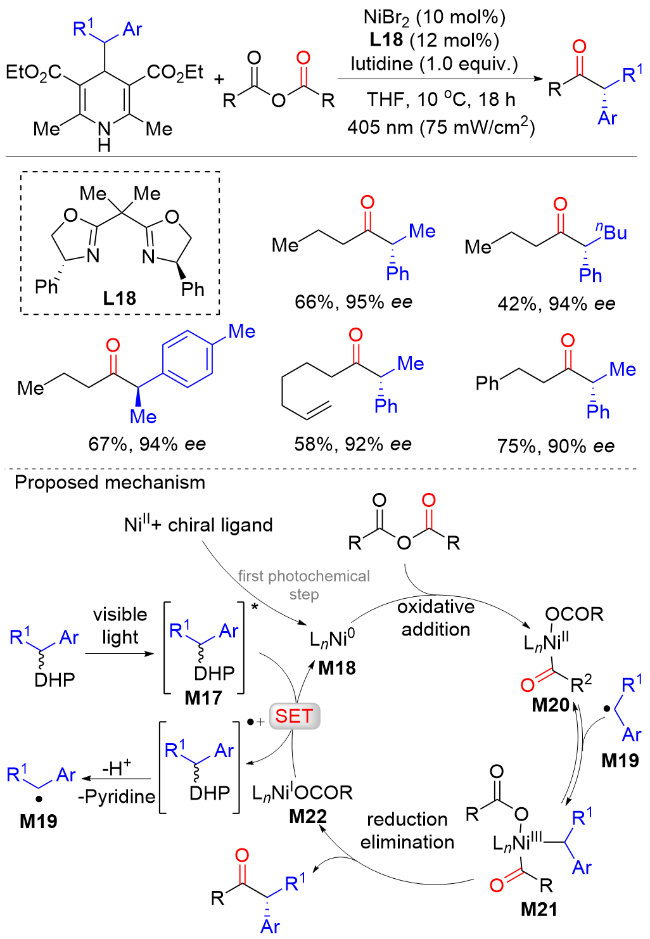
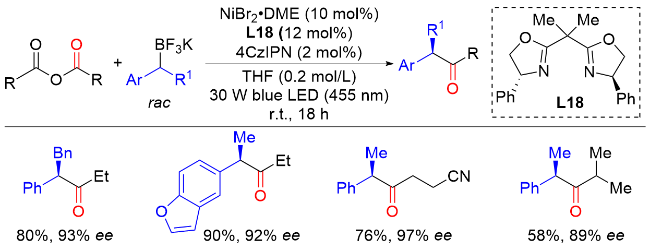
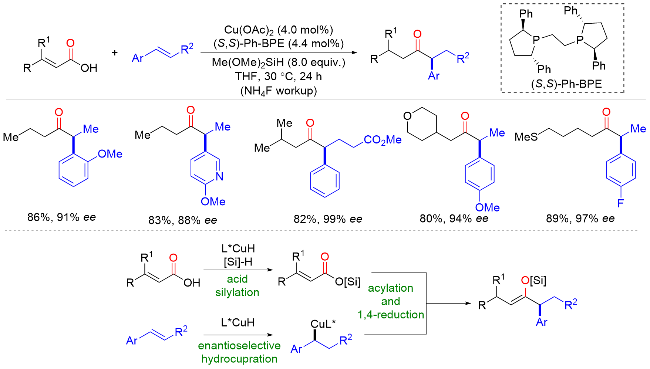
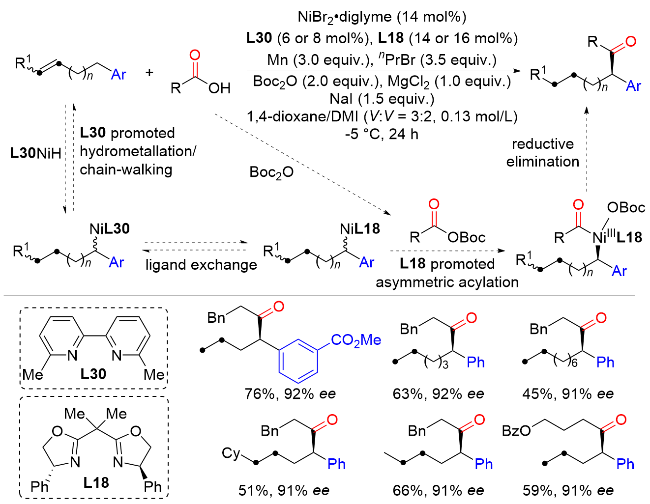
2022年, 朱少林课题组[48]提出了配体接力催化策略, 通过组合两个不同类型的配体来分别促进串联反应中的不同步骤, 不同步骤间通过中心金属动态的配体交换来实现催化接力, 成功实现了镍氢催化下优异区域和对映选择性的远程不对称酰基化反应合成手性α-芳基酮(Scheme 46). 该反应从丰富的起始原料烯烃和羧酸开始, 在温和的条件下有效地获得各种手性α-芳基酮化合物, 具有广泛的底物范围和出色的官能团容忍性, 兼容醚、卤素、酯、硼酸酯、醛, 以及各种杂环, 如呋喃、噻吩、吡啶和吲哚. 机理实验表明, 配体L30控制镍氢链行走, 而配体L18控制该反应的不对称酰基化过程. 该双配体接力策略为发展新的不对称催化体系提供了新的解决思路.
7 其他类型反应
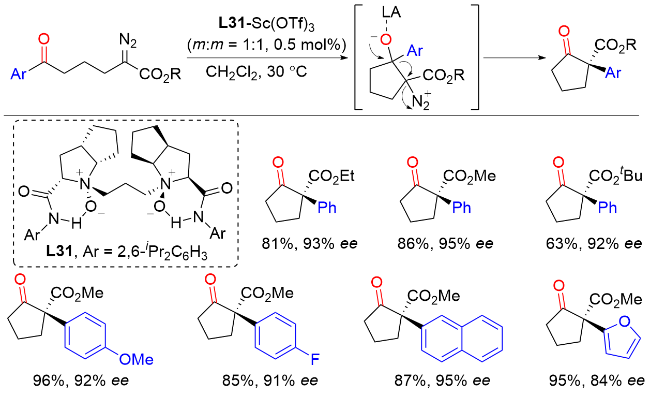
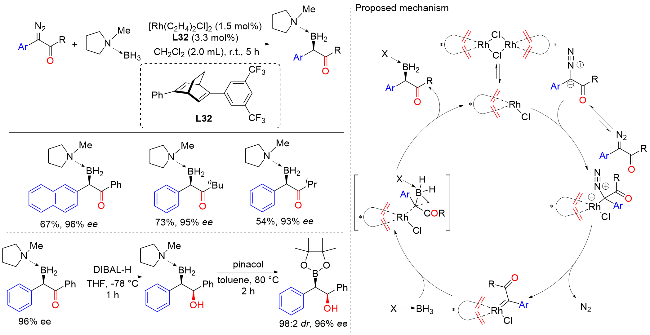
2015年, 徐明华课题组[50]报道了铑催化α-重氮羰基化合物的不对称B—H键插入反应, 高效合成了各种含α-羰基的手性有机硼烷(Scheme 48). 其中, 新开发的C1-对称手性双环[2.2.2]辛二烯配体在Rh(I)催化的C—B成键反应中表现出了显著的效率, 从而实现了较高的对映选择性. 此外, 该类有机硼烷化合物是有机合成中用途广泛且潜在的中间体, 可通过相应的转化得到具有两个手性中心的β-硼醇化合物, 具有良好的非对映选择性和对映选择性. 反应机理表明, 双(铑/二烯)配合物解离生成活性单铑催化剂, 该催化剂与重氮化合物反应生成Rh(I)-类羰基中间体. 这种高活性类羰基化合物通过协调的过渡态插入到胺硼烷加合物的B—H键中, 然后得到相应的有机硼烷产物并再生催化剂.
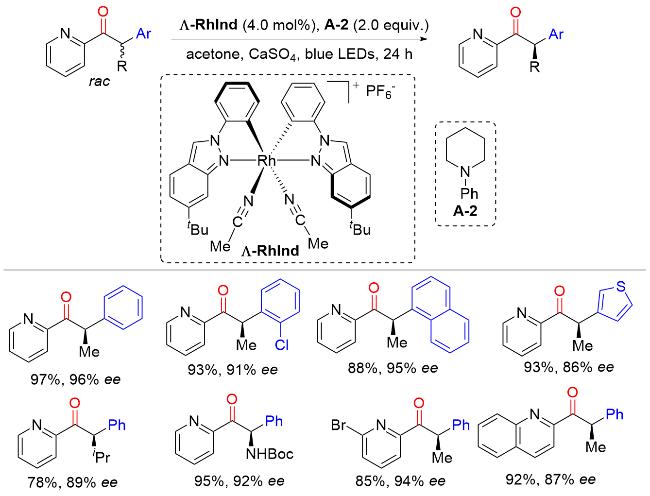
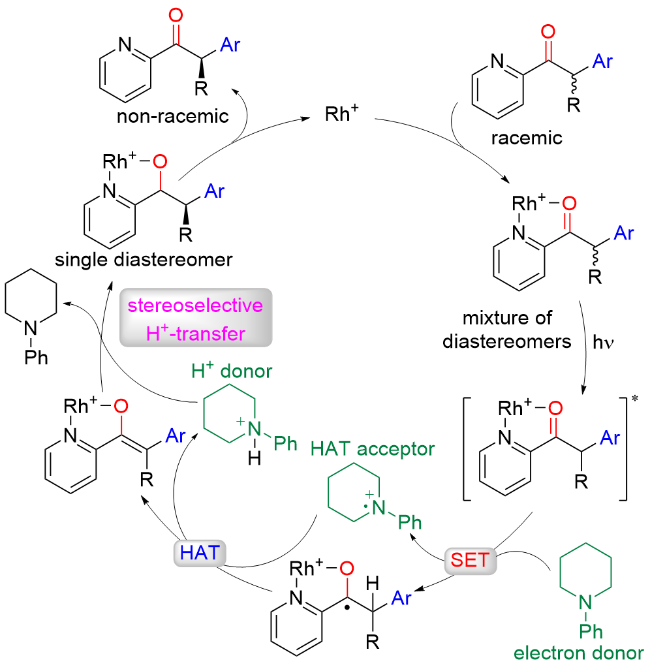
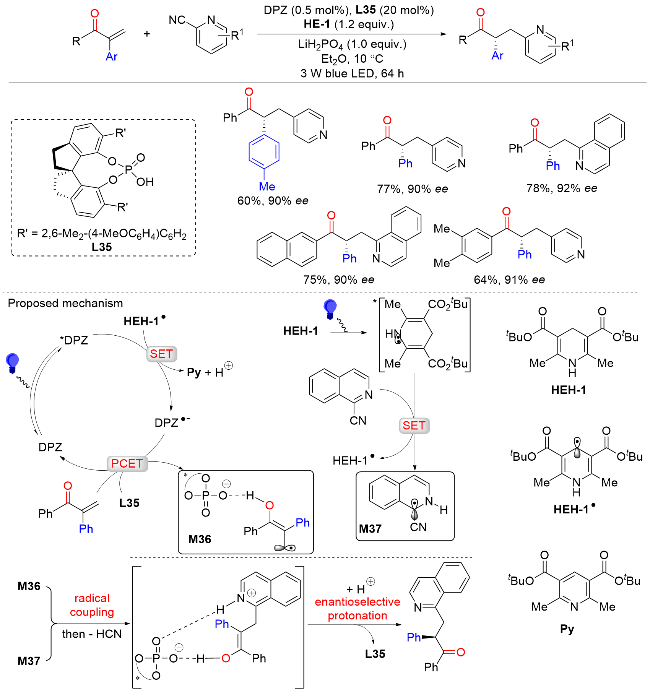
2021年, Meggers课题组[53]报道了在光催化剂和简单叔胺的存在下, 将外消旋α-芳基吡啶酮通过去外消旋化的策略转化为手性α-芳基吡啶酮(Scheme 51). 该反应具有较好的底物范围, 适用于各种芳基、烷基以及吡啶取代的α-芳基吡啶酮. 此外, 喹啉也可以替代吡啶, 以92%收率及87% ee值得到相应的目标产物. 遗憾的是, 使用吡唑或咪唑代替吡啶时不能得到相应的手性酮化合物. 机理实验和密度泛函理论计算表明, 该反应通过光氧化去质子化生成烯醇中间体, 然后再进行不对称质子化. 其中, 手性铑催化剂具有光氧化还原催化剂和手性路易斯酸催化剂的双重功能, 叔胺催化剂在催化循环中具有单电子还原剂、氢原子受体和质子源等三重作用(Scheme 52).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
8 总结与展望
综上所述, 手性α-芳基酮作为一种重要结构单元, 其催化不对称合成已经受到广泛重视并取得了重大发展. 本文针对催化合成手性α-芳基酮策略进行了总结, 包括酮α位C—H键不对称芳基化反应、外消旋α-芳基酮的不对称官能团化反应、烯醇的不对称芳基化反应、α-溴代酮与有机芳基金属试剂的不对称交叉偶联反应、酰基亲电试剂的不对称苄基化反应及芳基烯烃的不对称氢酰基化反应等. 这些反应策略可以实现从简单易得的原料出发, 经历不同的反应历程构建手性α-芳基酮类化合物, 显示出了巨大的合成潜力以及广阔的应用前景. 手性α-芳基酮的催化合成存在的主要局限和挑战包括: (1)反应类型相对单一, 更多类型的反应尚未得到充分开发; (2)反应需要使用预先生成的有机金属试剂或其它相对复杂的反应原料等. 因此该领域仍有待进一步发展, 未来的研究重点将集中在以下几个方面: (1)发展新的催化合成策略来实现手性α-芳基酮的多样化合成, 提高反应的效率和对映选择性; (2)发展从简单易得的原料出发, 快速、高效地合成手性α-芳基酮的催化方法; (3)结合多种催化策略, 如光催化、电催化和金属催化等策略来实现手性α-芳基酮的高效合成; (4)深入研究反应机理, 并推进α-手性芳基酮类化合物在目标分子合成、生物医药及功能材料分子等方面的应用.
(Zhao, C.)