


Biologically active compounds known as isothiocyanates (ITCs), characterized by R—N=C=S functional group, are found in cruciferous vegetables like cauliflower, broccoli, watercress, and cabbage. They have anti-tumor properties.[1-3] The biologically significant welwitindolinone and hapalindole alkaloids that have been extracted from different algae species serve as examples of them in both natural products and pharmaceutical substances.[4] Notably, the N=C=S functional group is present in glucosinolates, which are secondary metabolites present in nearly all plants and serve as a precursor for many ITCs.[5] As a defensive strategy, plant tissue injury increases the activity of the myrosinase enzyme, which causes glucosinolates to degrade and releases compounds like sulfora- phane, allyl, benzyl, or phenethyl ITC.[6] Particularly, sulforaphane demonstrated neuroprotective activity in the management of Parkinson’s and Alzheimer’s diseases, which are neurodegenerative conditions.[2,7] Moreover, ITCs express significant antiproliferative activity as well,[3,8] and the anti-microbial nature of certain ITCs makes them useful in food preservation.[9] They have also recently been used as covalent warheads in chemical biology and medicinal chemistry applications to identify cysteine or lysine residues.[10-11] Notably, they are frequently employed as intermediates in organic synthesis because of their high and adaptable reactivity.[12] ITCs are employed in polymer chemistry, participate in cycloadditions that produce a variety of heterocycles, and react easily with nucleophiles.[13]
In recent years, several new synthetic routes have been developed for the synthesis of ITCs, using fluorine-con- taining reagents, such as Langlois reagent (F3CSO2Na),[14] Ph3P+CF3CO2- (PDFA)/S8,[15] (Me4N)SCF3,[16] CF3SiMe3/ S8 or AgSCF3,[17] and BrCF2CO2Na/ S8.[18] But the most common methods are Staudinger/aza Wittig tandem reaction, in which R—N3 (azides) react with PPh3 (triphenylphosphine) and then with CS2 to produce ITCs (Scheme 1, route a).[19-22] ITCs are also synthesized by reacting R—NH2 with thiophosgene as a reagent for the transfer of the thiocarbonyl moiety in the presence of a base (Scheme 1, route b).[23-24] However, due to the toxicity of triphosgene and its sensitivity to functional groups, this reagent is increasingly replaced by surrogates, such as di-(2-pyridyl)thionocarbamate,[25] 1,1-thiocarbonyldiimida- zole,[26] or 1,1-thiocarbonyldi-2-(1H)-pyridone.[27] Final most common method is a two-step, one-pot procedure. In the presence of CS2 and base, primary amines are converted to ITCs passing through dithiocarbamate intermediate (Scheme 1, route c).
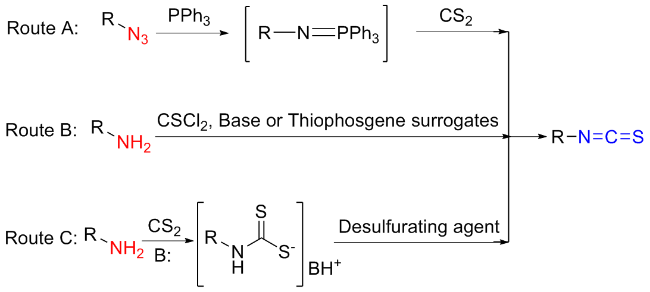
1 Synthetic approaches towards the synthesis of isothiocyanates
1.1 Synthesis of isothiocyanates from primary amines
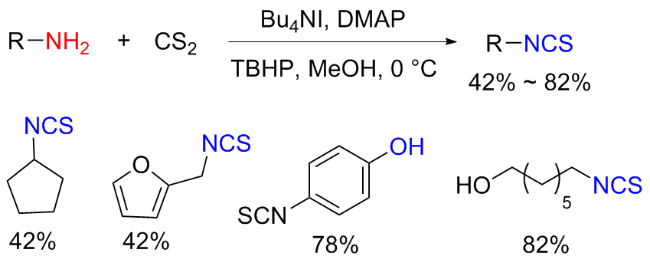
Rong et al.[28] synthesized isothiocyanates by reacting various aryl, benzyl, alkyl, and hydroxyl primary amines with carbon disulphide in the presence of dimethyl aminopyridine as a catalyst, tetrabutylammonium iodide as an excess base, and tert-butyl hydroperoxide as an oxidant. The range of findings that was achieved was 41%~82%.
Similarly, Janczewski’s research team[29] produced around thirty ITC compounds with a fair or very excellent yield (25%~97%) using primary amine and carbon disulfide. Using an organic base (Et3N, 1,5-dizzabicyclo[5.4.0]- undecen-5-ene (DBU), or N-methylmorpholinium (NMM)) and carbon disulfide via dithiocarbamates, synthesis was carried out in a “one-pot”, two-step process (Scheme 3).
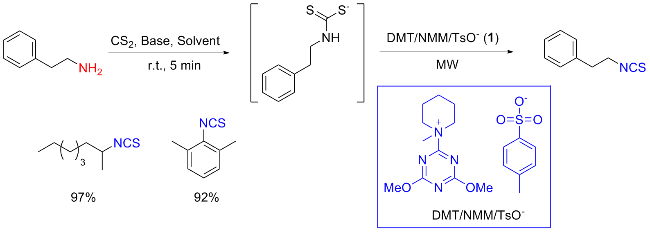
A desulfurization reagent was 4-(4,6-dimethoxy-1,3,5- triazin-2-yl)-4-methylmorpholinium toluene-4-sulfonate (DMT/NMM/TsO-), where it made use of a variety of substrates by employing primary amines, both aromatic and aliphatic. With aliphatic the maximum yield was 97%, and with aromatic it was 92%. The yield was ascribed to the lone pair of amine groups’ delocalization.
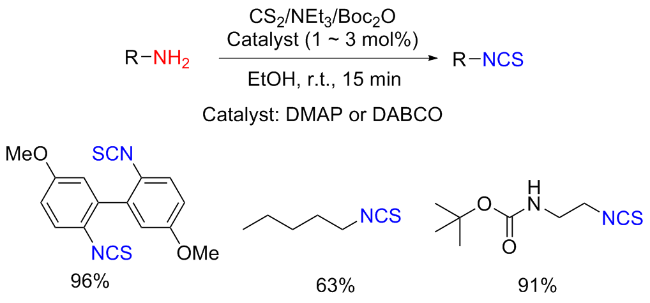
Furthermore, in 2008, utilizing di-tert-butyl dicarbonate (Boc2O) and 1~3 mol% of 4-dimethylaminopyridine (DMAP) or 1,4-diazabicyclo[2.2.2]octane (DABCO) as a catalyst, alkyl and aryl amines were smoothly converted to the respective isothiocyanates through the dithiocarbamates in good to outstanding yields. It is now possible to prepare isothiocyanates quickly, cleanly, and with excellent yields and purity, without the need for additional workup, thanks to a mild and chemoselective technique. With aliphatic and activated aromatic substrates, the reaction can be completed in 15 min; however, longer reaction durations are required for deactivated arylamines to create the dithiocarbamate entirely and avoid side reactions, such thiourea production or amine Boc-protection (Scheme 4).[30]
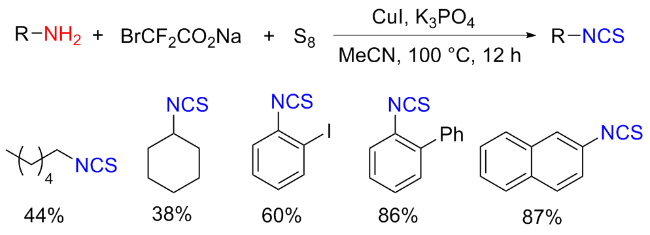
Without organophosphine, the Wei research group[18] discovered a synthetic method for isothiocyanation of amines using sulphur, sodium bromo-difluoroacetate, and a copper catalyst. It is a cheap, safe, and odorless way to synthesize ITCs in a single pot, making it easy, efficient, and capable of withstanding high functional group tole-rance. This procedure produced aryl and alkyl isothiocyanates in moderate to excellent yields from a range of primary amines (Scheme 5).
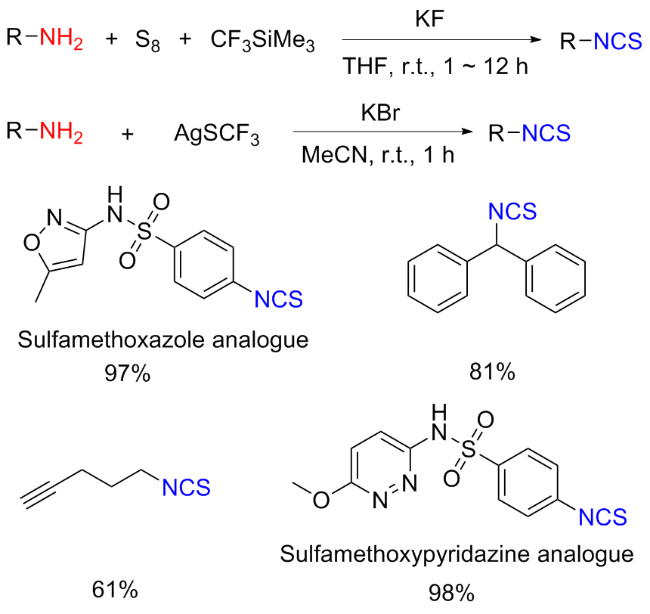
In 2019, Zhen and colleagues[17] conducted reactions between thiocarbonyl fluoride, which was obtained from inexpensive, easily accessible, and often utilized CF3Si- Me3, elemental sulphur, KF, and primary amines at room temperature in tetrahydrofuran (THF). The results yielded a diverse range of isothiocyanates in moderate to good yields. Both reactions exhibit good tolerance to functional groups and a wide range of substrates. Furthermore, AgSCF3 produces quantifiable isothiocyanates with late-stage applications when it interacts with primary amines under KBr at room temperature (Scheme 6).
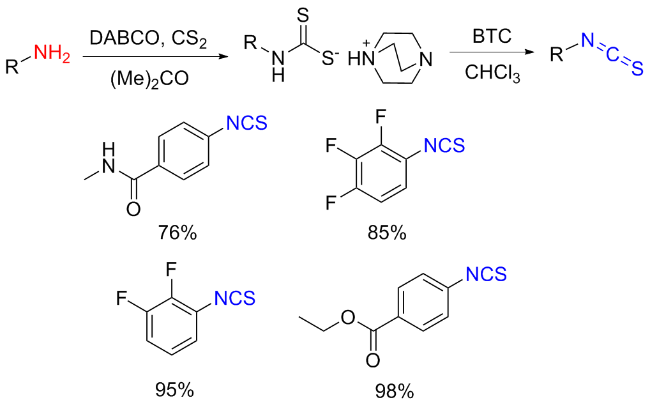
Using triphosgene as a dehydrosulurization reagent and (CH3)2CO, CS2 as a co-solvent, Liu et al.[31] established a technique for the formation of isothiocyanates from aryl and alkyl primary and dithiocarbamates. By utilizing several solvents and reaction conditions, the reactions were optimized. The discovered approach has the benefit of having mild reaction conditions, a high yield, and excellent compatibility across functional groups, making it a versatile synthetic method for synthesizing ITCs (Scheme 7).
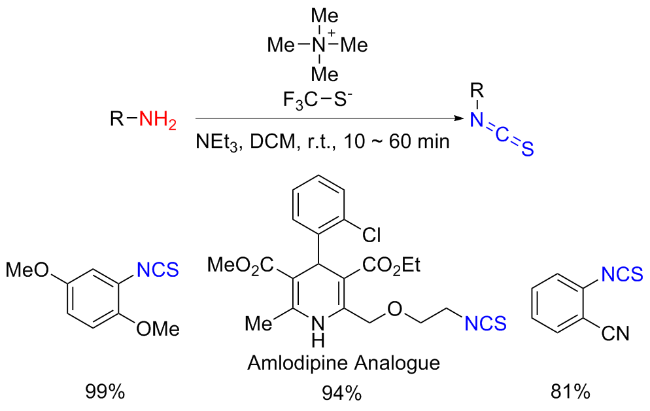
In addition to these synthetic methods, Scattolin and collaborators developed a procedure that uses non-volatile, non-toxic liquids to prepare isothiocyanates. They used the solid (Me4N)SCF3 reagent to create ITCs from primary amine, and this proved to be a fast, effective, and selective transformation process. The method benefits include fast speed, efficiency, high functional group tolerance, operational simplicity, and late-stage application. In addition to these benefits, its byproduct is a solid that is readily removed through filtration (Scheme 8).[32]
Whereas the in situ generated aryl dithiocarbamic acid (ArNHCSS-H+) exclusively produced the thia-Michael adduct (ArNHCSSCH2CH2COOMe), treatment of the carried out or the in situ produced aryl/alkyl dithiocarbamate triethylammonium salt (ArNHCSS-Et3NH+) with methyl acrylate in a water-soluble medium produced only arylisothiocyanate (ArNCS). Two possible mechanisms that depend on the reaction media pH and the type of counter cation can explain this varied reactivity. In a dithiocarbamate salt, the thiocarbonyl sulphur (C=S) atom, which has a large orbital coefficient and capable of undergoing 1,4- addition to the Michael acceptor, is softer than the thiol/thiolate sulphur (SH/S), regardless of the counter cations (Table 1).[33]
Table 1 Reaction of dithiocarbamate triethylammonium salt with methyl acrylate in aqueous buffer at different pH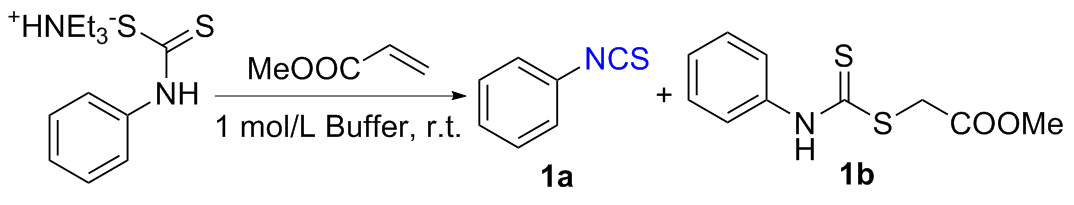 |
| Buffer | Initial pH | Final pH | Yield/% | |
|---|---|---|---|---|
| 1a | 1b | |||
| K2HPO4+KH2PO4 (1 mol/L) | 6 | 6.45 | Trace | 91 |
| 7 | 7.44 | 5 | 88 | |
| 8 | 8.84 | 15 | 77 | |
| 9 | 9.45 | 65 | 22 | |
| 10 | 10.18 | 82 | 8 | |
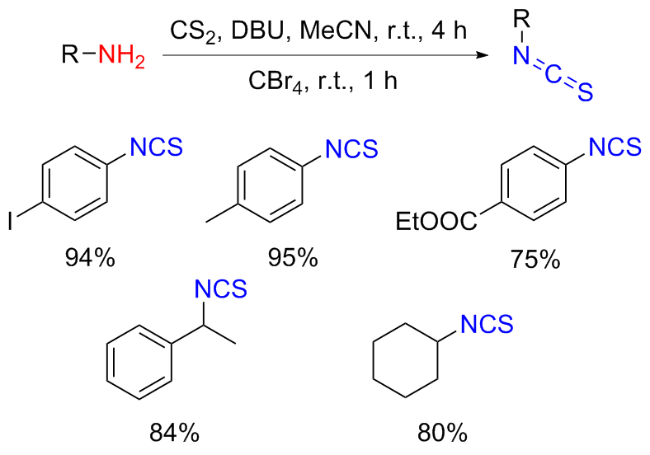
Techapanalai et al.[34] successfully synthesized isothiocyanates from the corresponding amines in a one-pot process by employing carbon tetrabromide as a desulfurizing agent. The best yield among those shown in Scheme 9 was obtained by in situ synthesis of dithiocarbamate salts from amines followed by desulfurization using carbon disulphide. They optimized the reaction with different substituted aryl amines, such as electron-withdrawing, electron- donating, halogens, and some aliphatic amines.
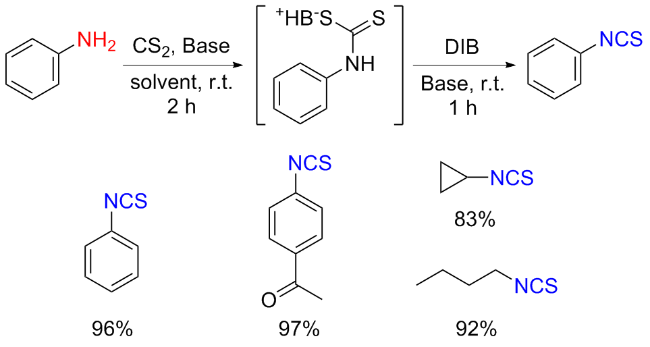
Diacetoxyiodobenzene was used as a desulfurizing agent during the synthesis of ITCs, which was carried out in micellar conditions in water. Under ideal reaction circumstances, functional group tolerance was also investigated. Every amine with electron-donating or electron-with- drawing properties undergoes the reaction to generate the corresponding isothiocyanates in moderate to good yields.
1.2 Synthesis of Isothiocyanate from isocyanides
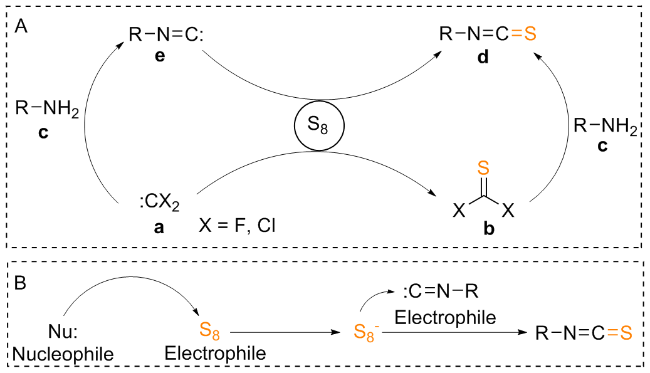
The sulfuration of isocyanides directly leads to ITCs (Scheme 11). Aromatic isocyanides react with sulfur to form isothiocyanates (ITCs) upon refluxing in benzene for 3 d with moderate yields obtained.[36] On the other hand, aliphatic isocyanides do not undergo any reaction.[37] This has led researchers to discover that catalysis or other types of activation, particularly nucleophilic additives, are necessary for an efficient, valuable, and comprehensive methodology. The most effective substitute for integrating the sulphur atom into the product is elemental sulphur. Sulphur, which acts as an electrophile because of its vacant d-orbitals, is attacked nucleophilically by in situ produced carbene functionalities (a). Using this method, primary amines (c) combine with thiocarbonyl surrogates (b), which are often dihalogenides to produce ITCs (d). Otherwise, ITCs are produced by reacting with sulphur under heat conditions or with external additions in isocyanides (e), where the terminal carbon atom may function as a carbene (Scheme 11a). Notably, it was also claimed that adding sulphur to formaldimines produced ITCs. However, this technique is hardly ever employed nowadays. The idea that a switched mechanism also exists, involving a nucleophilic sulphur anion (Sx-) and the carbene of the isocyanide (e) acting as an electrophile, is supported by the easy activation of sulphur by nucleophilic additives, such as aliphatic amines and hydroxyl, sulphide, and cyanide anions, and the correspondingly milder conditions compared to thermal activation (Scheme 11b).
1.2.1 Catalysis
Chalcogens or transition metal catalysts, i.e., selenium,[38] tellurium,[39] molybdenum,[40-41], or rhodium,[42] facilitate the synthesis of ITCs.[43-44] Unlike sulphur, selenium interacts with isocyanides in refluxing THF more quickly in the presence of a base, producing isoselenocyanates that can quickly transform into ITCs with sulphur (Scheme 12).[38] As demonstrated by Fujiwara and colleagues, selenium is required as an additive in the reaction, albeit only at a catalytic loading of 5 mol%. Afterward, they demonstrated that the same tellurium catalyst exhibited higher catalytic activity on aliphatic derivatives, achieving higher yields with a significantly lower catalyst loading of 0.02 mol%.[39]
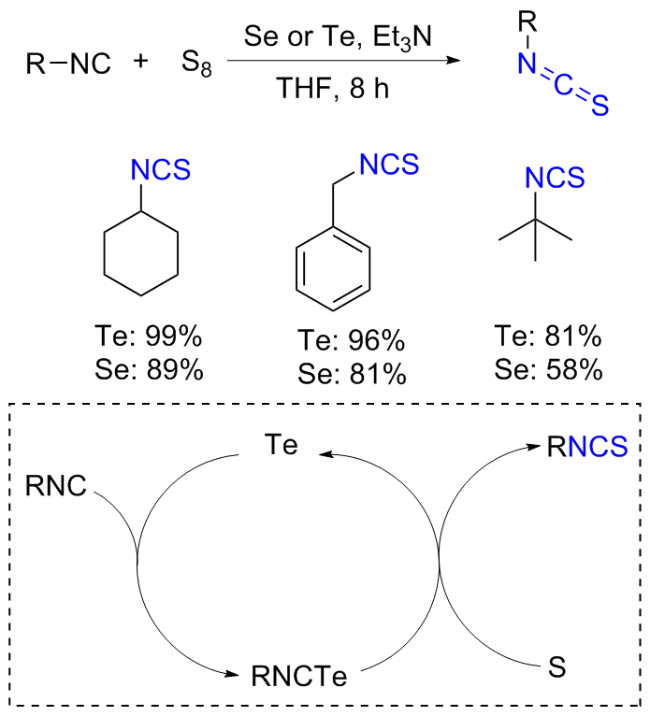
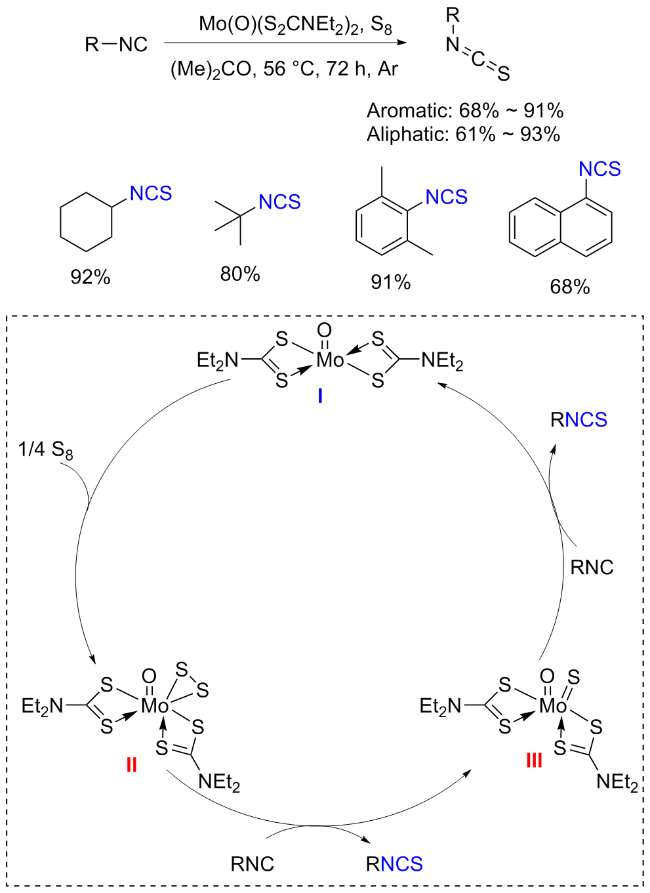
Adam and associates[40,45-46] developed a base-free method using a molybdenum catalyst, which had previously used to episulfidate alkenes and allenes with sulphur, to get around the toxicity of chalcogens. It took 3 d for the reaction between isocyanides (Scheme 13) and sulphur in the presence of molybdenum catalyst to reflux acetone and produce ITCs in good to outstanding yields.[40] Sulfuration may provide the molybdenum disulfur complex, the active sulfur-transferring agent in the initial stage of the process. Disulfur compound is applied in stoichiometric proportions, which causes ITC in just 2.5 h, demonstrating its role in the reaction (Scheme 13).
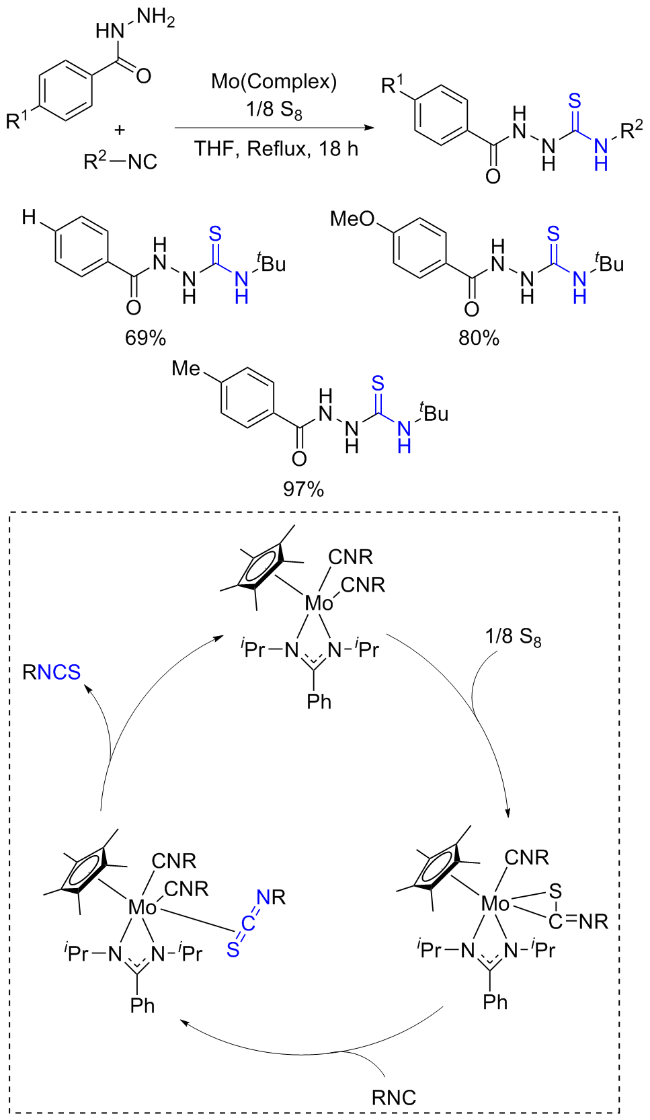
The work of Sita and associates[41] further supports the catalyst involvement in the sulfur-to-isocyanide addition. Through ligand exchange, they created bis(isocyanide)-Mo complexes, which then converted into (S,C)-ITC-molyb- denum complexes using sulphur (Scheme 14). Based on X-ray crystallography, a sulphur-linked complex is likely a crucial chemical step. Based on isonitrile as the starting point, 1H NMR studies at 50 ℃ in benzene-d6 with a 5% catalyst loading revealed that ITCs were produced in 16 h in the presence of isocyanide and sulphur.
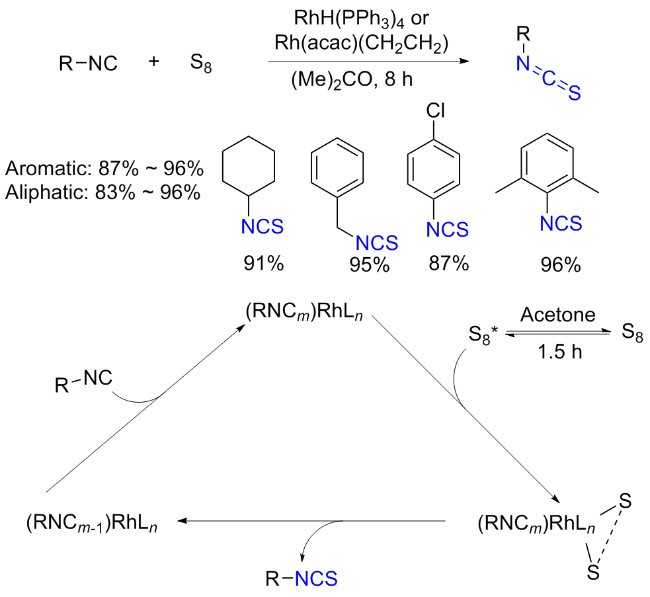
Following molybdenum, rhodium was shown to have catalytic activity in the sulphur processes during the synthesis of 1,4-dithiins from cyclic alkenes, diaryl sulphides, and alkene episulfidation.[47-49] Rh(acac)(CH2=CH2)2 and 1 mol% RhH(PPh3)4 were used by Yamaguchi and colleagues[42] to convert isocyanides to ITCs in refluxing acetone (Scheme 15). Interestingly, they found that refluxing sulphur in acetone for 1.5 h before usage reduced reaction times. Sulphur most likely undergoes thermal activation by producing polysulfides, which is followed by catalyst-in- duced sulphur atom exchange.[50] Specifically, the use of organic tri- and tetrasulfides in the isocyanide reaction also resulted in the synthesis of ITC.
1.2.2 Nucleophile-induced transformation of isothiocyanides to ITCs
The most frequent way to activate Sulphur is to split the octasulfur ring using nucleophiles.[51-57] Under mild circumstances, cyanide, hydroxyl, and sulphide ions can generally break S—S bonds homolytically (Scheme 16A) or heterolytically (Scheme 16B), producing reactive linear polysulphide anion chains of varying lengths and radical anions.[58-59] Notably, (hetero)aromatic amines are typically not nucleophilic enough, whereas nucleophilic aliphatic amines are quite successful at activating sulphur.[60] Although primary and secondary amines can function in ambient circumstances, their application is restricted due to their involuntary reaction to in-situ produced ITCs. To stabilize the linear polysulphide chains, tertiary amines require more severe conditions to activate sulphur and perhaps the presence of a proton source.[61]
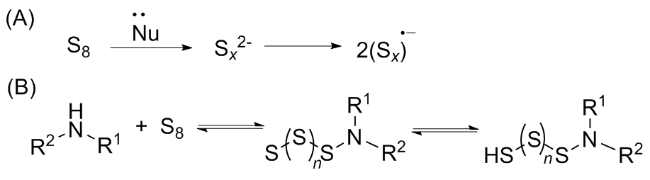
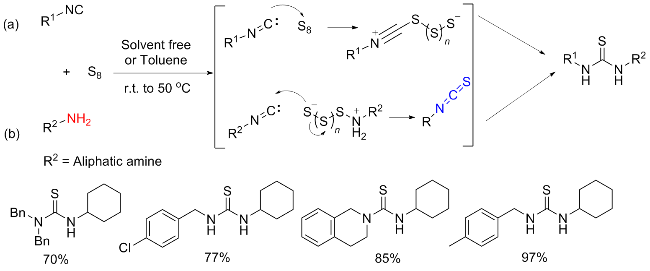
Al-Mourabit and colleagues[62] developed a three-step process for synthesizing thioureas, which begins with sulphur, aliphatic amines, and isocyanides. They proposed two mechanistic paths. One pathway goes through an intermediate that results from the nucleophilic attack on sulphur and contained a structural constituent of nitrilium (Scheme 17a). The thioureas are then produced by the electrophilic adduct nitrilium interacting with an aliphatic amine. Conversely, the aliphatic amines have the potential to produce nucleophilic polysulphide anions first from sulphur (Scheme 17b), which would reverse the reactivity with sulphur acting as the nucleophile and isocyanide (Scheme 17a) as the electrophile. Thiourea could then be produced by a straightforward addition reaction between the in situ synthesized ITCs and aliphatic amine. Path b (Scheme 17b) is supported by the mild conditions because the reaction would require much higher heat activation in the absence of exogenous additives.[36-37]
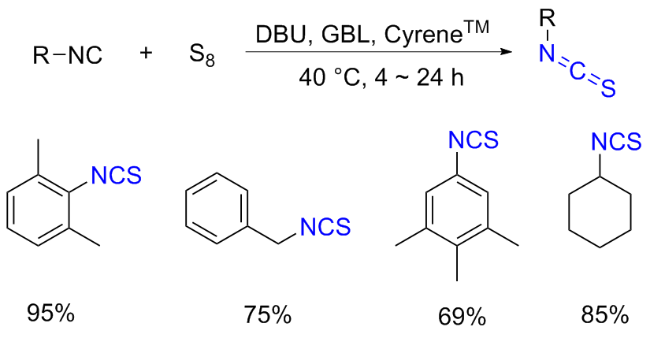
Ultimately, Meier and associates[63] have released their refined technique for producing ITCs from sulphur and isocyanides with just a 2~4 mol% base in renewable solvents (Scheme 18). They examined several tertiary amines in the process, such as DMAP, 1-methylimidazole (NMI), Et3N, DABCO, DBU, and 1,5,7-triazabicyclo[4.4.0]dec- 5-ene (TBD), and it was discovered that, in general, more substantial conversions were correlated with increased basicity. They eventually used the discovered approach to synthesize a small library of ITCs, demonstrating the technology broad applicability. Regarding sulphur as a nucleophilic partner in the reaction with the electrophilic isocyanide, they put out the same mechanistic arguments.[63]
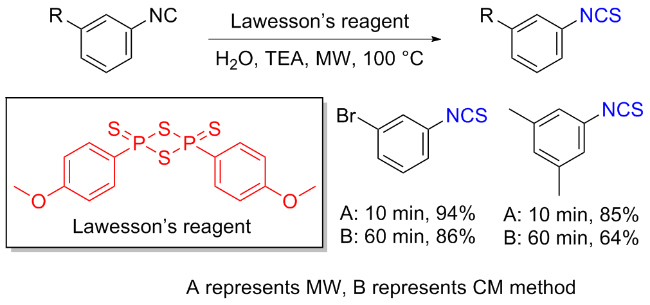
The Salami research group[64] used Lawesson’s reagent as a sulphurization agent and amine bases such as triethyl amine in a catalytic amount to synthesize isothiocyanates from isocyanides. This process includes microwave exposure of reactants in the presence of water. The researchers also used a conventional approach to carry out the synthesis of ITCs, although the microwave-assisted reaction produced a higher yield than the traditional method did (Scheme 19).
1.3 Other synthetic methods for isothiocyanates
As research improves the quality and opens up new avenues for targets, the main sources for the synthesis of ITCs are primary amines and isocyanides. A novel route via Staudinger/aza from azide, the related primary deoxyisothiocyanato sugars are produced in high yield by the Witting reaction of primary azidodeoxy sugars with triphenylphosphine-carbon disulphide. One benefit of the approach used to synthesize ITCs is that it limits the migration of O-N acyls or prevents the production of dimeric carbodiimides. Furthermore, triarylphosphine supported by polymers can effectively substitute triphenylphosphine, reducing the purification stage to a straightforward filtration procedure. From the appropriate azide precursors, the method also permits the synthesis of 5-deoxy-5-isothio- cyanato sugars, a yet unidentified class of chemicals (Scheme 20).[65]
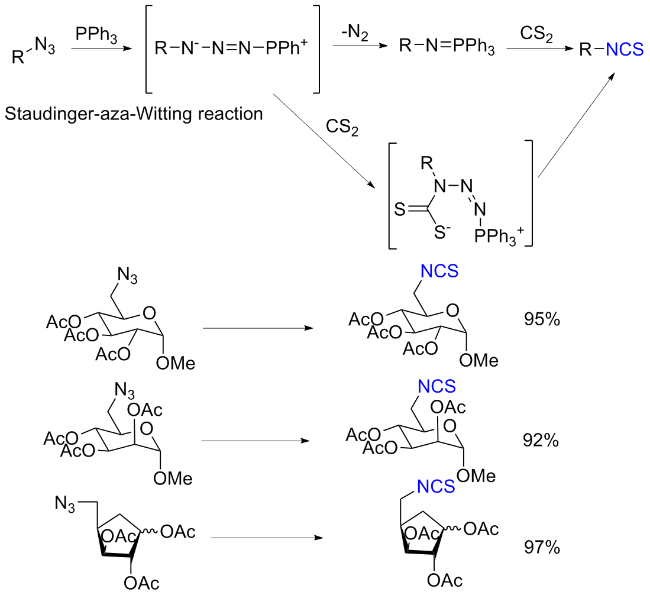
Similarly, Santhosh et al.[19] developed the synthesis of ITCs. The method for obtaining Nβ-protected aminoalkyl isothiocyanates from Nβ-protected aminoalkyl is unified. The availability of the precursors and, particularly in amino acid chemistry, the behavior of the other reactive groups towards them determine the kind of protocol to access isothiocyanates.
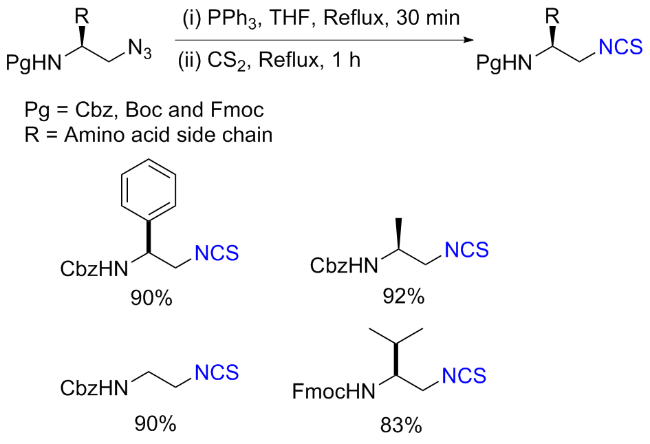
Luckily, neither of these variables was a worry because the current approach cleared the way to access title compounds without compromising the benzyl and tertiary butyl groups in the side chains, the Boc, Cbz, and Fmoc protective groups, or the precursors azides, which were synthesized easily by standard protocols. The study benefits include the elimination of the requirement for the amine title compound, configuration retention, ease of handling, and simple purification (Scheme 21).
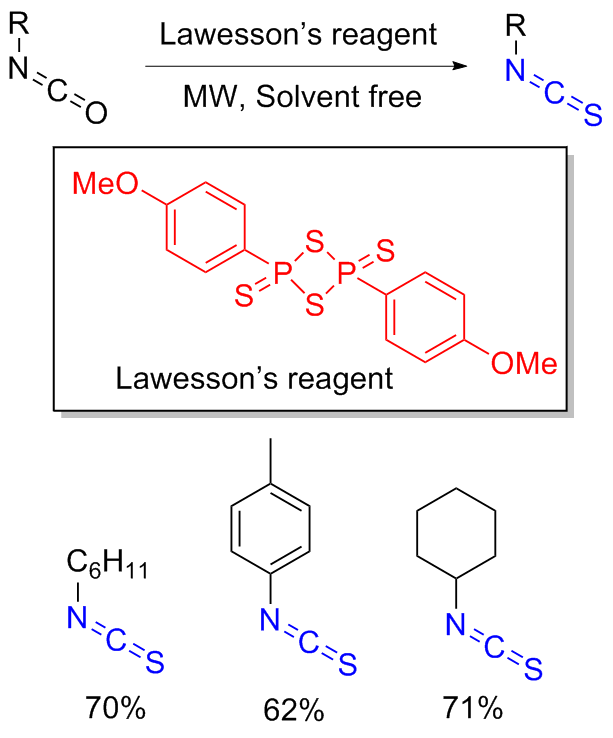
Valette et al.[66] established a solvent-free, microwave- assisted synthetic method for creating isothiocyanates from isocyanate by substituting sulphur for oxygen using Lawe- sson’s reagent. Thanks to the straightforward and quick technique, the work-up is swift, clean, and doesn’t require much time. They produced aryl and alkyl isothiocyanates in their study, albeit with varying yields (Scheme 22).
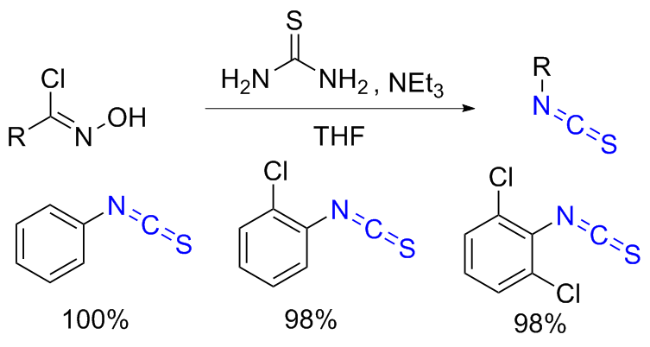
Although it is usual to obtain isothiocyanates from amines, aldehydes are the first step in this process. This method has rapid reaction times, straightforward workup, and quantitative yields. It was accomplished by adding triethylamine to a hydroximoyl chloride and thiourea solution in tetrahydrofuran (Scheme 23). For one to five minutes, this mixture is mixed at room temperature. Even though urea is created as a byproduct, it is easily extracted using water and diethyl ether. No additional purification is necessary. Alkyl and aryl isothiocyanates can be produced using this process, and all of them yield ≥98%.[67]
2 Synthetic applications of isothiocyanates
Isothiocyanates are the thio-analogue of isocyanates. It has diverse synthetic applications in the synthesis of different heterocyclic and non-cyclic compounds such as thiophene,[68] thiourea,[69] and some of its applications in heterocyclic compounds are overviewed (Figure 1).[70-71] Beside these, Sharma[72] and Yang[73] have provided detailed reviews of the ITCs applications in the synthesis of thiazetidines, thiophenes, imidazoles, thiazoles, oxazoles, isothiazoles, oxathiolanes, dithiolanes, dioxolanes, triazoles, thiadiazoles, oxadiazoles, tetrazoles, thiatriazoles, pyridines, thiopyrans, oxazines, thiazines, pyrimidines, triazines, thiadiazines, oxadiazines, triazepines, benzimidazoles, benzothiazoles, quinazolones, thiadiazoles and various bicyclic and polyheterocyclic compounds with ring nitrogen and sulfur.
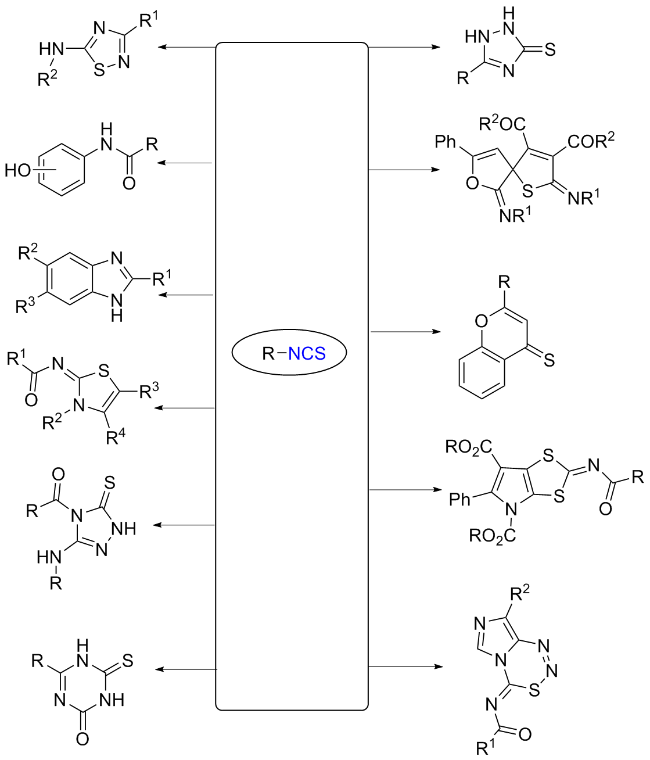
2.1 Cycloaddition reactions
ITCs are a useful synthon for creating a variety of heterocycles because of their highly electrophilic and nucleophilic centers. In many different organic syntheses, these heterocycles serve as intermediates. Sorting these ITCs according to their different cyclization modes will allow us to study their reactivity. It is discovered that the cyclized product yields distinct heterocycles depending on the cyclization mechanism. We have covered its reactivity with this distinction in the following section.
A significant and topical structural motif found in both natural goods and pharmaceutically active substances is 2-aminobenzothiazole.[74-76] Furthermore, several compounds with this structural motif are used to treat various illnesses, including cancer, tumors, and tuberculosis.[77-80] Scientists constantly look for effective ways to synthesize 2-aminobenzothiazoles. The most popular of these techniques is the use of 2-halo anilines as a beginning precursor.[77,79]
In this context, Karthikeyan et al.[78] presented a metal- free, one-pot technique via electron-deficient 2-haloanili- nes using aromatic isothiocyanates to synthesize 2-amino- benzothiazoles. Mechanistically, targeted benzothiazoles are produced by intramolecular cyclization of the thiourea produced in situ by the SNAr process (Scheme 24).
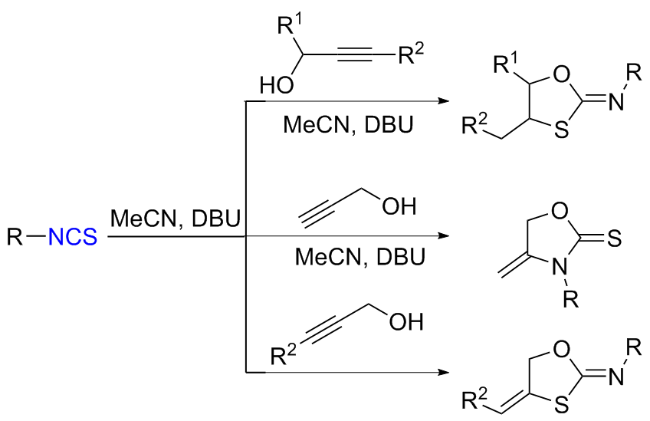
In 2020, Bera et al.[80] published a DBU-mediated synthesis of 1,3-oxathiol-2-ylidenes and 4-methyleneoxazo- lidine-2-thiones by combining ITCs and propargylic alcohols. The propargylic alcohol substitution pattern governs this 5-exo-dig cyclization. The terminal alcohols favor the N-nucleophilic assault that produces 3-substituted 4-meth- ylene oxazolidine-2-thiones. On the other hand, from secondary and primary propargylic alcohols, respectively, internal propargylic alcohols undergo exclusive S-nucleo- philic cyclization to provide (Z)-1,3-oxathiol-2-ylidenes and (Z)-N-4-ethylidene-1,3-oxathiolan-2-ylidenes (Scheme 25).
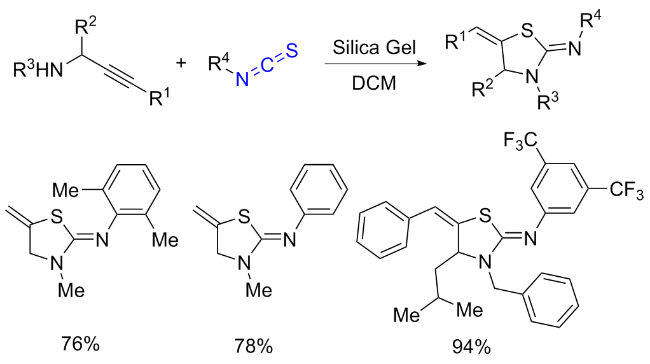
The Lovely group[81] demonstrated the synthesis of a wide range of thiozolidines by tandem thioacylation-hy- drosulfenylation of propargylamine aided by silica gel. Although thioacylation proceeds quickly at ambient temperature, careful observation of the reaction showed that the ultimate cyclization only occurred during chromatographic purification or in the presence of silica gel. The process generates excellent to good yields of 2-amino- thiazolidines quickly and is tolerant to a wide range of substitution patterns, internal and terminal alkynes, and isothiocyanates (Scheme 26). Clausen et al.[82] report another efficient technique for the synthesis of 2-imino-4- methylenethiazolidines from isothiocyanates and secondary propargylic amine. The reaction has several notable characteristics, including the ability to occur in aqueous medium, the absence of additives, and gentle conditions. The synthesis of a functionalized ether lipid allowed for more investigation of the reaction compatibility.
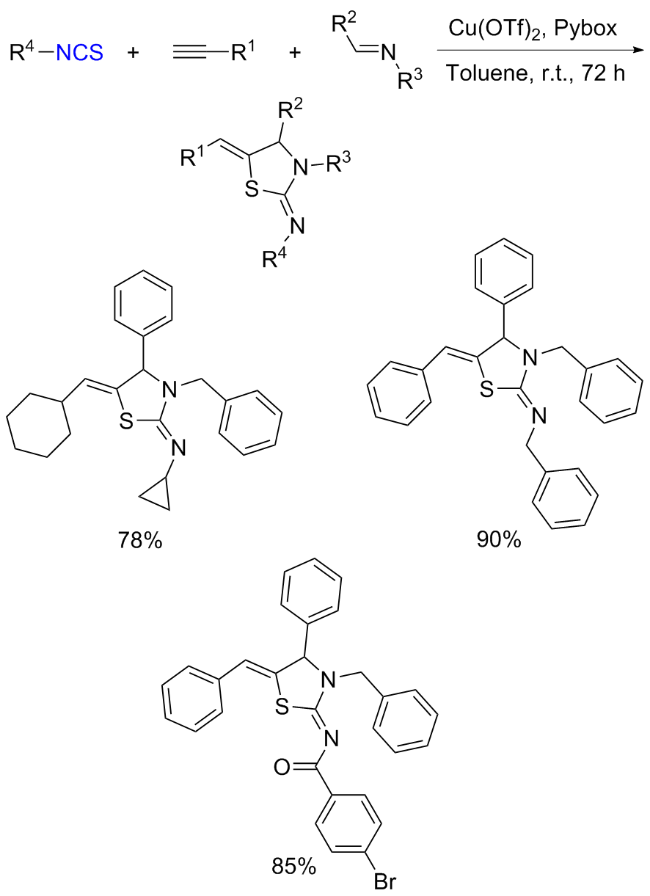
The Dethe group[83] used tandem alkynylation/hydro- thiolation to create enantiopure thiazolidine-2-imine from acetylenes, imines, and isothiocyanates. The chiral copper-pybox catalyst is responsible for catalyzing this multi-component reaction. The product exhibits enhanced enantioselectivity and regioselectivity upon the adoption of a copper-pybox catalyst system (Scheme 27).
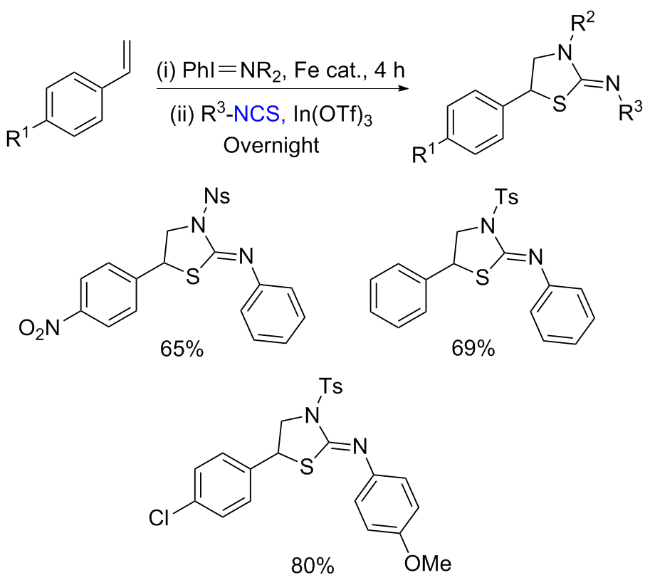
Coin and Latour group[84] described the synthesis of 2-iminothiazolidines from styrene and phenyl isothiocyanate using a domino ring opening cyclization reaction (DROC) as an effective method. This one-pot approach combines the domino ring-opening cyclization (DROC) and nitrene transfer processes in the telescopic reaction methodology. The first step in the suggested synthesis process involved aziridinating styrene using an iron catalyst and a nitrene precursor (PhI=NTs). To create 2-imi- nothiazolidines, cumulene phenyl isothiocyanate and Lewis acid In(OTf)3 were introduced to the same reaction vessel after PhI=NTs had been completely consumed. Using different styrene and phenyl isothiocyanate substituents, a wide range of 2-iminothiazolidines were created with consideration for their potential use in medicine (Scheme 28). Nevertheless, a minor excess of phenyl isothiocyanate is required to improve the yield of 2-minothiazolidines, since the ring-opening of aziridine produced several by-products.
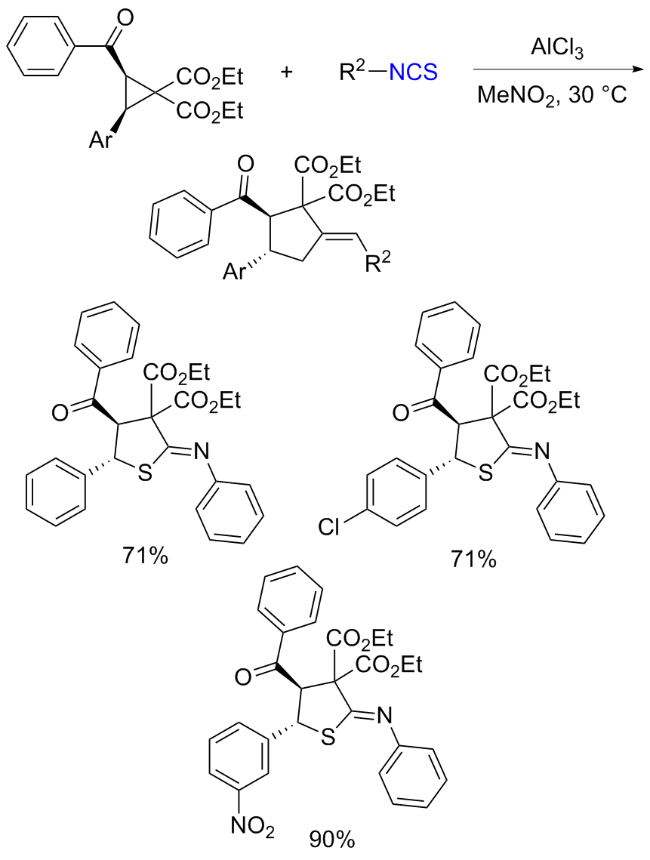
An AlCl3 catalyzed [3+2] annulation of donor-acceptor cyclopropanes using isothiocyanates has been developed by Yang and colleagues[85]. They selected cis-2,3-disub-stituted cyclopropane 1,1-diesters as the 1,3-dipole and aryl isothiocyanates, and the matching dipolarophile for this transformation. The annulated product, poly-substi- tuted 2-iminodihydrothiophenes, was produced in moderate to good yields. The reaction was favored in polar solvents such as CH3NO2 (Scheme 29).
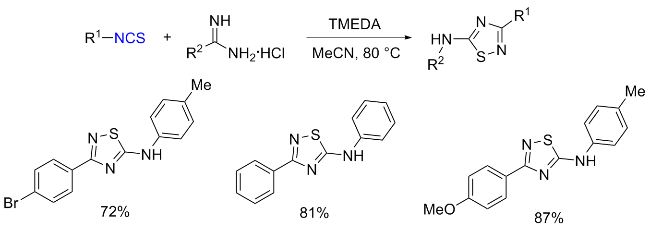
An effective green technique for the synthesis of 5-amino-1,2,4-thiadiazoles from amidine hydrochloride and isothiocyanates was devised by Yang and Zhou’s group[73]. The procedure for creating C—N bonds for uses of iodine or any transition metal catalyst. The reaction fate was examined at optimized conditions using different substituted phenyl isothiocyanate, and it was discovered that electron-donating substituents are more effective in the reaction than electron-withdrawing ones. However, under mild reaction conditions, several functional groups are readily tolerated and produce the expected products with good yield and strong regioselectivity (Scheme 30).
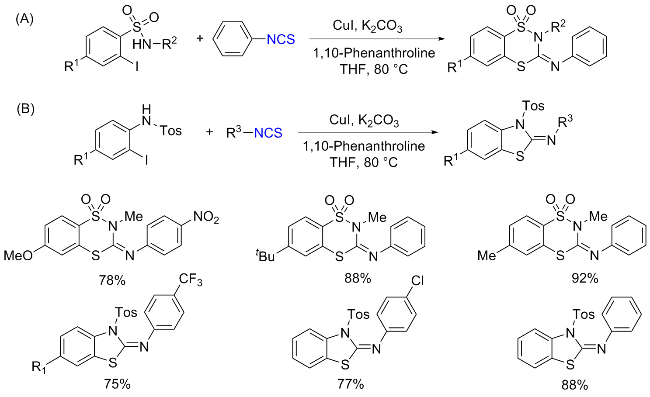
Using 2-iodo-sulfonomides and Cu-catalyzed annulation of isothiocyanates, Swamy et al.[86] were able to construct benzodithiazines and benzothiazolylidene-anilines. This one-pot cyclization of substituted sulfonamides is achieved with a broad functional group tolerance and high stereoselectivity. When the 2-iodo-N-tosyl system was used, the same technique was applied to create benzo[d]thiazol- 2(3H)-ylidene-anilines. A wide functional group for aryl isothiocyanates is likewise tolerated by this reaction, producing the required product in moderate to good yields (Scheme 31).
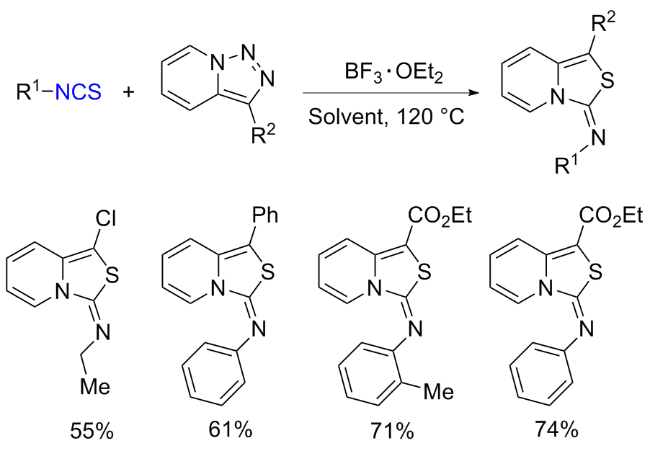
Adimurthy et al.[87] reported a denitrogenative transannulation of pyridotriazoles using isothiocyanates, catalysed by BF3•OEt2. Iminothiazolo-pyridines are produced by this metal-free transannulation in an excellent to moderate yield when solvents (dichlorobenzene-dichloroethane) are combined. Both esters and the aryl-modified substrate (for R1) produce the matching annulated products in good yields under ideal circumstances. Under this approach, various substituents containing isothiocyanates that donate and withdraw electrons interacted well with both substrates (Scheme 32).
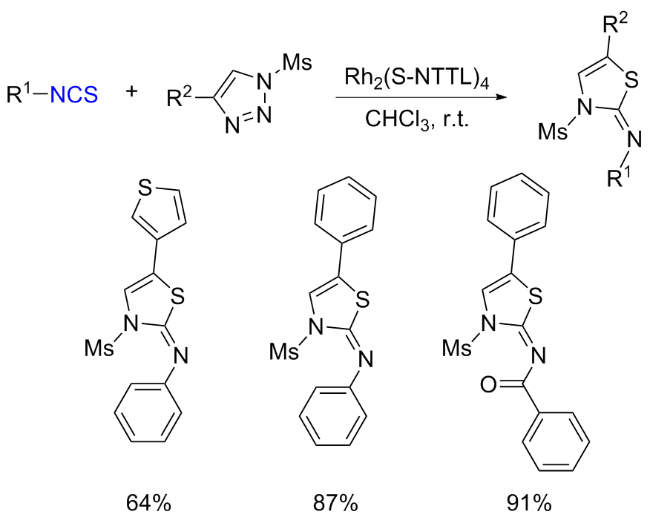
Fokin et al.[88] discovered another denitrogentaive trans-annulation of a similar kind for the synthesis of thiazoles. For this [3+2] cycloaddition synthesis, the easily accessible 1-mesyl-1,2,3-triazoles and isothiocyanates were utilized. Rh(II) salt acted as the catalyst for the reaction, which moved forward through an azavinyl carbene intermediate (Scheme 33).
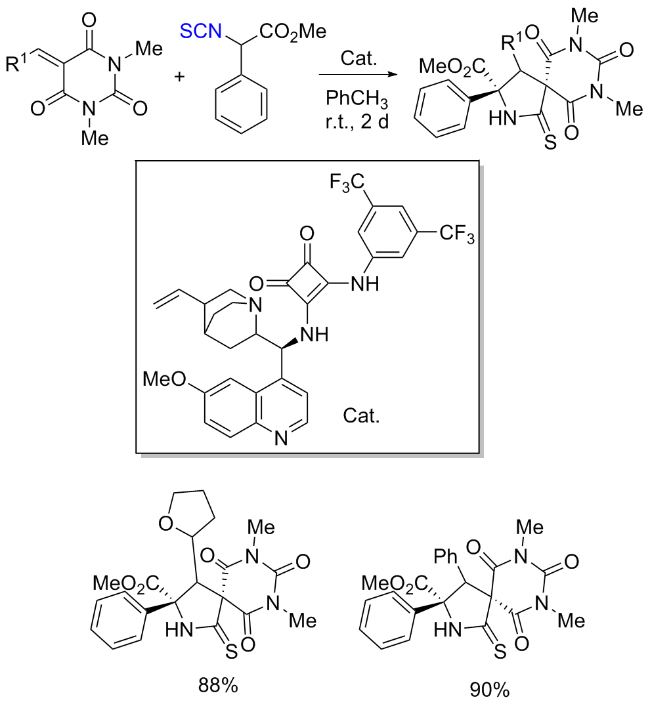
Under conditions of differential reactivity, the electrophilic site in ITCs involving azomethine linkage may result in the formation of diverse heterocycles. Various five- and six-membered heterocycles containing one or more heteroatoms have been formed in a cascade manner, depending on the type of nucleophile and initial isothiocya- nate.[89-94] Numerous techniques for synthesizing spiropyrrolidine skeletons have been published due to their biological significance. The central structure of spiropyrrolidine frameworks opens up significant possibilities for creating new medications.[95-96] The Albrecht group[97] stated in 2018 that they had synthesized pyrrolidine-based heterocycles with enantioselectivity utilizing isothiocyanates produced from α-substituted α-amino acids and olefinic barbiturates or Meldrum’s acid derivative. The cascade approach starts with cyclization and moves on to a Michael addition. Excellent stereocontrol and a good yield of targeted heterocycles were produced (Scheme 34).
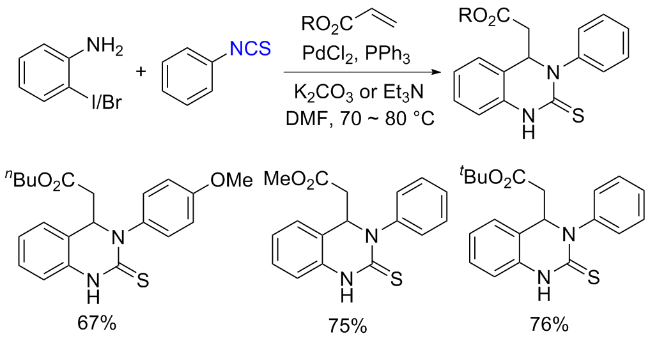
An effective Pd-catalyzed tandem method was created by Verma et al.[98] to synthesize highly functionalized tetrahydroquinazolines, which were obtained from o-halo- anilines, acrylates, and isothiocyanates (Scheme 35). The first step of the reaction is the Pd-catalyzed intermolecular Heck coupling between o-haloani lines and acrylates. This is followed by the intramolecular chemo-selective aza- Michael addition and the in situ synthesis of thioureido compound. The approach is helpful for the synthesis of substituted tetrahydroquinazolines because it can accommodate different functional groups in isothiocyanates, o-haloanilines, and acrylates.
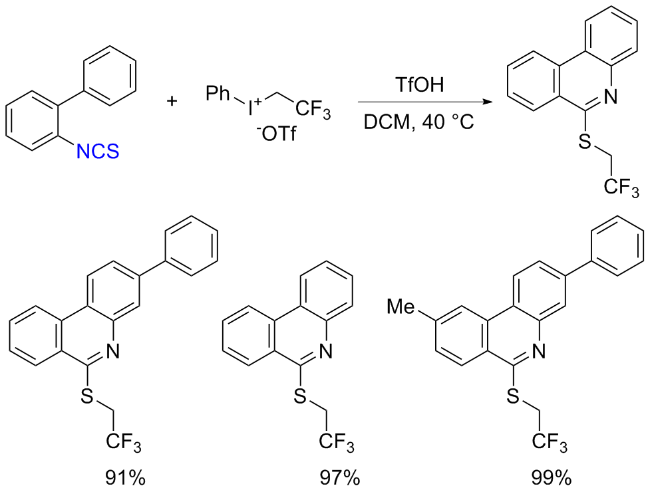
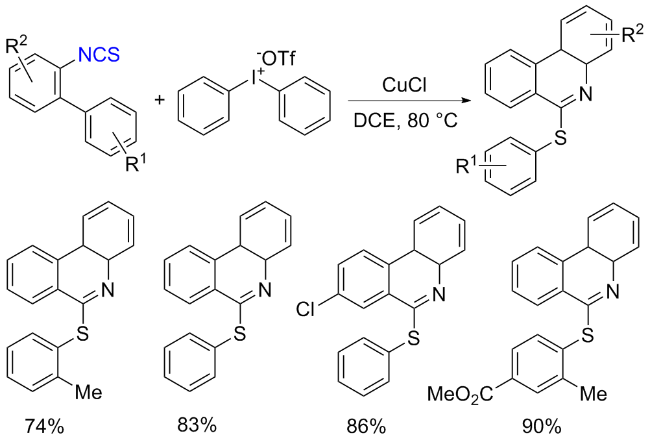
Zhang et al.[99] reported a TfOH-promoted synthesis of aryl(2,2,2-trifluoroethyl) iodonium salts and 2-isothiocya- nato biaryls that yield trifluoroethylated phenanthridine. This convenient cascade trifluoroethylation/cyclisation was facilitated by TfOH as its absence resulted in only traces of the desired product. When the fluorinated phenanthridine and isoquinoline derivatives were synthesized from isothiocyanates, aryl(2,2,2-trifluoroethyl) iodonium salts were a potent electrophilic source of trifluoromethylation. According to the controlled experiments, TfOH initiated the production of phenanthridine-6(5H)-thione, the first step in the process. The NCS nitrogen is protonated by the acid, creating a cation resulting in phenanthridine-6(5H)-thione. The final product is finally produced by fast trifluoromethylation of this thioamide intermediate at the sulphur center by aryl(2,2,2-trifluoroethyl)iodonium salts (Scheme 36).
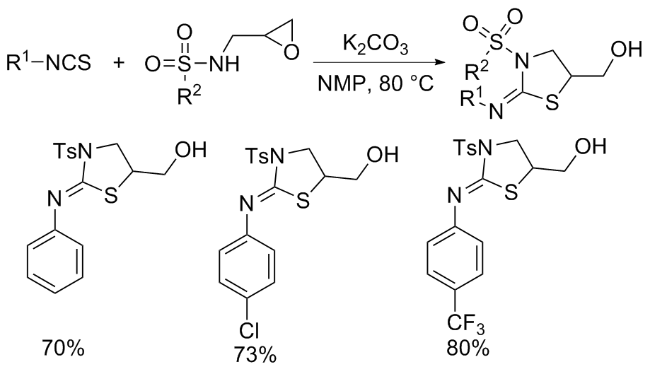
According to a technique described by Anitha and Swamy[100], epoxides exhibit aziridine-like behavior and expand their rings when exposed to isothiocyanates in the presence of a base. Isothiocyanates regioselectively open up sulfonamides containing epoxides to create iminothiazolidines. The nice thing about this reaction is that it didn’t require the addition of any Lewis acid to isolate the regio selective product in good to moderate yields (Scheme 37).
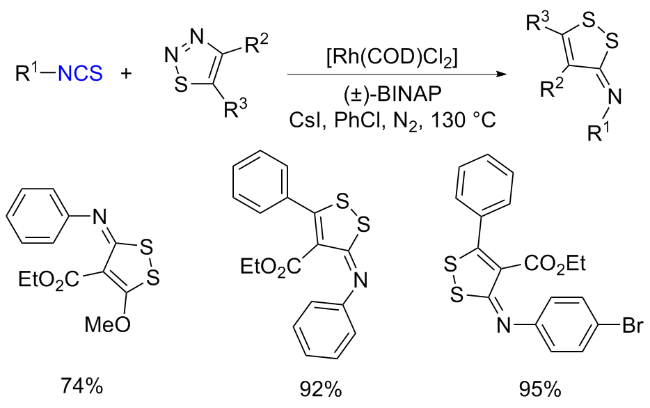
Yang et al.[101] described a Rh-catalyzed [3+2] oxidation of α-thioacyl carbenes with aryl isothiocyanates, obtained from 1,2,3-thiadiazoles. Because the more nucleophilic C—S acts as the reactive site rather than the less nucleophilic C—N bond of the isothiocyanate, the reaction proceeded in an umpolung, stereospecific, and regiospecific manner. In contrast, 1,2,3-thiadiazoles participate in this Rh-catalyzed redox-neutral [3+2] annulation as masked S-electrophilic thia-1,3-dipoles. The synthesized product’s exocyclic C—N bond and 1,2-dithiacycle backbone increase its bioactivity and emphasize the importance of this step-economical protocol. By synthesizing a library of product 3H-1,2-dithiol-(Z)-3-imines and 3H-1,2-dithiol- (Z)-3-thiones by altering substituent both in isothiocyanate and 1,2,3-thiadiazole, the substrate scope and functional group tolerance in the process were verified (Scheme 38).
Chen’s group[102] developed a Cu-catalyzed procedure to synthesize thio-substituted phenanthridines from 2-biaryl isothiocyanates. In this reaction, they used diaryliodonium salts as an electrophilic aryl donor that forms tandem C—S/C—C bonds. The process scope was investigated using a variety of substituted biaryl isothiocyanates. It is easy to convert ortho- and para-substituted 2-biaryl isothiocyanates to the required product. Metasubstituted isothiocyanates, on the other hand, produced two irreversible positional isomers. Additionally, this process was effectively expanded to produce alkaloid trisphaeridine in a high yield (Scheme 39).
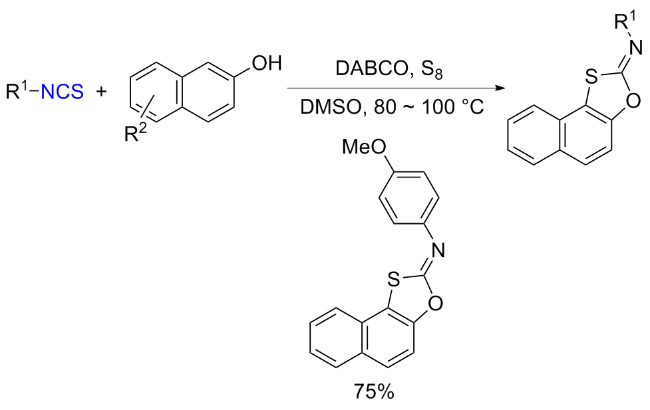
An effective cyclocondensation process utilizing aryl isothiocyanates and 2-naphthols was devised by Retailleau and Nguyen[103] to synthesize 2-imino-naphtho-1,3-oxa- thioles. DABCO is utilized as a basic catalyst to produce desired compounds in moderate to good yields when atomic sulphur is present. Since the reaction needs more phenyl isothiocyanate when atomic sulphur is absent, it was discovered that atomic sulphur had a significant impact on the reaction route. This may be because, in the absence of atomic sulphur, isothiocyanate has an extra role as a dehydrating agent (Scheme 40).
2.2 Nucleophilic addition reactions
Isothiocyanates have a lot of applications in organic synthesis. Its legacy can be attributed to its structure which bears two nucleophilic sites and an electrophilic site. The nucleophilic sites include N and S of RNCS group, while the carbon in the middle works as the best electrophilic center. The R-group of RNCS can favor the reactivity of RNCS or unfavorable in some cases as well (Figure 2).[71]

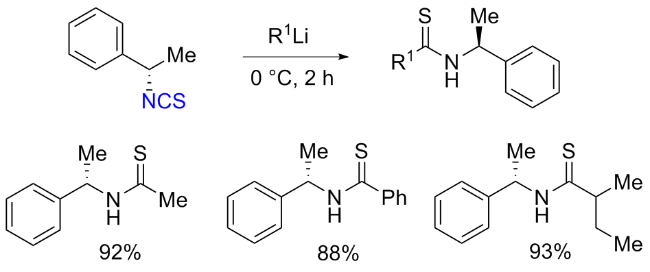
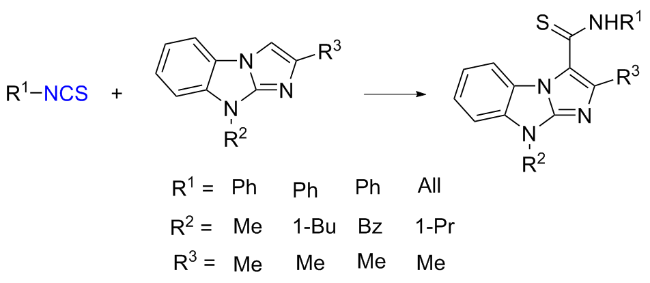
Functionalized organometallic reagents, such as Grignard reagents and organolithiums, can undergo nucleophilic addition to isothiocyanates to prepare amide-type compounds that are not always effectively attainable through traditional chemistry. In 2012, Pace and their colleague[104] synthesized thioamide using a Grignard reagent and organolithium compound which worked as a nucleophile to attack the electrophilic center of isothiocyanate (Scheme 41). They synthesized several thioamide using different RLi compounds in better to excellent yields. Anisimova and colleagues[105] synthesized thiocarbamates using an electrophilic substitution reaction of diazole compounds (Scheme 42). Extending the synthetic applications of isocyanate, de la Vega-Hernández et al.[106] synthesized thioformamide from isothiocyanate through the partial reduction of ITCs with the in-situ generated Schwartz reagent (Scheme 43).
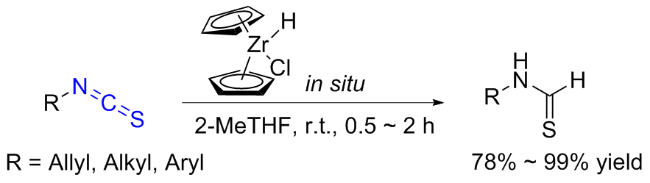
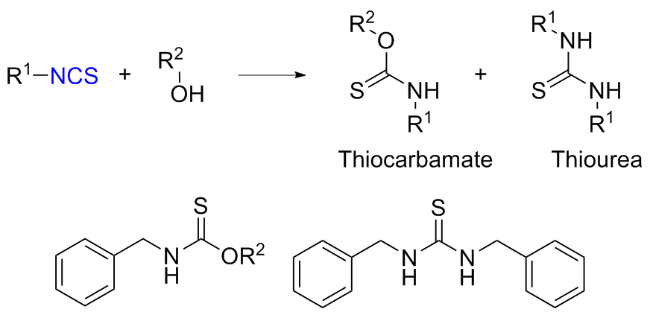
Taking advantage of alcohol nucleophilicity, Perveen’s group[107] has synthesized thiocarbamates and thiourea by reacting to different short to long-chain alcohols. This is a very straightforward, cost-effective, and one-step synthesis of N-aryl-O-alkyl carbamates. When isothiocyanates were treated with alcohols, such as n-hexanol, n-heptanol, and n-octanol, without the use of a solvent, a single product was formed. However, when the same reactions were carried out with small-chain alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, n- amyl alcohol, and isoamyl alcohol, two products of carbamate and thiourea were formed. This reaction indicates that isothiocyanate is susceptible to nucleophilic attack (Sche- me 44).
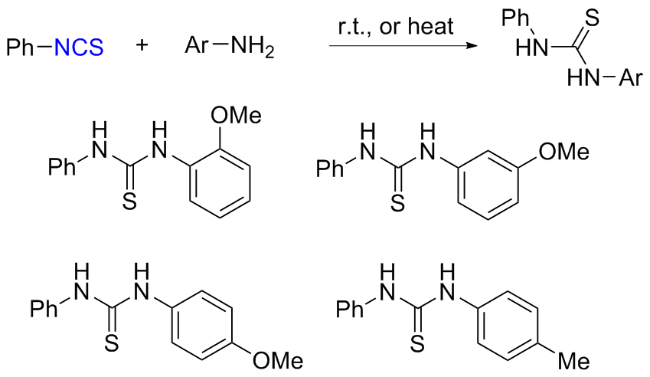
Taking advantage of the electrophilic center of isothiocyanate, a variety of nucleophiles can be reacted to form an addition product. Begum et al. [108] synthesized unsymmetrical thiourea to test their cytotoxic (in vitro), phytotoxic (in vitro), acetylcholinesterase, and butrylcholinesterase activities (Scheme 45). Similarly, Viana’s research group[109] synthesized trisubstituted thiourea derivatives to test their anti-leishmanial activities. They reacted secondary amine with isothiocyanate at reflux in DCM (Scheme 46).
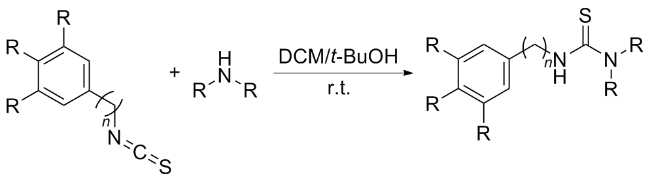
Using ZnO nanoparticles as a photocatalyst, Koohgard et al.[110] carried out the formation of unsymmetrical thiourea from isothiocyanates, which is a moderate, effective, and eco-friendly method. Using N-ZnO as a photocatalyst and visible light irradiation, they produced unsymmetrical thiourea derivatives in moderate to good yields by reacting tertiary aromatic and aliphatic amines with phenyl-isothio- cyanate. Through C—N bond breakage, this technique offers a route to the tertiary aromatic and aliphatic amines (Scheme 47).
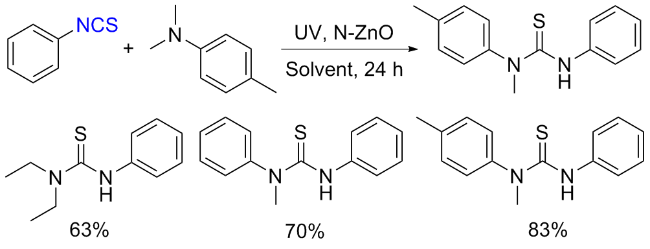
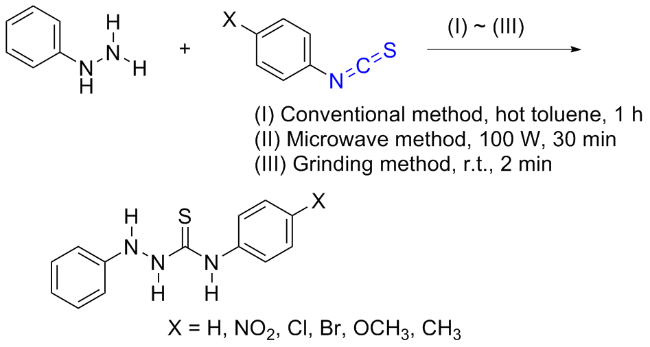
To block the tyrosinase enzyme, Sousa-Pereira et al.[111] synthesized biologically active N-aryl-2-phenyl-hydrazine carboxyamides. Three methods of mixing, microwave irradiation, and mechanical grinding were used to synthesize hydrazinecarbothioamide from the reaction of phenylhydrazine and isothiocyanates (Scheme 48). The latter method yielded a quantifiable yield. In addition to this, phenylthiourea compounds were synthesized for the allosteric inhibition of pyoverdine maturation enzyme PvdP tyrosinase by the Wibowo group[112] in 2020 (Scheme 49).
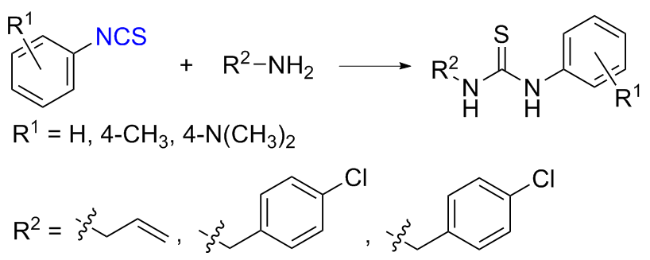
2.3 Isothiocyanate’s sulphur replacement
Functional small-molecule chemistry frequently uses fluorinated small molecules, and N-difluoromethyl (N- CF2H) substances are especially interesting because of their distinct and undiscovered physiochemical characteristics. N-Difluoromethyl compounds were synthesized by our research group[113] from thioformamide, which is a byproduct of isothiocyanate partial reduction (Scheme 50a).
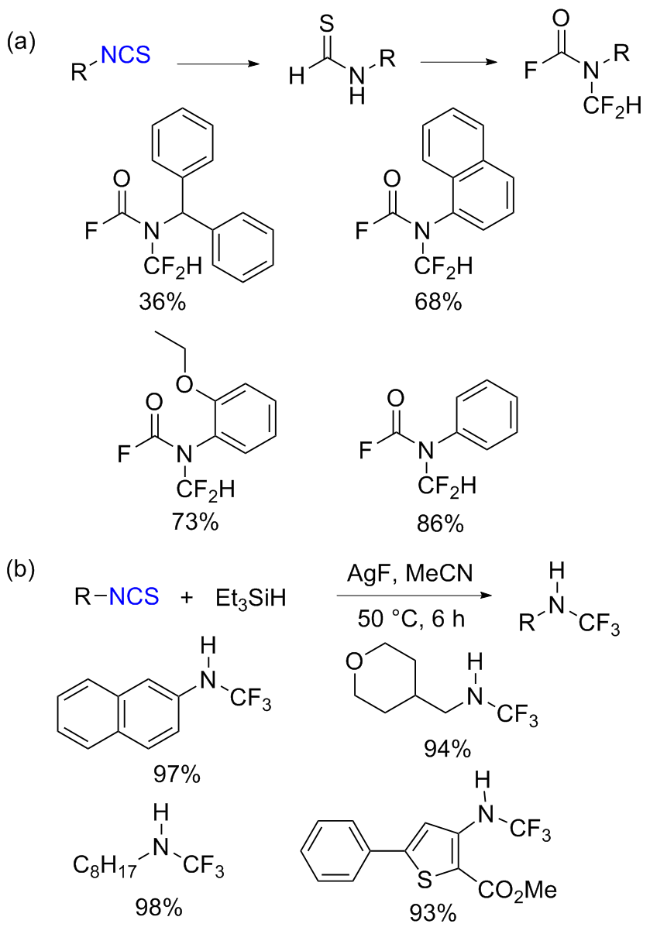
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Similarly, our research group[114] synthesizes N-trifluo- romethyl sulfonamide from isothiocyanates and sulfonoyl halides. First, N-trifluoromethyl secondary amines were synthesized from isothiocyanates using silver fluoride and triethylsilane. The secondary amines were then reacted with transformed sulfonoyl halides. The Yi group introduced a highly efficient and general method for the syn-thesis of secondary trifluoromethyl amine by reacting iso- thiocyanates with silver fluoride and triethylsilane in acetonitriles solvent at 50 ℃. The results of the designed syn- thesis are straightforward, simple, and obtained in excellent yield under mild reaction conditions (Scheme 50b).[114]
3 Conclusions
The synthesis of isothiocyanates (ITCs) has witnessed significant advancements, encompassing both classical and novel methodologies that offer greater efficiency, selectivity, and sustainability. Isothiocyanates have demonstrated immense potential across diverse scientific fields. Their applications span from potent anticancer agents to biofumigants in agriculture, antimicrobial agents in food preservation, and building blocks in pharmaceutical development. Furthermore, ITCs hold promise in environmental remediation and industrial processes due to their chemical reactivity and versatility. The continuous exploration of their mechanistic properties, particularly in cancer prevention, antimicrobial efficacy, and anti-inflammatory effects, highlights their therapeutic potential. Future research efforts should focus on expanding green synthesis methods, optimizing catalytic systems, and elucidating the full scope of ITCs bioactivity in complex biological systems. As isothiocyanates continue to attract interest across multiple disciplines, their role in addressing global challenges, such as sustainable agriculture, food security, and health, becomes increasingly significant. These advances not only enhance the practicality of isothiocyanate synthesis but also solidify their role as valuable compounds with wide-ranging applications.
(Cheng, F.)