


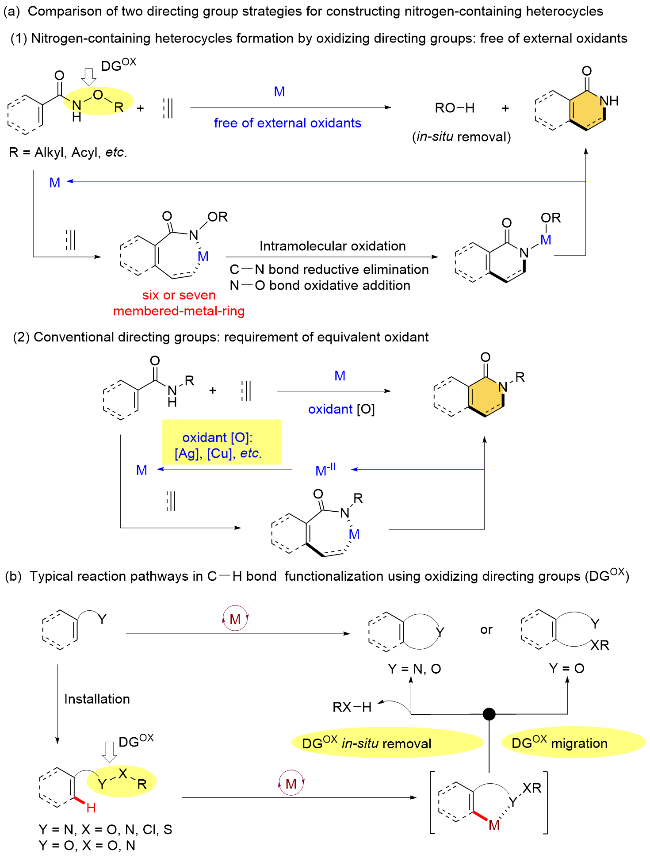
C—H键是自然界中最常见的化学键之一, 普遍存在于有机化合物中. C—H键的选择性活化官能团化反应策略可以为药物、材料等功能分子的逆合成提供全新的思路, 并有望精简传统有机合成路线, 目前已经成为构建复杂目标功能分子的一条捷径[1]. 总结已有的C—H键活化反应文献可以发现, 该领域仍存在以下两个问题: 首先, 由于分子中通常存在多个化学环境相似的 C—H键, 一般需要在底物中引入导向基团(DG), 通过与金属催化剂的配位导向来选择性活化特定C—H键[2]. 然而, 导向基团的引入不仅限制了产物结构的多样性, 而且通常在反应后还需要额外的脱除步骤. 因此, 如果能使底物中的导向基团在反应后原位去除, 或者参与到 C—H键转化, 并且进入最终的产物分子, 无疑将大幅提高反应效率. 其次, 许多过渡金属催化的C—H键官能化反应需要加入等物质的量的氧化剂(如金属试剂、过氧化物等)用于实现催化循环中电子的守恒[3]. 这些氧化剂的加入增加了反应变量, 不利于反应条件的探索, 使反应体系复杂化, 并影响反应的收率与分离难度. 更重要的是, 许多手性配体与催化剂无法兼容强氧化条件, 也在一定程度上限制了相关不对称C—H键官能团化的反应类型与适用范围.
为了解决上述问题, 研究人员发展了许多重要的策略. 其中, 氧化型导向基团策略(Oxidizing directing groups, DGox)提供了一种较为理想的解决思路[4]. 它既可以作为导向基团来实现特定碳氢键的选择性活化, 还可以作为“分子内氧化剂”驱动过渡金属的催化循环. 因此, 该策略不仅使得导向基团在反应后自动脱除(或参与到产物骨架的构建), 同时也避免了额外氧化剂的使用. 吴养洁和崔秀灵课题组[5]率先发展了该策略, 在无需外加氧化剂的条件下实现了喹啉N-氧化物与烯烃的氧化型Heck交叉偶联反应. Fagnou和Glorius等课题 组[6]将该策略应用于异喹诺酮类化合物的合成中, 通过 C—H键活化一步实现苯甲酰胺类衍生物与炔/烯烃等不饱和碳氢化合物的环化反应(Scheme 1, a-1). 与常规的非氧化型导向基团相比(Scheme 1, a-2), 氧化型导向基团策略避免了外部氧化剂的使用, 且在完成分子内氧化剂功能后自动脱除, 使得反应体系更加简洁. 在以上先驱工作报道后, 氧化型导向基团策略获得了广泛关注和快速发展, 除了上述N—O键[7]可作为分子内氧化剂之外, 包含其他化学键(如N—N键[8]、N—S键[9]、N—Cl键[10]、O—O键[11]、O—N键[12]、C—N键[13]及C—S 键[14]等)的氧化型导向基团也相继被报道, 并且在过渡金属(Rh、Ru、Co、Pd等)催化C—H键活化转化中应用广泛, 主要反应类型包括导向基团参与的成环反应, 以及官能团迁移插入反应等, 为复杂功能分子的构建提供了一种高效的策略(Scheme 1, b)[15].
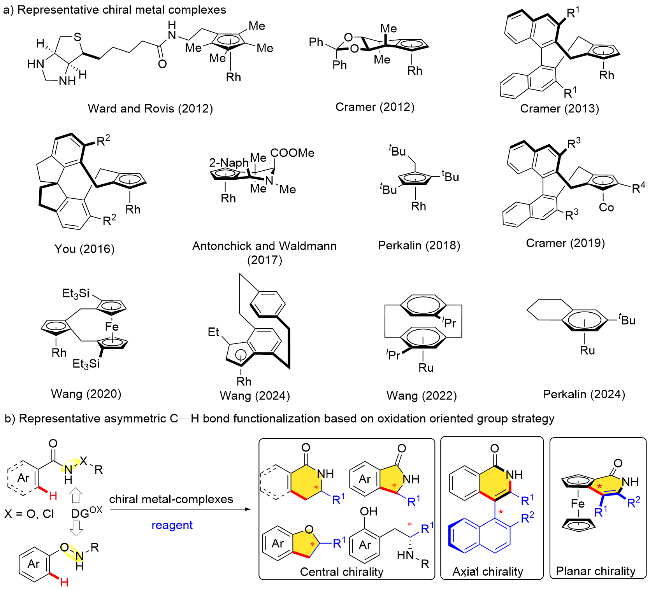
另一方面, 手性是天然化合物的重要特性之一, 具有光学活性的杂环化合物除了是重要的有机合成中间体, 还广泛存在于天然产物分子、药物分子以及生物大分子骨架中. 因此, 如何简单、高效、快速地获得手性杂环化合物一直是合成化学和药物化学等领域的研究热点[16]. 近年来, 过渡金属催化的不对称C—H键官能团化反应引起了化学家们的广泛关注, 已成为有机合成化学最前沿的研究方向之一[17]. 其中, 基于氧化型导向基团策略的不对称C—H键官能团化反应, 由于具有上述多种合成优势而成为该研究领域的一个重要分支.
总的来说, 开发新型的手性金属催化剂是该领域研究的重点内容(Scheme 2, a). 早在2012年, Ward、Rovis以及Cramer等[18]就分别发展了几例新型的手性环戊二烯铑配合物, 利用苯甲酰胺衍生物分子中N—O键片段作为分子内氧化型导向基团, 高对映选择性地实现了不对称C—H键活化反应, 快速构建了相对复杂的手性分子骨架. 自2012年以来, 特别是近五年来, 一些新型手性配体和金属催化剂被设计合成出来[17b,19], 为含氧化型导向基团底物的不对称转化反应提供了优异的手性催化剂. 目前, 相关的不对称C—H键活化反应发展迅速, 已经先后实现了轴手性、中心手性以及平面手性等多种手性元素的高效构建[19s-19t,20](Scheme 2, b). 此前已有一些综述性文章从其他角度(比如过渡金属催化剂、手性配体等)对其中部分反应进行了总结和评述[19], 但专门针对基于氧化型导向基团策略的不对称C—H键转化反应仍缺少较为全面的综述类报道. 鉴于近年来该领域的快速发展及其在手性功能分子高效构建中的重要性, 亟需对相关不对称转化反应体系进行适时总结, 分析其存在的问题和挑战, 并对未来研究方向进行展望. 本综述将详细介绍自2012年首例氧化型导向基团参与的不对称C—H键转化报道以来的相关研究工作, 根据氧化型导向基团类型分不同小节进行陈述, 而在每个小节中将根据反应底物以及反应类型分别进行介绍和评述, 相信这将为新型不对称C—H键官能化反应策略的发展提供有益的参考.
1 N—O键氧化型导向基团参与的不对称C—H键官能团化反应
含有N—O键的氧化型导向基团发展最早, 也是目前反应类型最丰富的氧化型导向基团之一. 本小节主要讨论含N—O键氧化型导向基团参与的底物C—H键活化与烯烃、炔烃或者重氮化合物的不对称转化反应. 在手性金属配合物的催化下, 上述转化通过分子间或分子内的不对称转化可以构建复杂手性含氮杂环化合物(如异喹诺酮、吲哚酮及二茂铁并吡啶酮等).
1.1 不对称C—H键官能团化/[4+2]环加成反应
1.1.1 N-取代苯甲酰胺衍生物与烯烃的分子间[4+2]环加成反应
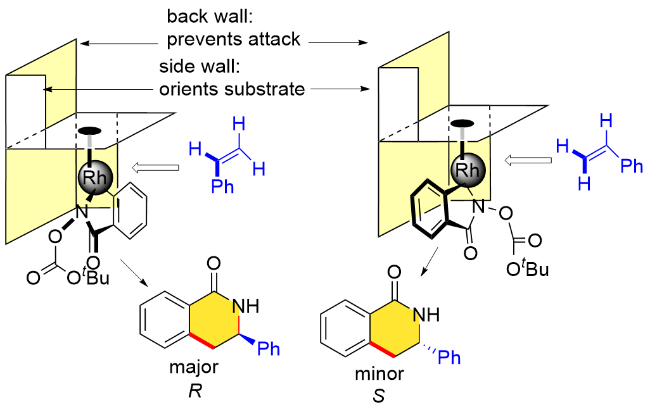
作者还给出了该不对称环化反应中可能的立体控制模型(图1). 首先, Cpx配体的手性环境控制着环金属化过程中底物的朝向, 配体的后壁则迫使烯烃从前方进攻, 从而有利于生成R构型产物.
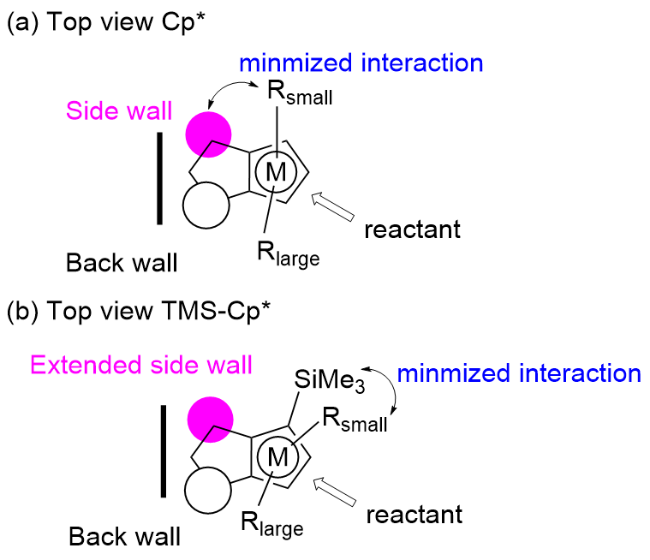
上述两个工作开启了手性单茂铑催化剂在氧化型导向基团参与的C—H键不对称转化中的先河. 在后续研究中, 科学家们进一步优化催化剂, 发现通过调节Cp配体的电子和空间位阻特性可以显著提高手性金属催化剂的反应性能. 比如, 作者将位阻较大的三甲基硅基(TMS)引入到Cp配体中, 使得“侧壁”得到延伸, 尽管对手性骨架影响不大, 但它对烯烃分子进攻环金属中间体的方向起着重要的作用(图2), 从而提高反应的对映选择性.
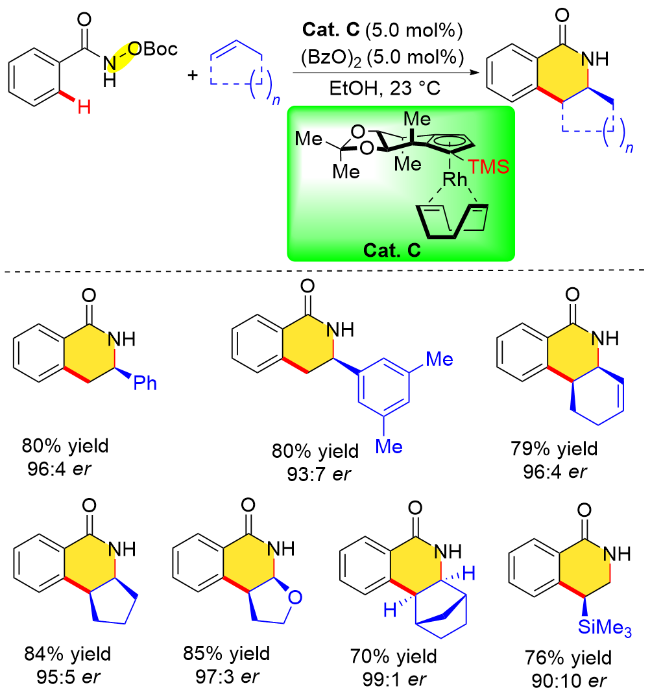
此外, 作者对该反应立体选择性产生的可能原因进行了阐述(Scheme 7). 他们认为, Cp配体的手性环境使得底物中的Boc基团朝向空间位阻更小的方向, 而苯乙烯在靠近金属中心时其芳基取代基趋于远离配体骨架以避免发生空间位阻冲突, 这些因素有利于S构型产物的生成.
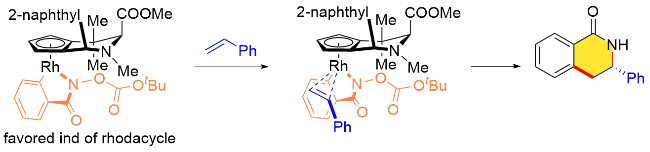
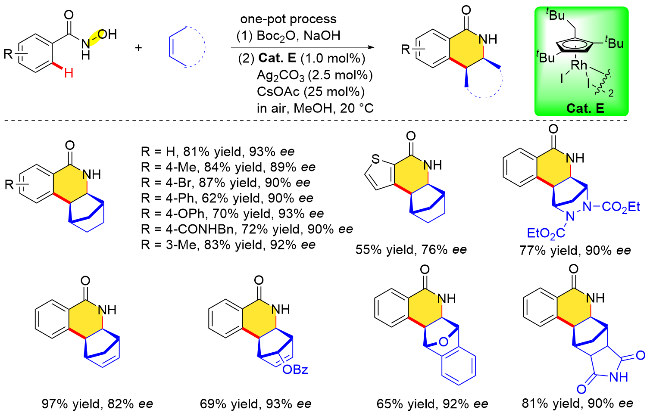
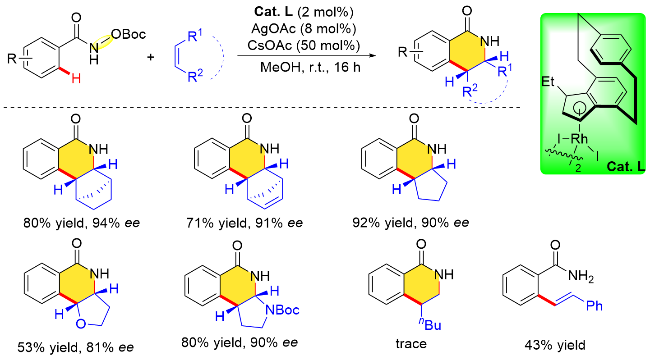
具有C2对称性的环戊二烯基配体来源更加丰富易得, 相对报道较多, 而基于环戊二烯基平面手性衍生的金属催化剂则发展较为缓慢. 2018年, Perekalin课题 组[23]发展了一种新型的平面手性铑催化剂, 由商用[Rh(cod)Cl]2和叔丁基乙炔经两步合成铑配合物外消旋体, 通过碘单质的氧化以及S-脯氨酸的重结晶拆分得到一对非对映异构体, 最后经酸化、碘代反应获得单一构型的平面手性环戊二烯基铑配合物Cat. E. 随后, 该课题组利用氧化型导向基团策略, 将上述平面手性环戊二烯铑配合物Cat. E应用在N-羟基苯甲酰胺和环状烯烃的对映选择性环合反应中(Scheme 8). 结果表明, 该反应可以在温和条件下进行, 底物适用性广泛, 并且以高的收率(53%~97%)及优异的对映选择性(76%~95% ee)合成了手性异喹啉酮类化合物.
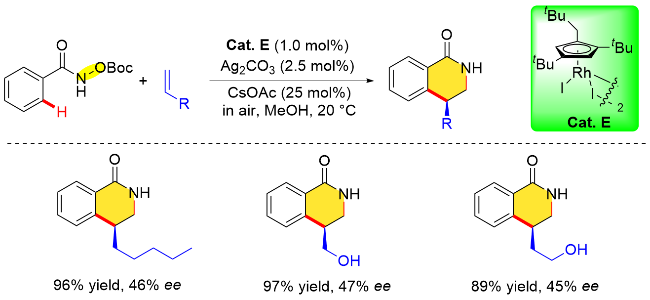
另一方面, 对于一些末端烷基烯烃, 如1-庚烯、烯丙醇或3-丁烯-1-醇等, 尽管该平面手性Rh配合物也能以89%~97%的收率得到相应的4-取代二氢异喹诺酮类化合物, 但对映选择性水平相对较差(45%~47% ee), 这可能是因为末端烯烃和环戊二烯配体之间的空间位阻相互作用不足(Scheme 9).
2021年, Perekalin课题组[24]报道了一例由四个叔丁基乙炔分子与铑金属前体经现场反应生成的大位阻环戊二烯基铑金属配合物, 并研究了其催化性能. 在Et3N的存在下, [Rh(cod)Cl]2可以与叔丁基乙炔发生反应生成一种特别的(tBu4Cp)Rh(cod)配合物[25], 其中的环戊二烯配体tBu4Cp由四个炔烃分子组装而成. 紧接着, 用氯或溴氧化该铑(I)配合物得到铑(III)配合物(tBu4Cp)RhX2 (X=Cl, Br). 最后, 作者将外消旋氯化物(tBu4Cp)RhCl2与(R)-苯基甘氨醇反应并拆分得到了单一对映体Cat. F. 上述平面手性环戊二烯铑配合物Cat. F可以实现N-新戊酰氧基苯甲酰胺和烯烃的对映选择性环合反应, 但对映选择性却并不理想, 比如在使用1-己烯和降冰片烯作为偶联试剂时, 对映体过量值分别为19%和58% (Scheme 10).
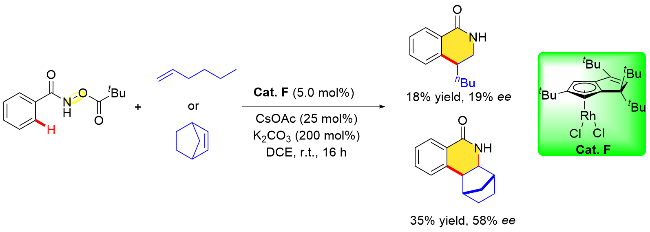
两年后, 上述课题组[26]在先前研究的基础上, 继续合成了一种烷氧基取代的具有平面手性的环戊二烯基铑配合物. 作者发现, 在AlCl3的存在下, [Rh(cod)Cl]2可以与叔丁基乙炔发生反应生成一种阳离子, 随后该阳离子与AlCl3水解生成的HCl进一步反应生成[(C5H2- tBu2CH2tBu)-RhCl2]2配合物, 在该实验过程中, 作者开发了醇类化合物与该阳离子[(C5H2tBu2=CHtBu)Rh- (cod)]+的亲核加成反应, 进而合成一系列环辛二烯铑配合物. 随后用卤素氧化该配合物, 并在L-苯基甘氨醇的存在下, 通过制备薄层色谱(TLC)分离出了对映体, 从而获得了相应的平面手性环戊二烯铑(III)催化剂[(C5H2- tBu2CH(OR)tBu)RhX2]n (Cat. G). 作者同样利用氧化型导向基团策略, 测试了Cat. G的性能(Scheme 11). 与两年前相比, 使用烷氧基取代的平面手性环戊二烯基铑配合物, 确实能提升N-新戊酰氧基苯甲酰胺和烯烃的对映选择性环合的对映体过量值.
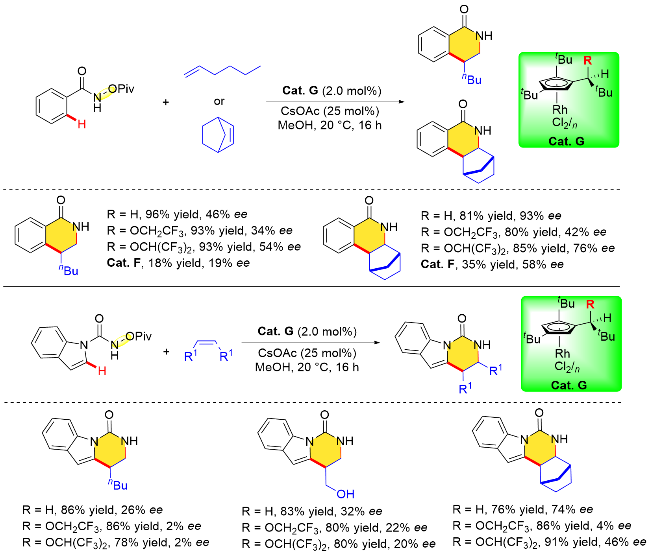
2022年, 汪君课题组[27]在Takahashi等[28]早期研究工作的基础上, 成功制备了一类结构易于修饰的平面手性环戊二烯基铑配合物Cat. H. 该平面手性铑催化剂Cat. H可以实现NOBoc苯甲酰胺邻位C—H键活化与降冰片烯的不对称/[4+2]环加成反应(Scheme 12), 以出色的收率和优异的对映选择性获得目标化合物. 在最优条件下, 作者发现N-OBoc苯甲酰胺底物具有良好的耐受性(收率为62%~90%, 对映性选择为90%~93% ee). 此外, N-叔丁氧羰基噻吩酰胺能使该反应顺利进行, 得到产物收率为92%, 对映体过量值为87% ee. 该工作进一步丰富了手性CpxRh(III)催化剂的种类, 有望在不对称C—H键活化等领域进一步得到应用.
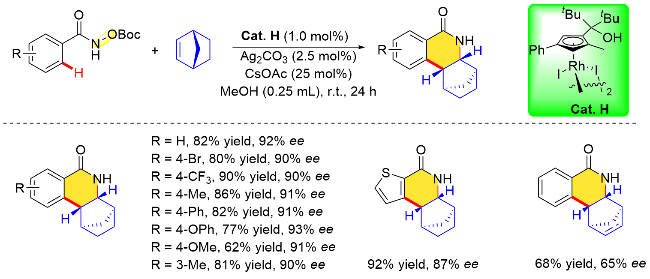

2023年, 汪君课题组[30]设计了一例手性3,3,3',3'-四甲基-1,1'螺联茚基环戊二烯(TMSCp)配体. 作者以3,3,3',3'-四甲基-1,1'-螺二茚-6,6'-二醇(6,6'-TMSPINOL)为初始原料, 经由五步反应成功制备了手性TMSCp配体. 该类配体具有合成方便、易修饰及成本相对较低等特点, 最终与[Rh(COD)OAc]或[Rh(C2H4)2Cl]2络合得到手性CpxRh(III)配合物, 并通过X射线晶体衍射得到其绝对构型. 根据先前Cramer等[18b]提出的模型, 作者推测这类手性配体的Cp环可能更加接近手性螺环的骨架, 有望带来优异的对映选择性控制. 当将此类手性催化剂应用于苯甲酰胺衍生物与降冰片烯分子的环化反应时, 作者通过调节Cp配体“侧壁”苯环上取代基的电子和空间特性获得Cat J, 成功地以出色的收率以及优异的对映选择性合成了异喹啉酮类化合物(Scheme 14).
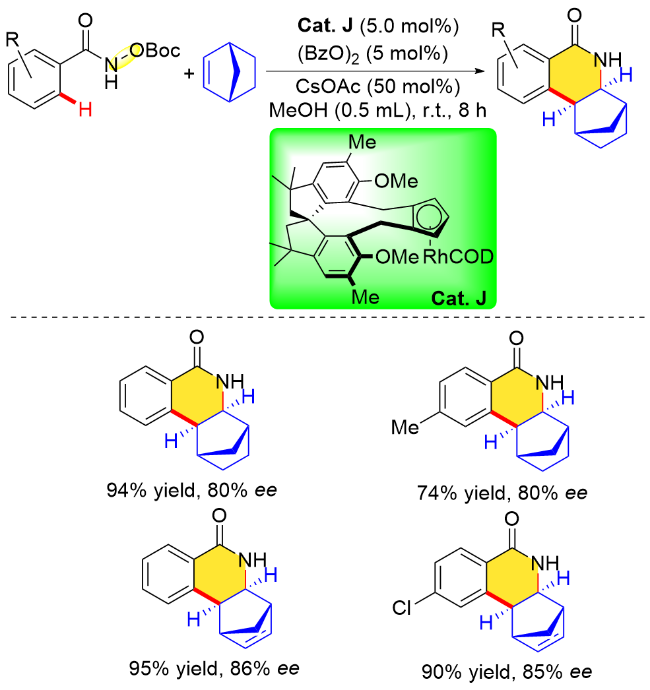
同年, 汪君课题组[31]在探索平面手性铑催化剂的过程中提出了一种新的设计理念和可靠的手性拆分方法, 并且成功制备了一类新型平面手性CppxRh(III)催化剂. 该设计理念是, 打破五甲基环戊二烯(Cp*)配体σv对称性, 即C(1)上的甲基由最小的取代基氢原子取代, C(2)和C(5)的甲基取代基保持不变, C(3)和C(4)的甲基取代基分别由大位阻取代基和中等位阻取代基取代. 基于此, 作者提出了一个立体控制模型, 通过C—H键活化的去质子化底物作为双齿配体与铑配位, 由于取代基之间存在较大的空间位阻差异, 偶联试剂应从中等空间位阻侧进攻环铑中间体, 从而获得较高的对映选择性. 另外, 作者认为首先将手性拆分试剂与Rh(I)配位, 络合物再与四取代Cppx进行配位得到手性催化剂的方法是十分有效的. 据此, 作者合成了一类四取代的新型平面手性CppxRh(III)催化剂. 随后, 作者利用氧化型导向基团策略, 将该平面手性铑催化剂应用在N-叔丁氧羰基苯甲酰胺与降冰片烯的对映选择性环合反应中[31], 结果表明, 该反应可以在温和条件下进行, 以高的收率(81%~90%)和优异的对映选择性(84%~90% ee)合成了手性异喹啉酮类化合物(Scheme 15).
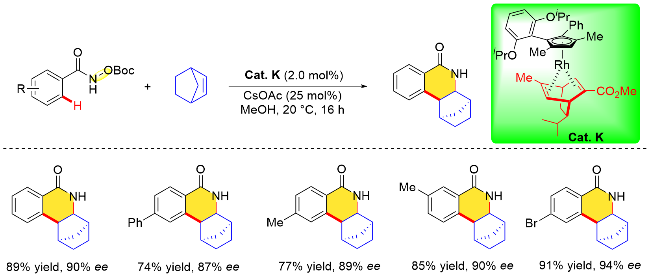
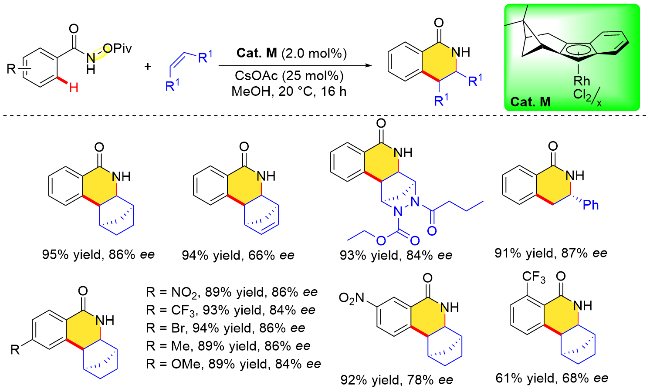
2023年, Loginov课题组[33]设计合成了一例由(−)-α-蒎烯衍生的手性茚铑催化剂. 该手性环戊二烯配体由对映体纯的(−)-α-蒎烯经多步反应得到, 其与RhCl3•3H2O在乙醇溶剂中80 ℃下反应16 h, 即可得到手性金属催化剂. 值得一提的是, 由于该配体的多环结构特点, 四氢芴铑配合物[(THFluverb)RhCl2]x无需进行手性拆分, 即可得到光学纯的金属催化剂(Cat. M). 作者利用氧化型导向基团策略测试了上述平面手性催化剂的性能, 结果表明, 在该手性四氢芴铑催化剂下, N-新戊酰氧基苯甲酰胺与烯烃分子的对映选择性环合反应在低催化剂负载量条件下, 反应30 min即可完成. 在考察底物范围时, 作者发现环状烯烃与末端烯烃均能很好地兼容该反应体系, 且N-新戊酰氧基苯甲酰胺对位取代基的电子耐受性良好, 以89%~94%的收率, 84%~86% ee的对映体过量值获得相应的手性异喹啉酮类化合物(Scheme 17).
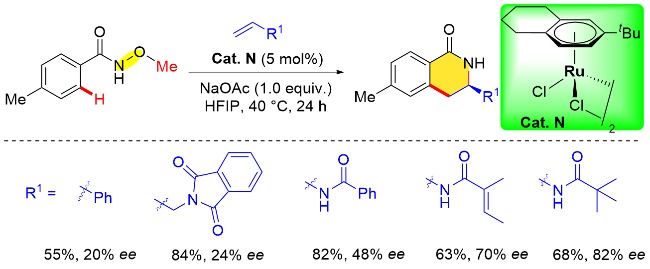
除了手性铑催化剂, 最近Perekalin课题组[19t]还设计合成了一种新型的平面手性钌催化剂. 首先, 四氢萘和叔丁基氯发生傅-克烷基化反应生成三取代的芳烃化合物, 该芳烃化合物在高温下与[(p-cymene)RuCl2]2进行芳烃配体交换形成钌配体的外消旋体, 最终在手性膦助剂的作用下, 经过拆分获得对映体纯的平面手性钌催化剂(Cat. N). 之后, 他们通过N-取代苯甲酰胺衍生物与烯烃的[4+2]环加成反应, 考察了该手性钌催化剂的性能. 研究结果表明, 端烯取代基的结构对反应的对映选择性有着显著的影响(Scheme 18). 结合此前Baidya等[34] 的工作, 作者发现使用含有酰胺结构的端烯更有利于反应对映选择性的控制, 而使用N-新戊酰氧基苯甲酰胺或者N-叔丁氧羰基苯甲酰胺作为底物时, 反应过程中会形成一种硝烯中间体, 从而导致催化剂分解, 并形成游离的苯甲酰胺阻碍反应的进行.
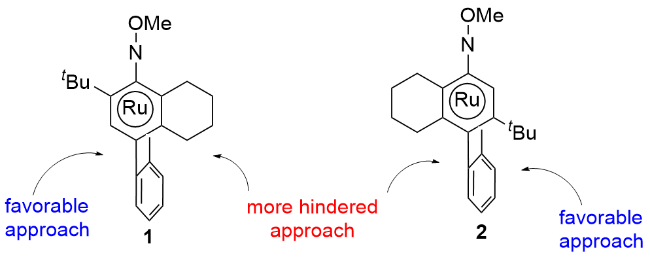
作者通过密度泛函理论(DFT)计算阐述了该手性钌催化剂的手性诱导机制, 并认为其立体选择性决定步骤应该是环金属中间体对烯烃的迁移插入过程. 由于该催化剂中芳烃配体的不对称性, 在形成五元环钌中间体时有两个稳定的构象(图3), 而当烯烃从左侧靠近中间体1是最有利的, 此时生成产物的绝对构型为R. 烯烃从中间体2的右侧靠近时需要克服相对较高的能垒, 是非优势反应路径.
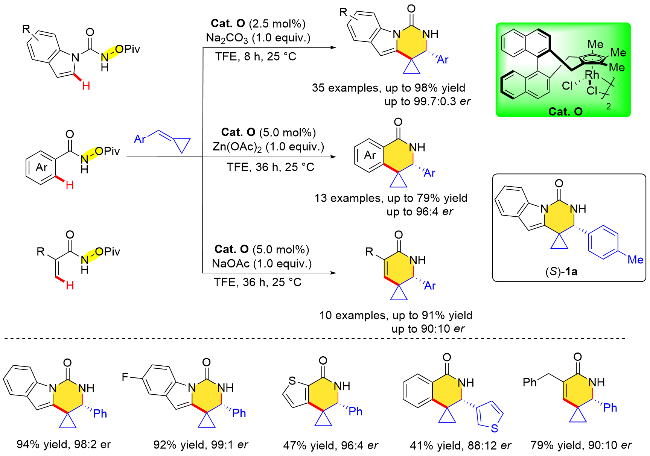
除了常规的苯甲酰胺衍生物与普通烯烃的不对称/ [4+2]环化反应, 易伟等[35]还利用手性CpmRh(III)催化剂Cat. O实现了含有N—O键氧化型导向基团的不同类型芳基酰胺(含吲哚、吡咯、苯、萘、噻吩骨架)以及丙烯酰胺等底物与亚甲基环丙烷类化合物(MCPs)的不对称C—H键活化/[4+2]环加成反应, 高对映选择性地完成了多种结构新颖的螺环丙烷骨架的构建. 结合实验和DFT计算研究, Cp环上的取代基和1,1-联-2-萘酚(BINOL)骨架的空间定位对目标产物的对映选择性有着显著影响. 此外, 研究团队通过药理活性筛选发现其中一个化合物(S)-1a对胰腺癌细胞系BxPC-3具有较好的抗增殖活性(IC50=1.7 μmol/L), 是潜在的新型结构抗胰腺癌先导化合物. 该方法为新型螺环丙烷类药物的合成提供了有效的制备方法(Scheme 19).
1.1.2 N-取代苯甲酰胺衍生物与炔烃的分子间[4+2]环加成反应
基于氧化型导向基团策略的不对称碳氢键活化环化反应除了能高效构建中心手性, 还可以通过改变反应底物构建轴手性联芳烃化合物. 李兴伟等[36]于2020年使用联萘基衍生的手性CpxRh配合物(Cat. P, Cat. Q), 通过含有氧化型导向基团的苯甲酰胺类衍生物与2-取代1-萘炔化合物以及大位阻二芳基乙炔的分子间C—H键活化/[4+2]环化反应, 成功构建了具有轴手性的异喹诺酮分子(Scheme 20). 作者研究发现通过调整手性CpxRh催化剂联萘环3,3'-位置的取代基, 苯甲酰胺类衍生物与不同结构炔烃的环化反应均能以较高的收率和出色的对映选择性获得目标产物, 具有优异的官能团耐受性. 值得注意的是, 当酰胺底物扩展到杂芳基(如噻吩、咪唑)酰胺时, 反应也能顺利进行.
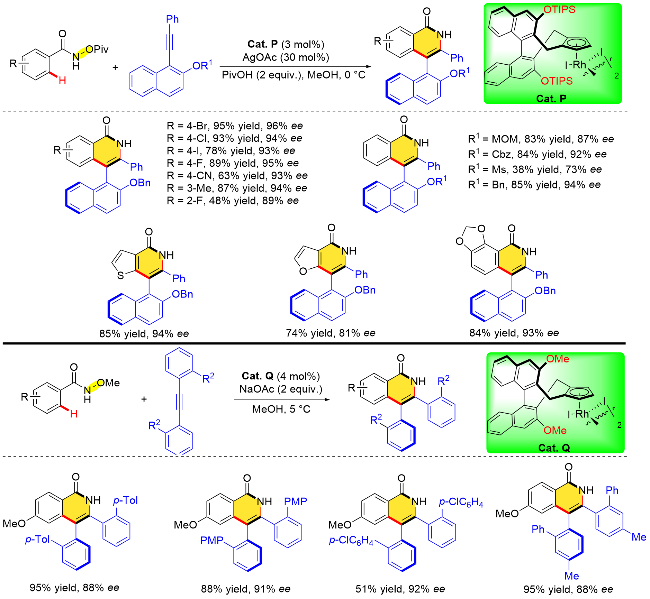
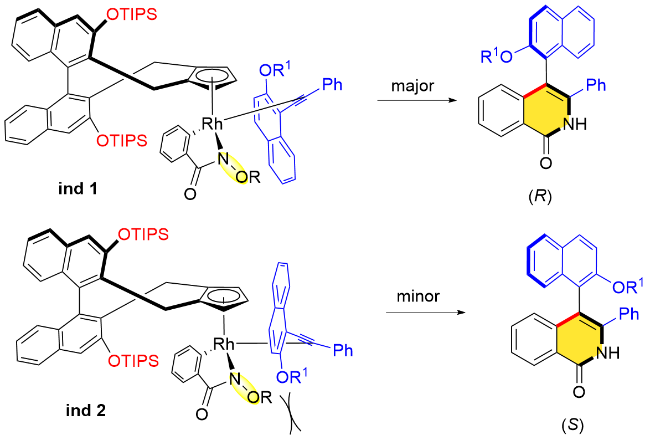
作者认为炔烃的选择性插入是决定对映选择性的关键步骤, 由于手性联萘基Cp配体“后壁”以及“侧壁”空间位阻的影响, 金属中心在与苯甲酰胺衍生物邻位C—H键的环金属化过程中锁定了环金属中间体的朝向. 随着炔烃分子的靠近, 空间上更小的萘环指向纸面内侧, 而OR1基团指向空间位阻较小的一侧(ind 1), 这种空间位置的排列使得萘环和N—OPiv导向基团之间的位阻冲突较小, 有利于生成R构型产物(Scheme 21).
相比于手性Cp铑催化剂, 手性钌催化剂由于缺少合适的手性配体, 发展较为滞后. 芳烃钌(II)催化剂对于涉及协同金属化去质子化(CMD)过程的多数C—H键活化反应十分有效, 设计开发含有手性芳烃配体的钌(II)催化剂是一种极具吸引力的策略. 汪君课题组在芳烃钌(ArRu)催化的基于氧化型导向基团的不对称C—H键活化方面取得重要进展, 2022年该课题组[37]发展了一例由[2.2]二聚对二甲苯衍生的C2对称手性芳烃配体, 通过与Ru的络合成功合成了相应的催化剂. 该催化剂的手性环境可以通过改变[2.2]二聚对二甲苯中上下两个苯环中R取代基的空间位阻和电子特性来调节. 利用该手性芳烃配体的钌(II)催化剂Cat. R, N-甲氧基苯甲酰胺邻位C—H键活化与炔烃的不对称环加成反应, 以高收率、高对映选择性构建了轴手性异喹诺酮类化合物(Scheme 22). 对手性芳烃配体钌配合物进行了X单晶衍射分析, 发现乙基取代配体的两个苄基碳的距离为0.4397 nm, 而异丙基取代配体的距离为0.4918 nm, 并且后者催化剂中两个苯环之间的二面角相较于前者显著增大(7.754° vs. 0.662°), 这说明配体中的两个异丙基之间存在强烈的空间位阻相互作用, 抑制了异丙基在金属配位处的旋转, 从而导致金属中心周围具有受阻的手性环境, 促进更好的对映选择性控制.
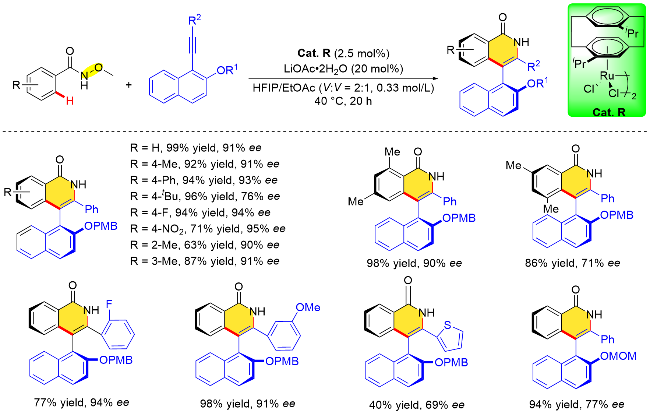
此外, 作者还提出了立体化学模型来解释手性诱导的方式. 首先, 由于[2.2]二聚对二甲苯配体中异丙基取代基和—CH2CH2—桥键等结构片段导致的空间位阻, 苯甲酰胺的甲氧基部分指向远离异丙基一侧, 而炔烃更倾向于从催化剂空间位阻较小的另外一侧进攻, 从而有利于生成S构型产物(图4).
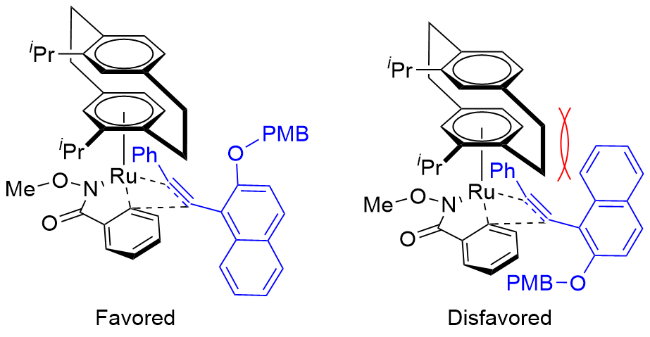
1.1.3 N-取代苯甲酰胺衍生物与烯烃或炔烃的分子内[4+2]环加成反应
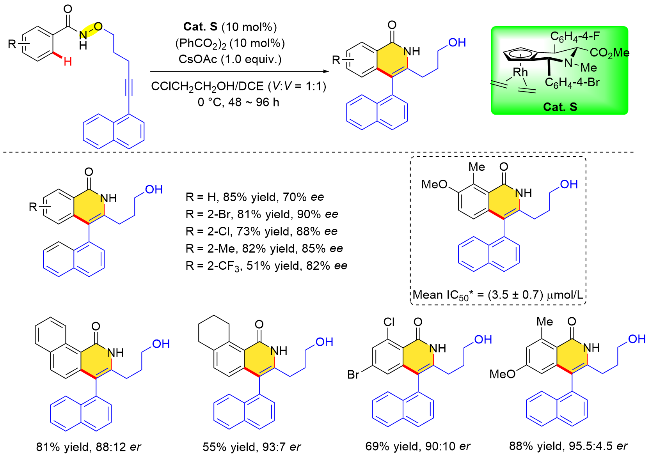
通过反应底物设计实现分子内的环化反应, 可以构建更加复杂、更易于转化的环状分子. Waldmann与Antonchick等[38]于2018年将氧化型导向基团与炔烃偶联试剂有机结合, 使用哌啶骨架衍生的手性CpJRh配合物Cat. S, 实现了分子内不对称C—H键活化与炔烃的 [4+2]环化反应, 高效地构建了轴手性4-芳基异喹诺酮类化合物(Scheme 23). 该反应对苯甲酰胺底物中芳环取代基的位置有一定的要求. 研究表明, 未取代的苯甲酰胺参与反应时对映选择性较差, 而当邻位有取代基时对映选择性可以显著提高. 萘环上的取代基则对反应影响不大, 并且可以扩展到其他π体系. 值得一提的是, 作者发现部分4-芳基喹啉产物还可作为一种新的非跨膜蛋白Smoothened (SMO)结合的刺猬信号(Hedgehog)通路抑制剂.
1.1.4 N-取代二茂铁甲酰胺与炔烃的分子间[4+2]环加成反应
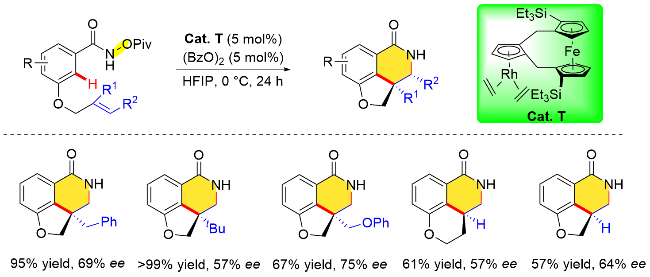
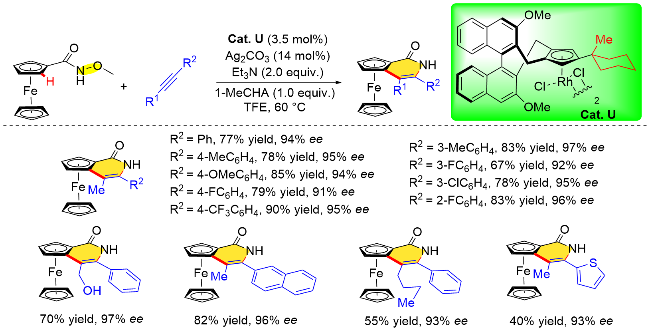
针对上述问题, 该课题组[19n]在后续的研究中发现, 对手性联萘基Cp配体的Cp环做进一步的修饰, 可以提高CpxRh催化剂的催化性能(Cat. U), 从而高效地构建平面手性二茂铁基吡啶酮类化合物. 研究表明, 在Cp环三号位安装了一个大位阻的取代基, 可以显著改善前期研究中手性调控不理想的问题, 并且反应可以在温和的条件下进行, 得到的平面手性二茂铁基吡啶酮具有较高的对映选择性(高达99% ee的对映体过量值). DFT计算表明, 炔烃的插入是决定对映选择性的关键步骤. 该反应对各种取代的1-苯基-1-丙炔均能适用. 然而, 当使用杂芳基炔烃时, 反应效率有所降低, 比如噻吩基取代的炔烃衍生物参与反应时, 反应收率大幅下降, 作者推测可能是由于杂原子对Rh中心的有害配位造成的(Scheme 26).
1.2 不对称C—H键官能团化/[4+1]环加成反应
1.2.1 N-取代苯甲酰胺衍生物与烯烃的分子间[4+1]环加成反应
2020年, 游书力课题组[41]发现在新型手性CpxRh配合物催化下N-OBoc取代苯甲酰胺衍生物可以与烯烃分子发生与众不同的不对称/[4+1]环化反应来构建手性3-取代异吲哚啉酮类化合物(Scheme 27). 该工作扭转了此前苯甲酰胺类衍生物与烯烃倾向于发生[4+2]环化反应的常规区域选择性, 为此类反应的发展提供了重要的补充. 该课题组首先利用Co2(CO)8介导的[2+2+1]环化为关键步骤, 设计了一系列可调控空间位阻和电子效应的手性联萘基CpxRh催化剂(Cat. O). 可以对此类BINOL衍生的Cp配体手性Cp环上的取代基(Me、iPr及tBu等)进行快速调整和修饰, 从而实现高对映选择性的转化反应. 实验发现, Cp环上只引入一个取代基 (R4=iPr或Ph)对反应结果的影响较小; 而当Cp环上引入多个甲基时(R3=R4=Me), 可以优异的收率、独特的区域选择性以及优异的对映选择性获得相应的手性异吲哚啉酮类化合物. 在此基础上, 作者考察了联萘基上的取代基(R2)的类型对反应结果的影响, 结果表明当(R2)位置为甲氧基或者苯基时, 预期的[4+1]环加成反应并没有发生, 反而得到了邻位烯基化的产物.
1.2.2 N-取代苯甲酰胺衍生物与重氮化合物的分子间[4+1]环加成反应
手性异吲哚啉酮片段广泛存在于生物活性化合物和天然产物中, 其高效合成受到了化学家们的广泛关注. 自2012年成功设计合成一例由D-甘露醇衍生的手性CpxRh配合物之后, Cramer等[42]致力于开发新型手性Cp配体, 探究其在催化氧化型导向基导向的不对称 C—H键转化反应中的性能. 2014年, Cramer课题组[43]利用2013年他们开发的手性催化剂(Cat. P), 实现了 N—OPiv酰胺氧化型导向基团参与的苯甲酰胺邻位C—H键活化与重氮化合物的不对称/[4+1]环加成反应, 成功地以优异的对映选择性构建了手性异吲哚酮类化合物(Scheme 29). 作者考察了N-新戊酰氧基苯甲酰胺底物的范围, 底物的芳烃上无论是供电子基团还是吸电子基团, 均能使反应顺利进行(收率为64%~92%, 对映体过量值为86%~93% ee), 对重氮化合物底物范围的研究表明, 重氮化合物含有烷基或芳基取代基以及醚或缩酮结构时, 反应仍能以优异的的收率和对映选择性顺利进行.
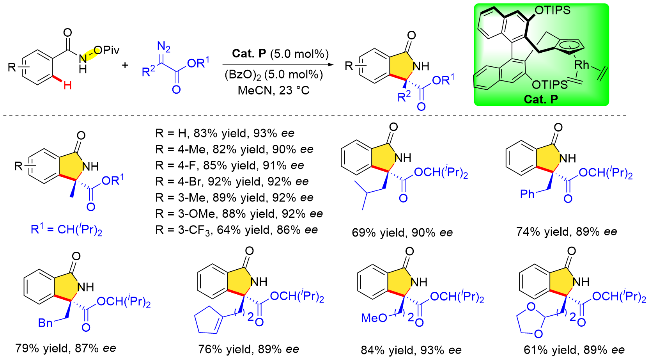
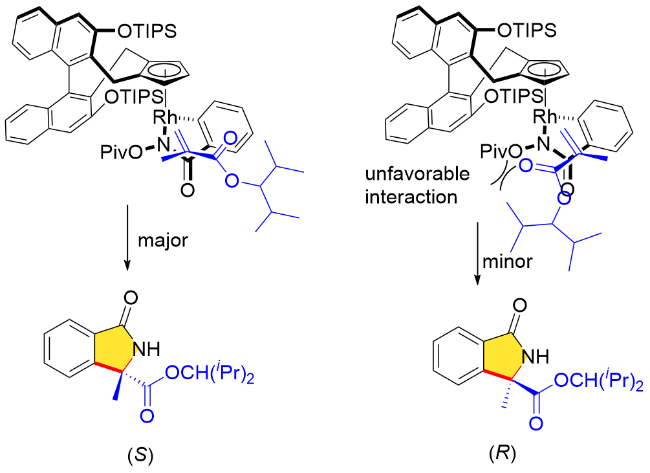
此外, 作者对该反应的对映选择性来源进行了合理的立体结构模型阐释(Scheme 30). 首先, 由于“侧壁”大位阻取代基(OTIPS)的影响, 五元金属铑环中间体中芳烃的位置指向位阻较小的一侧, 随后重氮化合物选择性的插入确定了产物的优势构型为S构型.
1.2.3 N-取代吲哚酰胺衍生物与重氮化合物的分子间[4+1]环加成反应
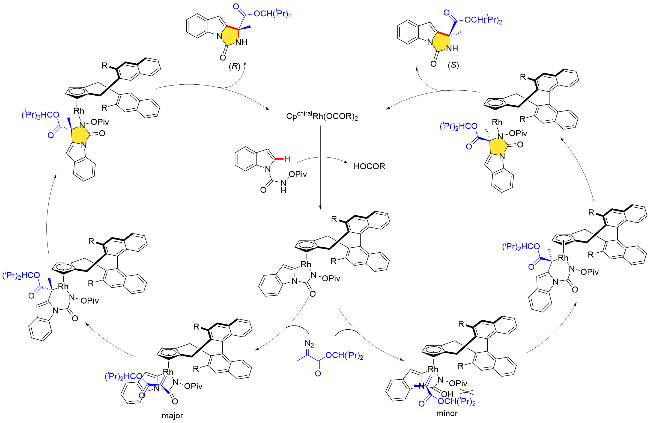
宋国勇课题组[44]在2017年报道了一例手性Cpx- Rh(III)配合物催化含有氧化型导向基团的吲哚分子与重氮化合物的分子间不对称C—H键活化/[4+1]环化反应, 高效地构建了手性1H-咪唑并[1,5]吲哚-3(2H)酮衍生物, 具有收率高、对映选择性优异、反应时间短和底物范围广等特点(Scheme 31). 作者发现该C—H键活化反应选择性地发生在吲哚分子C(2)位置, 而不是C(3)或C(7)位. 同时, 作者发现手性CpxRh配合物“侧壁”位置取代基的空间位阻对反应的活性以及对映选择性影响并不大. 在Cat. Q催化下, 作者探究了N-(新戊酰氧基)-1H-吲哚-1-甲酰胺衍生物底物的范围, 吲哚分子的C(5)位置具有吸电子或供电子或苯基官能团时, 反应均能顺利发生, 生成相应的环状产物收率高(80%~95%)、对映选择性优异(92∶8~95∶5 er). N-(戊酰氧基)-1H-吡咯-1-甲酰胺也是进行不对称C—H环化反应的合适底物. 然而, 2-重氮乙酸叔丁酯以及2-重氮乙酸异丙酯参与反应时, 相应的收率以及对映选择性略有下降, 作者推测可能是重氮化合物两个取代基的尺寸差异减小导致的[43].
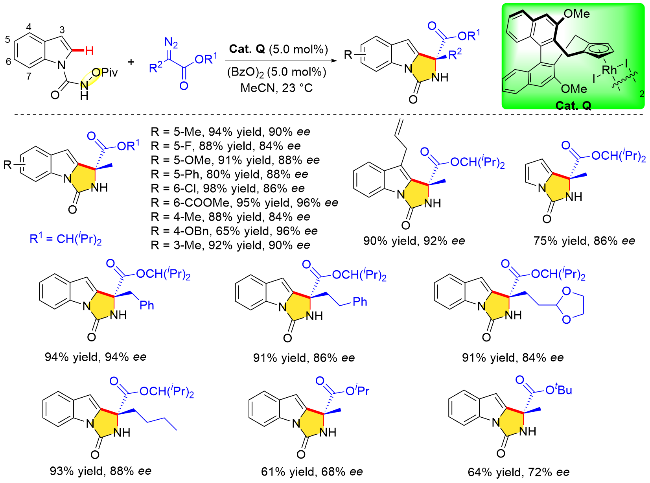
作者提出了吲哚分子不对称C—H键活化/环化可能的反应机理(Scheme 32). 首先由N-(新戊酰氧基)-吲哚甲酰胺与金属铑中心形成五元铑杂环中间体, 由于手性Cp环“侧壁”取代基位阻的影响, 这个环中间体形成之后, 吲哚指向空间位阻较小的一侧, 远离手性Cp环, 随后重氮化合物与金属中心的对映选择性配位形成金属-卡宾物种, 由于重氮化合物上酯基取代基以及吲哚分子内部大位阻特戊酯基的空间排斥效应, 优势立体构型在此时构建. 随着吲哚分子内部N—O键断裂和 C—N键的形成, 最终生成了目标化合物, 完成催化循环并释放催化剂.
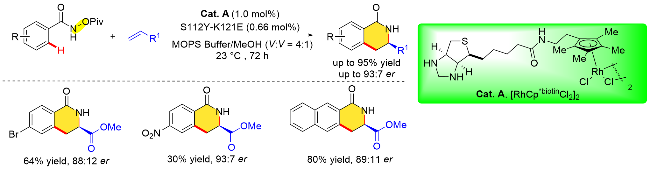
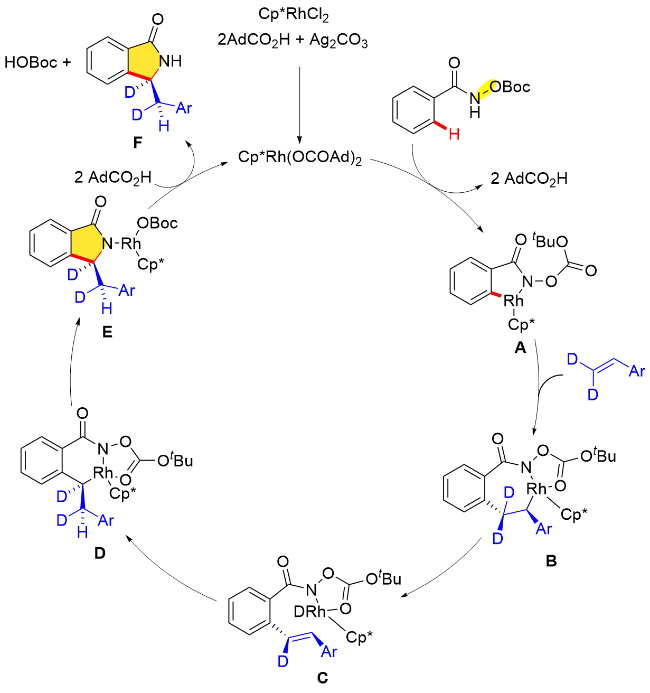
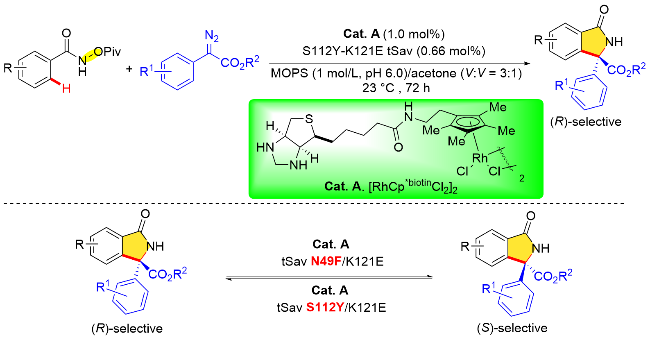
除此之外, 人工金属酶催化合成手性异吲哚酮的方法最近也取得了重大进展. 2024年, Bhaumik和Maiti 等[45]利用链霉亲和素人工金属酶合铑(III)(Cat. A)催化导向N-新戊酰氧基苯甲酰胺邻位C—H键活化与芳香族重氮酯化合物的不对称/[4+1]环加成反应, 成功地以极高的对映选择性(95∶5 er)获得了手性异吲哚酮类化合物(Scheme 33). 这个催化体系可以兼容多种不同的重氮酯, 如联苯、硫醚、硒醚、胺、烯烃和炔烃. 先前的研究表明S112和K121位置的关键基团在生物素化的铑催化剂中具有十分重要的作用[46]. 作者在探究了不同的Sav突变体之后发现, 在112或121位置引入脂肪族, 如丙氨酸基和异亮氨酸基, 能提高收率, 然而未增加反应的对映体选择性. 112位置的半胱氨酸基和121位置的蛋氨酸基都降低了产物的收率和选择性. 此外, 112位置的酪氨酸基和121位置的苯丙氨酸基可有效改善收率和选择性, 并且S112Y/K121E中酪氨酸和谷氨酸的协同作用对于反应的对映选择性至关重要. 此外, 机理研究表明C—H键的活化涉及定向的内球机制, 并且是不可逆的. 此外, 作者通过合理设计链霉亲和素与生物素化铑(III)催化剂, 利用突变体N49F/K121E实现了手性异吲哚酮反向立体选择性调控.
1.2.4 O-新戊酰基肟与重氮化合物分子间[4+1]螺环化反应
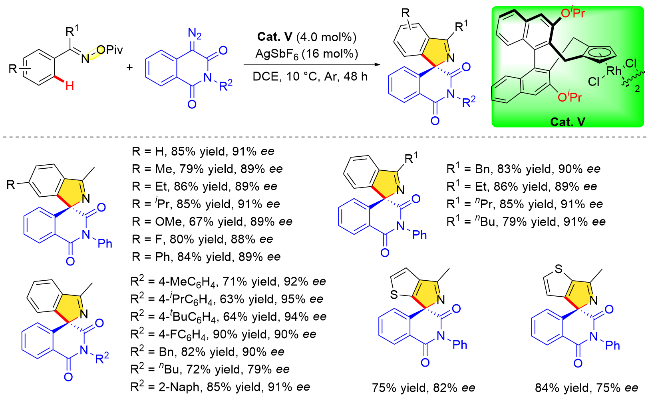
除了苯甲酰胺衍生物的不对称碳氢键转化反应, 2021年, 李兴伟课题组[47]还利用O-新戊酰基肟内部 N—OPiv片段作为分子内氧化剂, 使用联萘基衍生的手性CpxRh(III)配合物(Cat. V)作催化剂, 实现了其与α-重氮邻苯二甲酰亚胺的不对称C—H键活化/[4+1]螺环化反应, 成功构建了手性螺环亚胺类化合物(Scheme 34), 反应具有收率高及对映选择性优异等特点. 研究表明, 在现有氧化型导向基团辅助下, 开发多种底物类型可以实现新颖含氮杂环分子的不对称转化构建. 作者对O-新戊酰基肟的底物范围研究发现, 在其苯基对位引入各种供电子基团、吸电子基团和卤素基团, 均可以保持良好的收率和出色的对映选择性.
2 N—Cl键氧化型导向基团参与的不对称C—H键官能团化反应
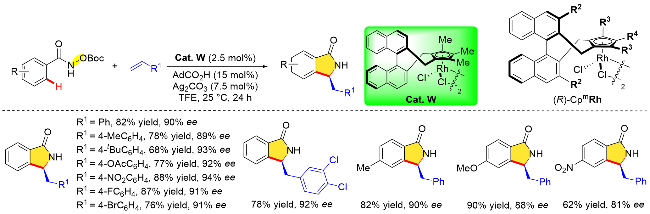
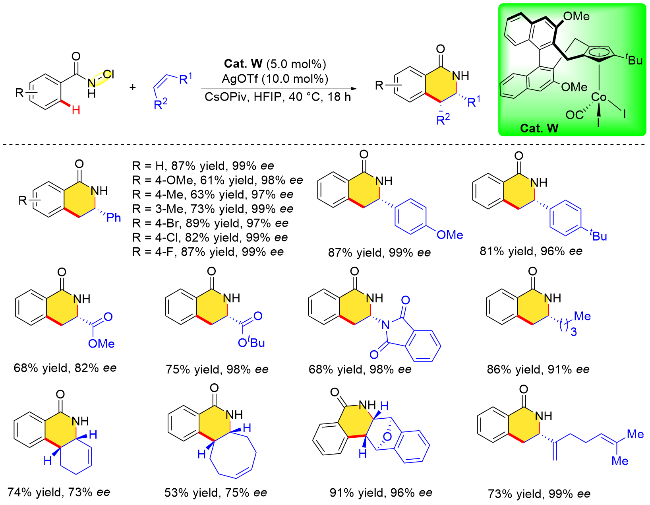
除了基于N—O键的氧化型导向基团, N-氯苯甲酰胺由于分子内的N—Cl键也具有高氧化能力, 可用作氧化型导向基团. 2017年, Zhu课题组[10a]在[CoCp*(CO)I]2]催化下完成了N-氯苯甲酰胺C—H键活化与炔烃分子或者烯烃分子的[4+2]环化反应, 其反应条件相比铑催化更加温和. 2019年, Cramer课题组[10f]同样以N-氯甲酰胺片段为氧化型导向基团, 在三取代的手性环戊二烯钴(III)配合物(Cat. W)催化作用下, 完成了N-氯苯甲酰胺与烯烃分子的不对称/[4+2]环加成反应, 成功地构建了手性二氢异喹啉酮类化合物, 反应具有优异的区域选择性以及出色的对映选择性(Scheme 36). 该手性钴(III)配合物同样也是通过手性联萘基Cp骨架与金属钴试剂络合而来的. 与贵金属铑的络合配位相比, 简单地将配体(CpxH)与Co2CO8混合, 然后用碘单质氧化即可获得目标配合物. 而修饰联萘骨架的3,3'-位置以及Cp环上的取代基可以获得一系列空间位阻和电子特性各异的手性CpCo(III)配合物. 在最优条件下, 该课题组研究了N-氯苯甲酰胺和苯乙烯底物取代基的范围. 无论供电子或吸电子取代基都不影响反应的对映选择性, 但供电子取代基对产物的收率略有影响(61%~63%). 值得注意的是, N-乙烯基邻苯二甲酰亚胺作为一种杂原子取代烯烃也能很好地适用该反应. 同时该反应还适用于环烯底物, 然而对映性选择性有所下降.
3 O—N键氧化型导向基团参与的不对称C—H键官能团化反应
3.1 烯醇衍生物的不对称C—H键官能团化
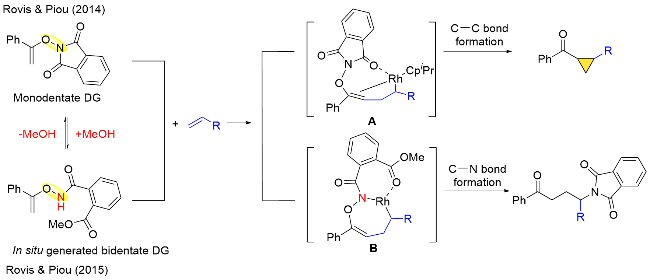
N-烷氧基酰胺或者N-酰氧基酰胺可以作为羧酸类底物的氧化型导向基团实现其衍生物的碳氢键转化, 逆转N—O键的排列顺序则可以实现含氧化合物(如烯醇、苯酚等)的碳氢键转化. 2014年Piou和Rovis[49]报道了Rh(III)催化的N-烯氧基邻苯二甲酰亚胺导向的烯基C—H键活化与烯烃分子的环丙烷化反应, 其中N-烯氧基邻苯二甲酰亚胺底物既能作为导向基团又能作为内部氧化剂活化烯醇的碳氢键. 该反应成功的关键是大位阻单取代异丙基环戊二烯基配体的设计. 不仅如此, 在2015年Piou和Rovis[50]发现通过改变反应条件, 同样的底物在Rh(III)催化下可以经导向烯烃C—H键活化, 与烯烃发生碳-酰胺化反应. 针对同一底物不同的反应结果, 作者给出了相应的解释: 在2014年的工作中, Rh原子是配位不饱和的, 单齿导向基团的存在使得烯烃和铑进行分子内的配位形成中间体A, 最终生成环丙烷化产物; 而在2015年的工作中, 同一底物在亲核溶剂甲醇中可以原位形成双齿导向基团, 此时中间体B是配位饱和的, 不利于分子内配位, 有利于发生还原消除, 从而实现碳酰胺化反应(Scheme 37).
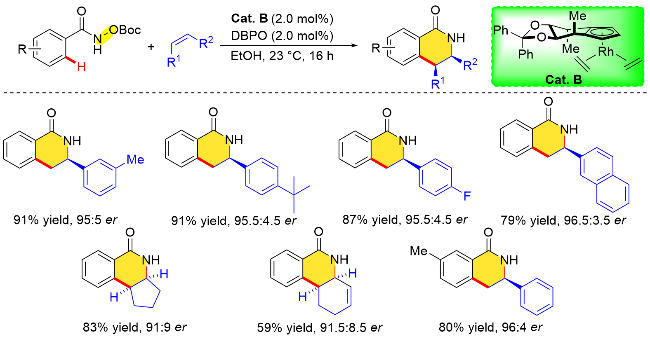
2019年, Cramer课题组[51]在手性CpxRh(III)金属配合物的催化作用下, 分别实现了上述两个反应的不对称转化. 该课题组利用N-烯氧基衍生物的氧化导向基团特性, 通过手性铑催化剂(Cat. X)催化邻位烯基C—H键活化与丙烯酸酯的对映选择性环丙烷化反应(Scheme 38). 在手性Rh催化剂的选用上, 作者首先采用其在2012年设计的第一代催化剂, 以71%的收率、>20∶1反式/顺式比率和优异对映选择性(93.5∶6.5 er)获得了手性环丙烷产物. 在后续的探究中, 作者发现增加催化剂配体“后壁”的位阻反而会使对映选择性降低. 作者在筛选一系列具有氧化型导向基团的底物后, 发现使用N-烯氧基琥珀酰亚胺参与反应时, 所需的反应时间更短, 收率更高, 并且对映选择性优异. 作者基于该对映选择性C—H键活化官能团化反应, 以三步反应39%的总收率和98% ee的对映体过量值, 获得了一种用于犬尿氨酸3-单加氧酶(KMO)的抑制剂UPF-648 (IC50=40 nmol/L).
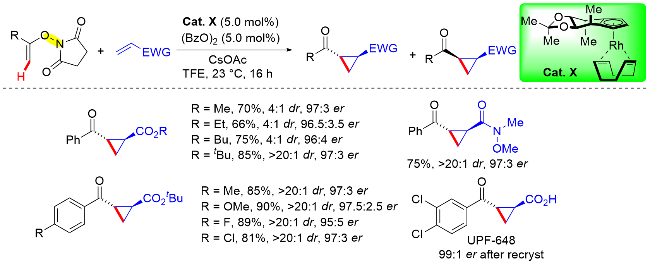
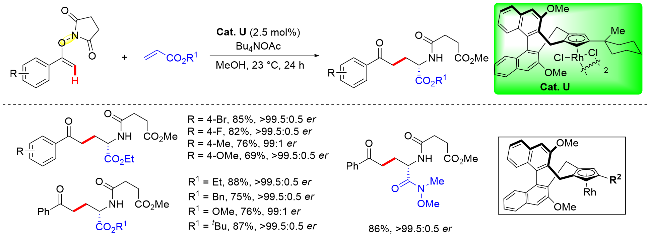
Cramer课题组[52]通过调整手性金属铑催化剂(Cat. U)以及反应条件, 于2020年实现了N-烯氧基琥珀酰亚胺底物导向的不对称烯基C—H键活化/碳-酰胺化反应(Scheme 39). 研究发现, 使用联萘骨架衍生的手性铑催化剂, 并且调整改变Cp环上取代基类型, 比如有大位阻取代基取代时, 能够以优异的对映选择性实现碳-酰胺化反应, 并成功地构建了具有优异对映选择性的非天然α-氨基酸衍生物. 此外, 作者发现在乙醇溶剂中, 碳-酰胺化反应不能发生. 值得注意的是, 相关的N-烯氧基亚胺型底物, 如邻苯二甲酰亚胺和3,4-二氯邻苯二甲酰亚胺衍生物也不能进行反应. 在优化条件下, 作者探究了丙烯酸酯的底物范围, 常用的丙烯酸酯(如甲基、苄基和丙烯酸叔丁酯)均能顺利参与反应, 产物具有极高的收率以及优异的对映选择性.
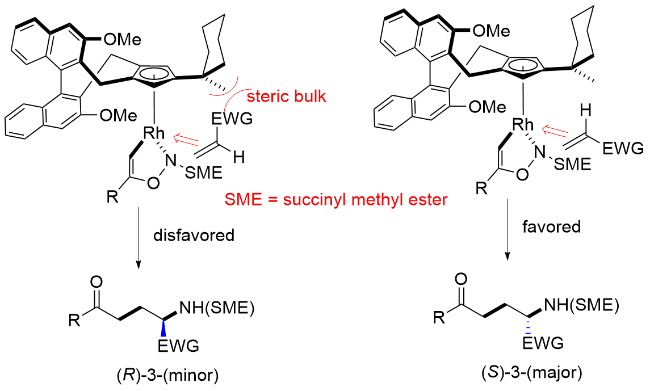
作者对优势构型的对映选择性形成的过程进行了立体结构模型阐释(Scheme 40). 由于手性Cp配体手性口袋控制的作用, 五元金属铑环中间体N-取代部分指向位阻较小的一侧, 随着烯烃选择性的插入确定了反应的优势构型为S构型.
3.2 酚类衍生物的不对称C—H键官能团化
3.2.1 与烯烃分子间的偶联反应
N-苯氧基酰胺也是过渡金属催化直接C—H键官能化反应中常用的一种具有氧化型导向基团的底物, 可以实现酚类化合物的碳氢键高效转化. 在该策略中, O—NHR通过O—N键的裂解以及裂解单元NHR片段释放来充当内部氧化剂. 利用这一反应性, 李兴伟等[53]使用环丙烯衍生物作为偶联伙伴, 以N-苯氧基对甲苯磺酰胺内部O—N键片段作为分子内氧化剂, 在手性CpxRh- (III)配合物(Cat. V)催化下高效地构建了手性环丙烷化合物. 研究表明, 环丙烯衍生物内部的羟基与酰胺组成双导向基团(DDGs), 顺利地促进1,2-二取代环丙烷产物的生成(Scheme 41). 进一步的机理研究表明, O—N键裂解可能通过Rh(V)类硝烯物种的形成而发生.
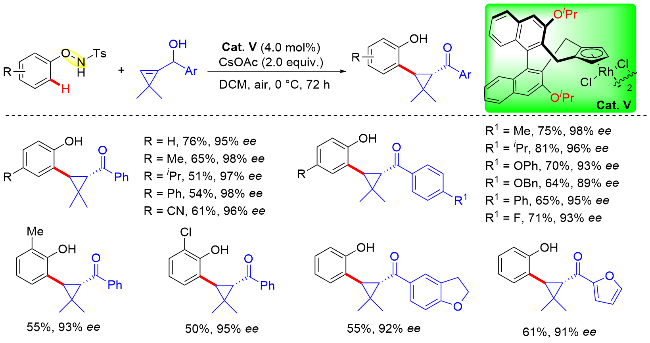
为了更好地理解反应机理的细节, 作者还进行了详细的DFT计算研究, 揭示了双导向基团(DDGs)的作用以及区域选择性、特殊反式-非对映选择性和(S,S)对映选择性的来源(Scheme 42). 其中, Ts和OH基团之间形成的分子内氢键在控制区域选择性方面起着了至关重要的作用. 另外, 烯烃选择性迁移插入五元环铑中间体是反应立体选择性的决定步骤, 作者研究了烯烃插入的不同立体选择性相对应的两种过渡态, 其相应的活化能差异为2.51~11.72 kJ/mol (10 ℃). 根据初步机理研究以及先前文献研究, 作者提出了一种可能的反应机制. 首先, 金属铑介导N-苯氧基对甲苯磺酰胺邻位C—H键活化, 形成五元环铑中间体. 随着烯烃的区域选择性和立体决定性迁移插入产生了七元环铑中间体, 该中间体经分子内氧化剂氧化加成、质子迁移以及碳氢键还原消除等步骤得到最终的环丙烷化合物, 而活性Rh(III)催化物种由质子化解离重新生成(Scheme 42).
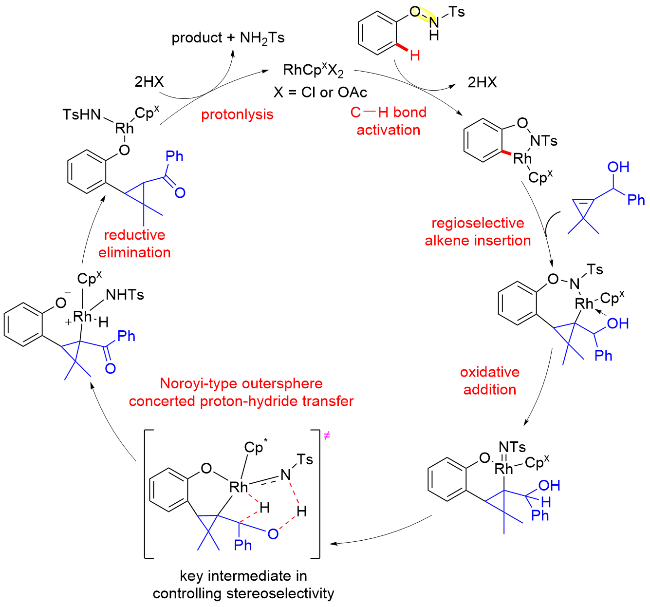
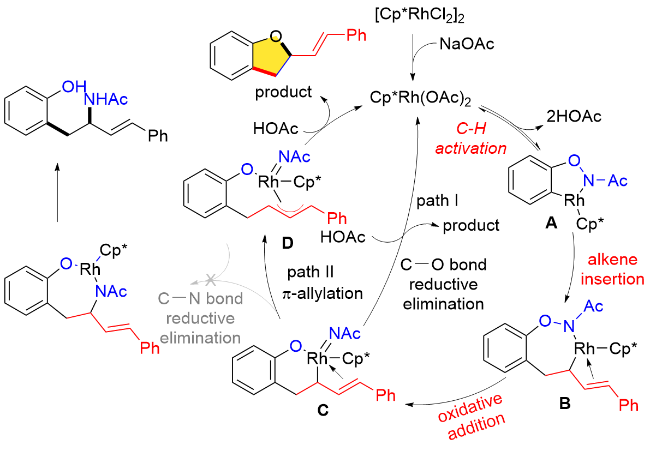
另一方面, NHR基团在C—H键官能化以及分子内氧化等基元步骤后, 如果能够直接迁移到产物中, 将实现100%原子经济性的转化, 更加符合绿色化学的理念. Cramer等[54]就利用N-苯氧基乙酰胺内O—NHAc片段作为氧化型导向基团, 使用三取代的手性环戊二烯钴(III)配合物(Cat. Y、Cat. W)作为催化剂, 通过邻位C—H键活化的方式, 在温和的条件下, 与丙烯酸酯以及双环烯烃完成了对映选择性的分子间碳-酰胺化反应(Scheme 43). 此外, N-苯氧基乙酰胺分子内乙酰基保护基还可以替换为三氟乙酰基. 该反应对于碳、氮或氧桥连的双环烯烃也有很好的适用性. 在反应过程中, 所用的原料最终均迁移进入产物中, 实现了100%的原子经济性.
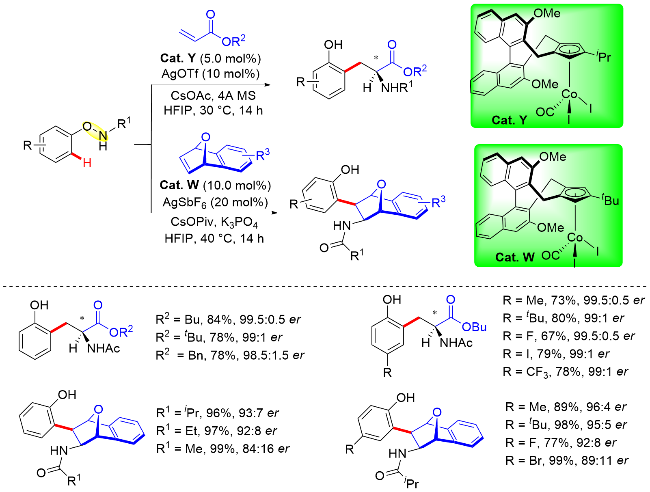
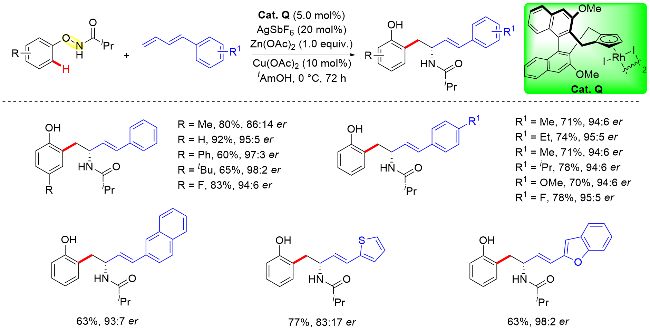
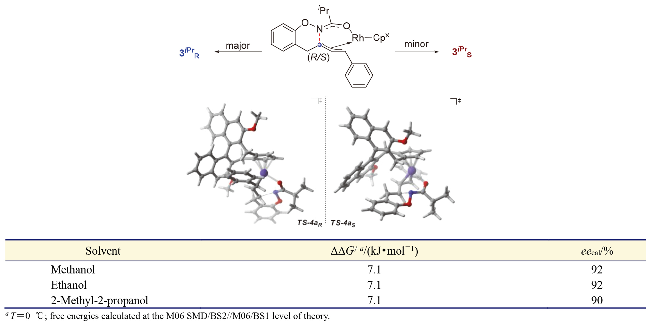
此外, 作者通过详细的DFT计算得出(R)-和(S)-对映体都可以通过烯插入/π-烯丙基化/分子内亲核取代级联反应生成, 并且亚胺对π-烯丙基铑的分子内亲核取代过程是反应立体选择性的决定步骤. 另外, 理论计算结果还表明TS-4aR和TS-4aS两种过渡态在各种反应溶剂中的能垒差异始终在7.11 kJ/mol (0 ℃)左右, 通过计算所得的理论ee值也与实验观察结果一致, 即(R)-产物具有良好的对映选择性(Scheme 45).
在上述研究工作基础上, 作者提出了一种特别的Rh(III)-Rh(I)-Rh(III)催化途径, 包括了烯烃插入/π-烯丙基化/分子内亲核取代等基元步骤. 作者推测, 环铑物种对共轭二烯烃的迁移插入所形成的π-烯丙基Rh络合物可以抑制β-H消除历程, 从而避免Heck类型产物的生成(Scheme 46).
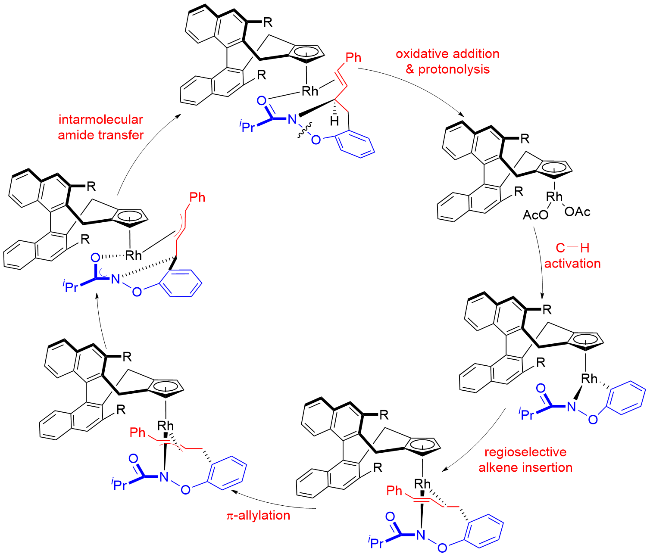
尽管该反应展现出新的化学选择性, 但立体选择性仍有待提高. 游书力课题组[58]一直致力于新型手性环戊二烯基铑配合物的合成及铑催化不对称C—H键官能团化反应研究. 针对上述反应存在的对映选择性控制水平不高的问题, 该课题组近期通过对手性CpxRh配合物结构的优化, 使用大位阻叔丁基取代的新型手性CpxRh配合物(Cat. Z), 大幅提高了该反应的化学选择性及立体选择性(Scheme 49). 作者对该反应底物普适性进行了探究. 对底物二烯烃分子中的芳环进行考察发现, 当苯环上有卤素或吸电子取代基取代时, 均能以良好的产率和优异的对映选择性获得相应预期产物(47%~76% yield, 92%~98% ee). 而供电子基团存在时, ee值有所降低, 可能是由于电子效应影响反应烯丙基铑中间体的稳定性, 从而影响对映选择性控制(70%~79% yield, 73%~89% ee). 然而, 当N-苯氧基酰胺苯环邻位被甲基或溴原子取代时, 由于位阻效应会导致反应收率较低, 但仍然保持较高的立体选择性.
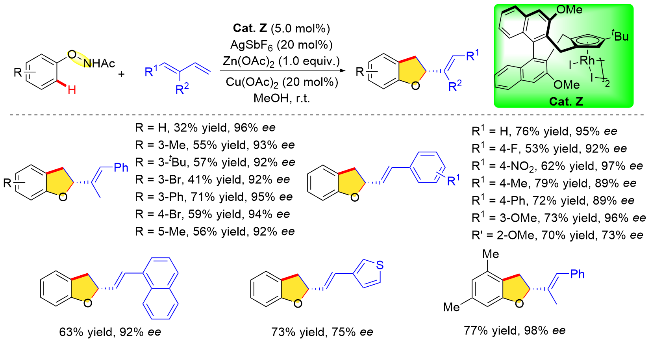
3.2.2 与炔烃分子间的偶联反应
同样的策略还可用于构建具有轴手性元素的非环状苯乙烯类化合物. 该类转化由于需要在克服较大的位阻效应的同时兼顾催化体系的活性以及烯烃产物相对较低的阻转能垒, 因此更具挑战性. 2022年, 李兴伟课题组[59]利用不对称碳氢键活化的策略, 在手性CpxRh- (III)催化剂(Cat. AA)作用下, 实现了两类含有可迁移导向基的芳烃与大位阻炔烃的偶联. 当采用N-氨酰基吲哚杂环作为底物时, 基于该底物中亲电型导向基团的特性, 可以与炔烃进行1,2-芳酰基化反应, 从而实现了手性C—N轴的构建. 值得注意的是, 该反应体系在消旋锌盐的辅助下, 可以取得优异的对映选择性. 当采用N-酚氧基酰胺作为底物时, 基于该底物内氧化型导向基团的特性, 可以与大位阻炔烃进行立体选择性1,2-芳胺化反应, 从而构建了手性C—C轴(Scheme 50). 两类反应均成功地构建了具有轴手性的非环状四取代烯烃, 具有条件温和、底物范围广、立体选择性优异和原子经济性100%等优点.
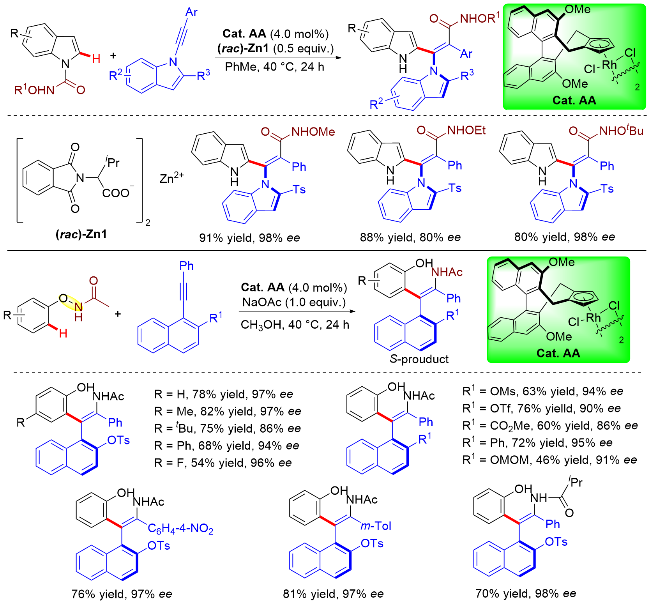
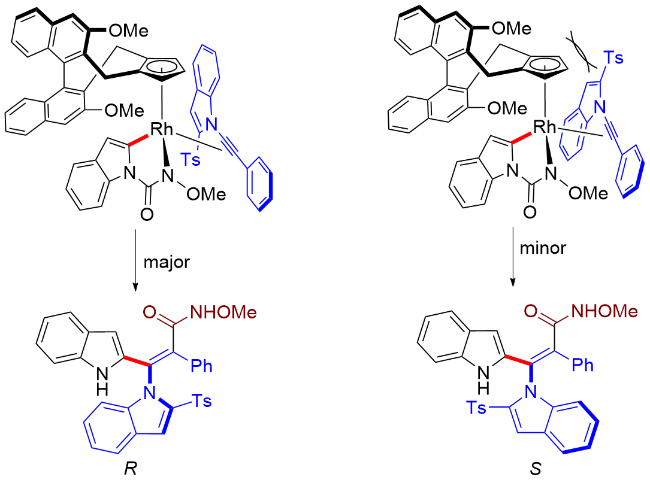
作者对N-甲氧基-吲哚甲酰胺和炔烃的偶联反应的对映选择性形成过程进行了立体结构模型阐释(Scheme 51). 在形成五元铑环中间体后, 所得的Rh(III)—芳基键进行区域选择性和对映选择性迁移插入到炔烃中. 在该过程中, 炔烃的Ts基团优选朝下以最小化与环戊二烯环的空间排斥, 由此得到主要的R构型产物.
4 C—S键氧化型导向基团参与的不对称C—H键官能团化反应
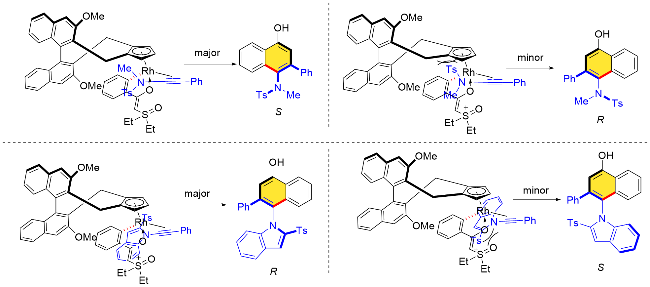
上述氧化型导向基团片段主要附着在底物芳烃侧链杂原子上, 比如N、O原子等, 随着N—O、N—Cl、O—N键的裂解, 杂原子将存在于最终的产物中. 开发碳原子连接的氧化型导向基团, 在过渡金属催化C—H键活化的过程中, 随着该氧化型导向基团内C—X键的裂解, 就能构建相应碳环骨架, 也是一种新颖的策略.
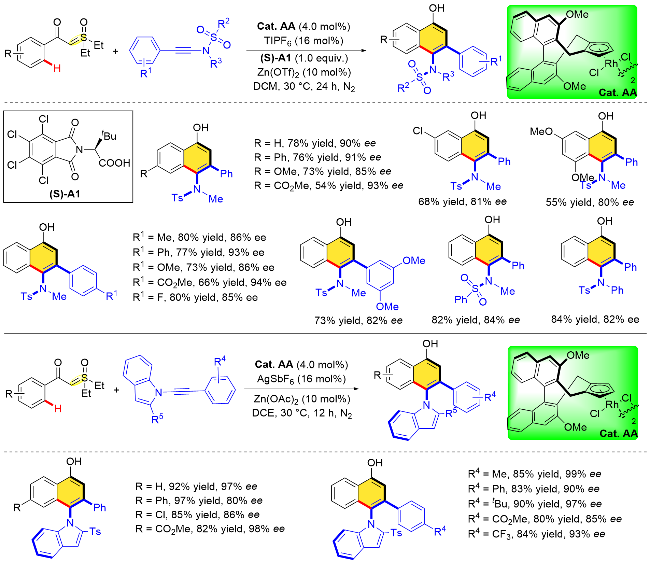
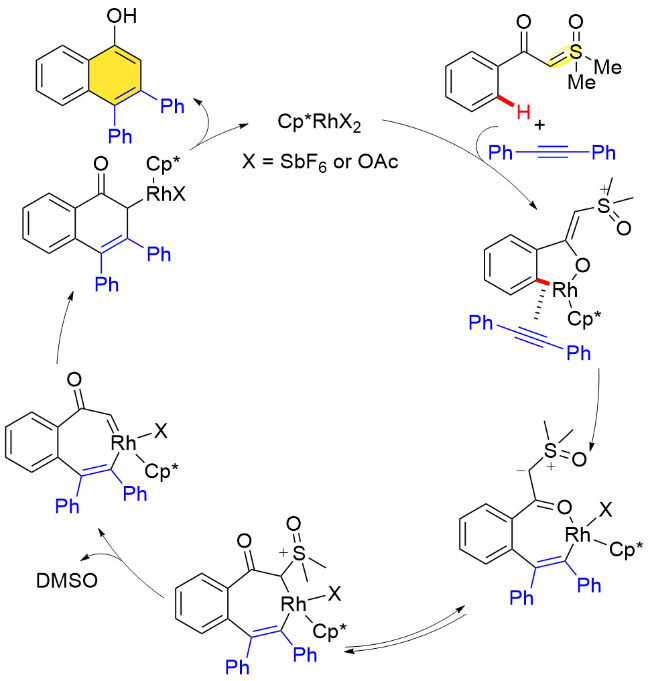
在手性版本的研究中, 通过相关机理探究实验, 作者认为该反应过程中C—H键活化步骤是可逆的. 为了更好地理解反应手性诱导的机制, 作者提出了可能的控制模型. 在C—H键活化过程中, 叶立德导向基团朝向外侧, 以减少与手性配体的空间排斥. 随后大位阻炔酰胺底物接近时, N-磺酰基朝下, 这种朝向可以使N-取代基与Cp环之间的空间位阻最小化, 最终生成S构型的主要产物. 类似地, 在二乙基硫叶立德和N-吲哚基取代的乙炔偶联的反应中, 吲哚基取代的乙炔底物接近吲哚朝下, 可能存在π-π相互作用, 从而生成R构型产物(Scheme 54).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
综上所述, 氧化型导向基团策略已成为过渡金属催化C—H键活化转化反应中的一种强有力工具. 它在转化中具有多重功能: 作为导向基团时, 可以提高C—H键官能团化的反应性和区域选择性; 作为内部氧化剂参与反应时, 避免了外加氧化剂的使用, 并且实现原位脱除或者参与反应, 大大提高了反应的原子经济性以及合成步骤的简易性. 在过去的十多年, 化学工作者们利用分子内自氧化策略, 设计合成了各种各样结构可调的手性配体及其相应的手性金属催化剂, 实现了多种氧化型导向基团参与的不对称C—H键活化转化反应, 高效地构建了具有多种手性元素的高附加值对映体富集的手性化合物.
其中, 手性环戊二烯配体作为催化不对称C—H键官能团化反应中的重要配体, 可在催化循环中稳定金属中心, 有效调控反应的化学选择性及立体选择性. 尽管如此, 仍然有几个重要的挑战有待解决: (1)利用分子内氧化型导向基团策略的对映选择性C(sp3)—H官能团化反应的研究较少. (2)手性环戊二烯配体合成步骤大多较为复杂, 部分合成反应条件严苛, 原料所需规格高且价格昂贵等, 仍需进一步简化合成反应的步骤, 同时发展新型手性配体也是迫在眉睫的工作. (3)分子内氧化导向基团的种类以内部N—O键为主, 仍需要拓展基于N—N键、N—S键、C—N键、O—O键等内部氧化剂底物分子的不对称转化研究. 目前手性环戊二烯基金属配合物仍主要集中在铑金属, 相对丰产廉价的其他金属配合物的合成及其应用, 仍值得本领域研究人员关注. 除了手性环戊二烯配体, 基于钌催化的手性芳烃配体的开发及其催化的其他不对称转化反应也具有研究前景.
(Zhao, C.)