


有机含硫化合物是生物体的重要组成部分, 通常以氨基酸、多肽、蛋白质和酶的形式存在[1]. 除此之外有机含硫化合物还广泛存在于药物、天然产物、活性生物分子和功能材料等领域中, 同时是有机合成化学中重要的前体与中间体[2]. 基于此类化合物的重要作用, 含硫骨架的合成一直以来受到化学家们的广泛关注. 其中 C—S键的直接构建为丰富有机含硫结构提供了快捷又简便的合成方法. 在过去的几十年研究中, 过渡金属(如钯、铜、铁、钴、和镍)催化/介导的C—S键活化已被认为是选择性构建新的C—S键的最有效方法之一[3]. 尽管取得了这些巨大研究进展, 这些反应仍然不可避免的受到金属-配体组合的影响, 或者需要高度官能团化的前体, 同时此类反应通常需要使用化学计量的氧化剂或还原剂, 过渡金属催化剂的中毒与残留也为此类反应在生物或医药方面的应用带来了一定的局限性.
近年来, 电化学合成作为一种新兴且环境友好的合成工具, 在有机合成领域得到了广泛的应用并引起了持续的研究兴趣[4]. 电化学催化中使用电子作为无质量试剂, 可以避免使用化学计量的氧化剂或还原剂, 从而减少废弃物的产生, 具有环境友好、操作简单和可持续性发展等特点. 相比于传统的热化学工作反应, 如烯胺的碳氢键芳基硫化、硫氰化以及二硫缩烯酮的芳基硫化反应等[5], 采用电化学催化合成方案可以弥补传统C—S键构建过程中的不足, 进一步扩大其在工业和制药领域的应用. 鉴于这一新兴合成手段在C—S键构建过程中的新突破和新应用[6], 本文将以不同种类有机含硫骨架的合成为分类依据, 介绍电化学条件下C—S键的形成及有机含硫化合物的构建最新的研究进展(Scheme 1).

1 电化学氧化巯基化构建硫醚类化合物
1.1 硫醚/硫酚起始的C—S键构建合成硫醚类化合物
C—S键作为各种生物活性分子和功能材料中的重要结构基元, 在过去的几十年里, 人们始终关注如何开发有效的方法来构建它们. 各种过渡金属, 包括钯、钴、铁、铜或镍催化的有机卤化物/三氟甲磺酸酯与硫醇/苯硫酚的交叉偶联反应是目前应用最广泛的构建C—S键的方法[7]. 相比于传统的偶联反应, C—H/S—H键脱氢交叉偶联是构建新的C—S键最直接有效的方法之一. 然而当使用硫醇或苯硫酚作为硫源时, 其能作为一种配体与过渡金属发生紧密结合, 从而使过渡金属催化剂中毒. 同时硫醇或苯硫酚还容易发生氧化得到亚砜或砜类副产物. 这些问题极大程度地阻碍了C—S键交叉偶联反应的开发与应用. 因此近年来, 随着有机电化学合成的快速发展, 化学家们设想使用电解来驱动氢析出, 从而解决上述问题, 避免使用过渡金属催化剂和氧化剂[8].
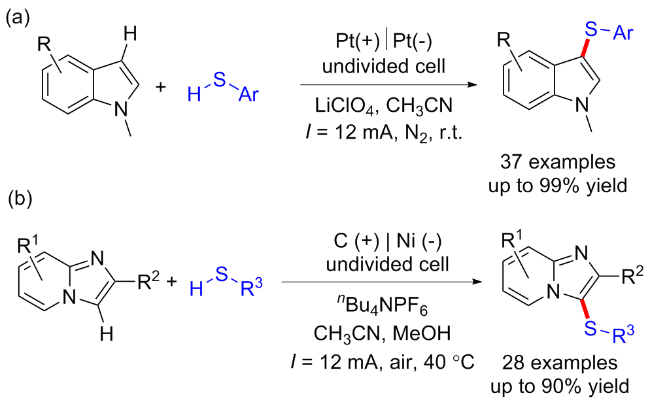
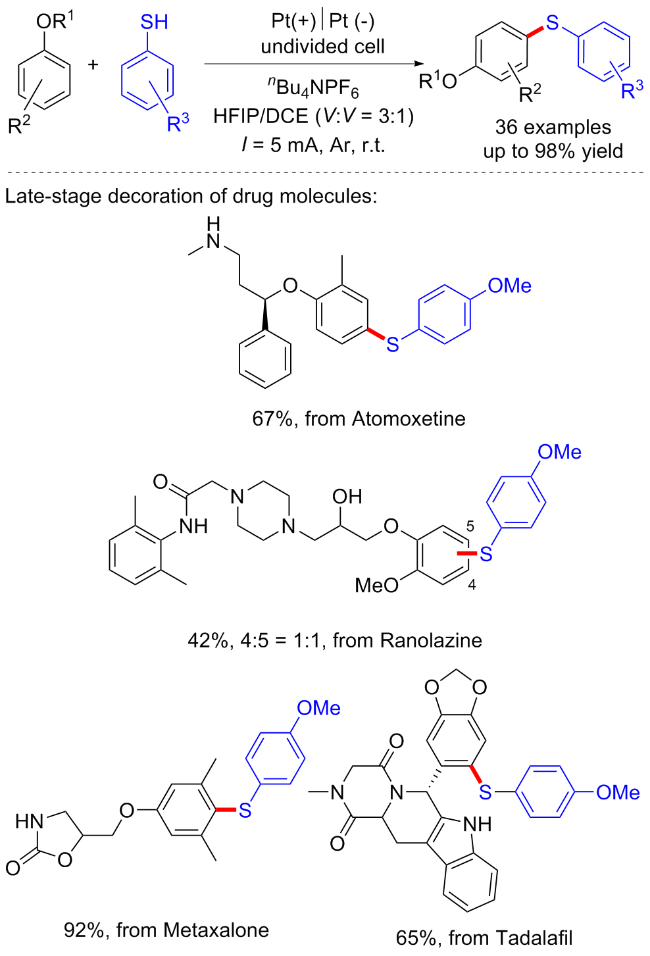
2017年雷爱文课题组[9]建立了一种环境友好的电催化脱氢C—H/S—H交叉偶联反应, 在无催化剂和氧化剂的条件下形成C—S键. 在未分隔的电解池中, 各种芳基/杂芳基的硫醇均能与富电子芳烃反应, 以24%~99%的产率得到对应C—S键偶联产物. 初步的机理研究表明, 芳基自由基阳离子中间体的生成是这一转化成功的关键(Scheme 2, a). 随后该课题组[10]选用咪唑并吡啶衍生物作为起始底物, 在电化学条件下实现了咪唑环的C—H键官能团化, 并产生氢气作为唯一的副产物. 其中芳基硫醇和脂肪族硫醇都表现出了良好的反应性(Scheme 2, b).
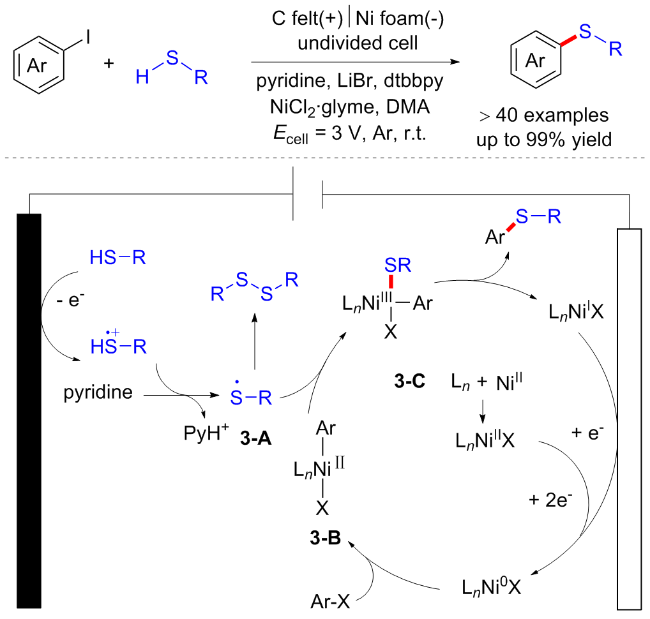
2019年潘毅课题组[11]报道了一例温和电化学条件下镍催化的芳基碘化物的Ullmann型硫醇化反应(Scheme 3). 该反应在未分隔的电解池中进行, 能兼容各类芳基或烷基硫醇以及芳基或杂芳基碘化物. 初步的机理研究表明, 硫醇在阳极上发生单电子转移(SET)氧化后与吡啶发生质子转移得到硫自由基3-A. 同时, Ni(II)在阴极还原产生Ni(0)中间体, 随后与芳基碘化物氧化加成生成Ni(II)中间体3-B, 中间体3-B捕获硫自由基3-A产生Ni(III)络合物3-C. 最后, 3-C发生还原消除得到目标化合物与Ni(I), Ni(I)也能够在阴极还原再生Ni(0), 继续参与催化循环(Scheme 3).
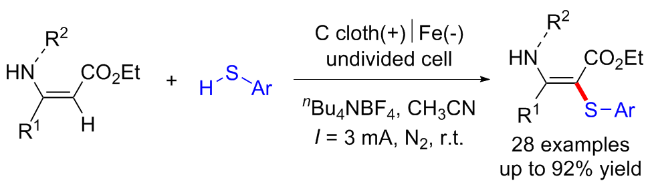
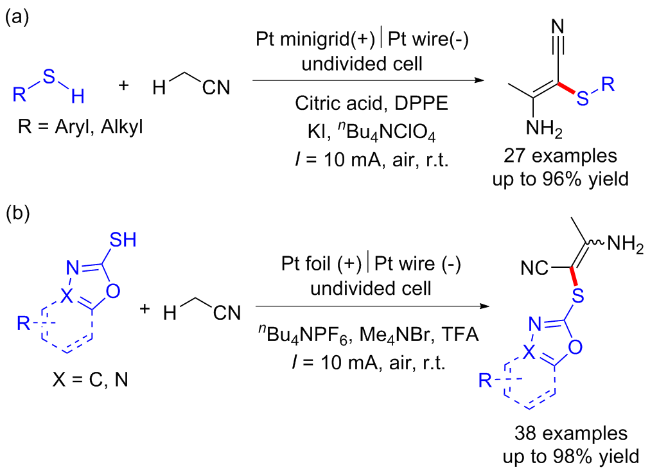
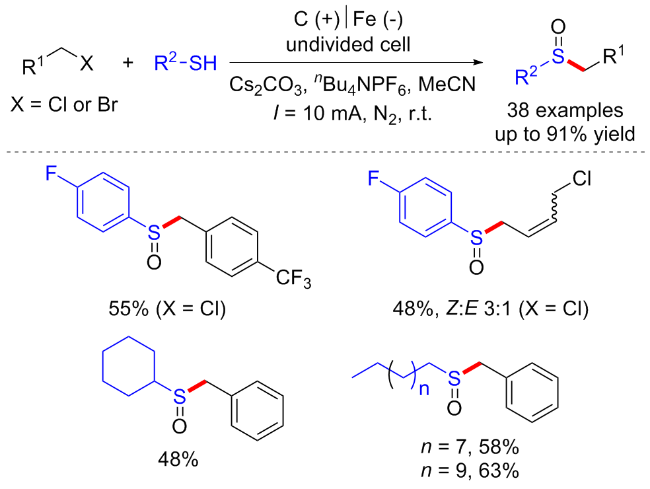
同年黄精美课题组[13]报道了乙腈与芳香族/脂肪族硫醇的电化学条件下C(sp3)—H键官能化反应, 合成了含硫的β-烯胺腈衍生物. 该反应以碘化钾作为氧化还原催化剂, 在阳极实现了乙腈C(sp3)—H键的活化并产生氰基甲基自由基, 从而实现了高立体选择性的C—S键的构建与烯胺化合物的合成(Scheme 5, a). 随后该课题组[14]于2022年建立了乙腈与杂芳基硫醇之间的电化学氧化C—H/S—H交叉偶联反应. 以Me4NBr为氧化还原催化剂, 对乙腈的C(sp3)—H和杂芳基硫醇的S—H进行氧化. 在无金属和无氧化剂的条件下, 以良好的产率和立体选择性得到了具有优异官能团耐受性的杂芳基乙烯基硫醚衍生物(Scheme 5, b).



1.2 二硫醚起始的C—S键构建合成硫醚类化合物
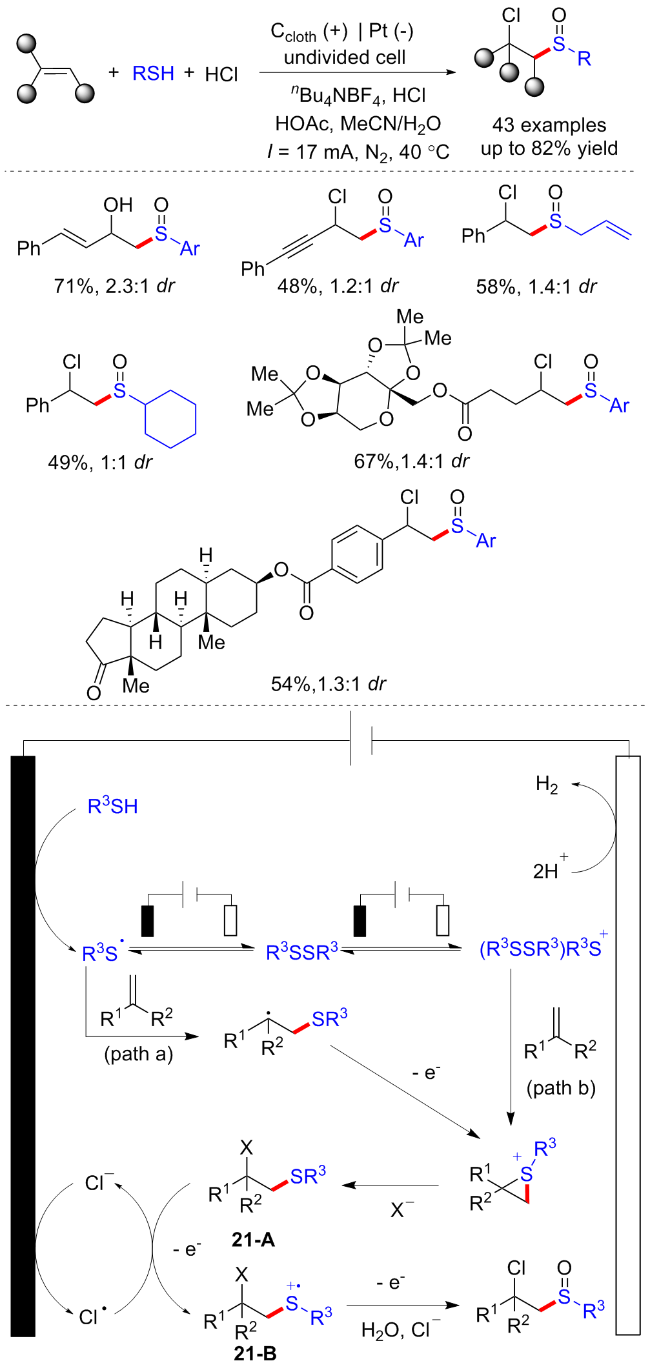
二硫化物衍生物已经成为构建C—S键的广泛研究的底物之一. 二硫化物能在电化学催化的条件下发生阳极氧化, 经历S—S键断裂以生成硫正离子, 可以通过对芳香族化合物进行亲电攻击以得到硫醚产物. 除此之外, 二硫化物经历S—S键断裂后还会生成硫自由基, 其另一种反应形式即为对硫自由基的原位捕获, 构建新的C—S键.
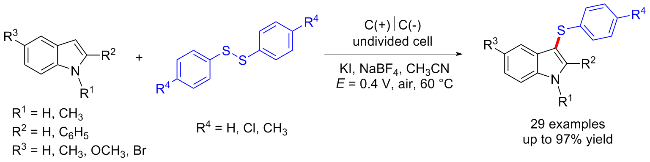


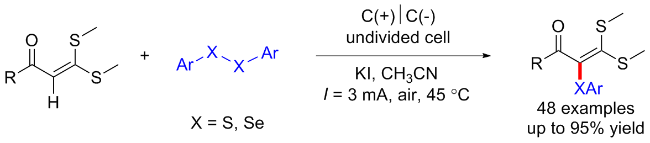
1.3 其他底物起始的C—S键构建合成硫醚类化合物
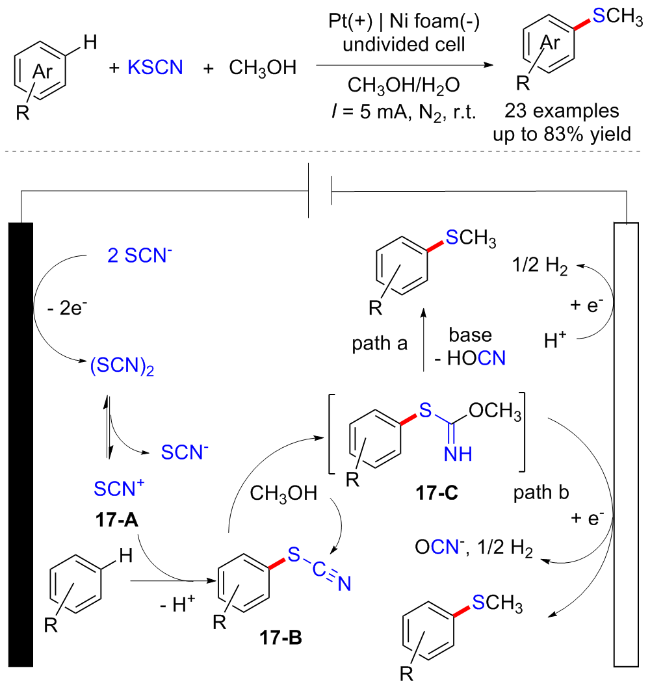
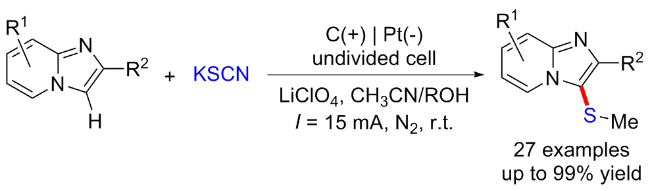
2020年, 赵明课题组[26]通过三组分交叉偶联策略实现了富电子芳烃的电化学诱导C—H键硫甲基化反应. 该方法使用KSCN作为支撑电解质和硫源, 甲醇作为甲基化试剂. 各种(杂)芳香族化合物, 如苯胺、苯甲醚和吲哚均适用于该反应, 以中等至优的产率得到相应的目标产物. 该反应在温和的条件下进行, 不需要任何金属催化剂、外源氧化剂和剧毒的硫试剂, 并且能够放大到克级规模. 机理研究表明, 最初, SCN在阳极通过单电子转移过程被氧化, 产生自由基后偶联得到(SCN)2, 随后生成亲电试剂SCN+中间体17-A并与芳烃发生亲电取代和去质子化得到芳基硫氰酸酯中间体17-B. 中间体17-B和甲醇的反应得到亚胺中间体17-C, 其可以在电化学条件下去质子化下并进行分子内重排, 得到所需的产物(Scheme 17).
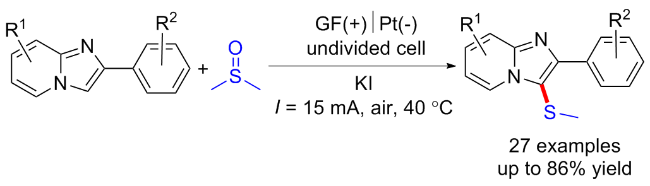
2 电化学氧化亚磺酰化构建亚砜类化合物
亚砜类化合物作为一类重要的有机合成结构单元, 在手性配体、生物活性分子和功能材料等领域具有广泛的应用前景[29]. 开发绿色、可持续的合成方法对它们的高效制备具有重要意义. 然而, 传统的通过硫醚氧化合成亚砜类化合物这一途径不可避免地受到过度氧化、过量氧化剂的使用和硫醚的繁琐制备的阻碍. 为了解决这些缺点, 近年来化学家们借助电化学这一新兴的绿色合成手段, 实现了结构多样化的亚砜类化合物的合成, 成功地避免了化学计量的氧化剂或还原剂的添加.
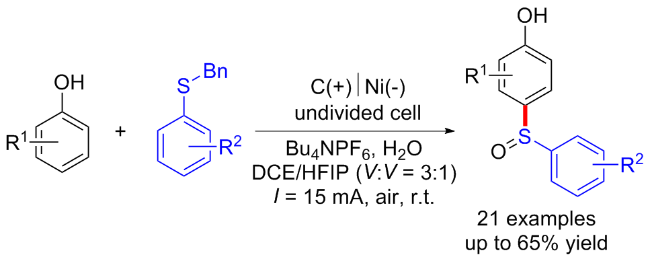
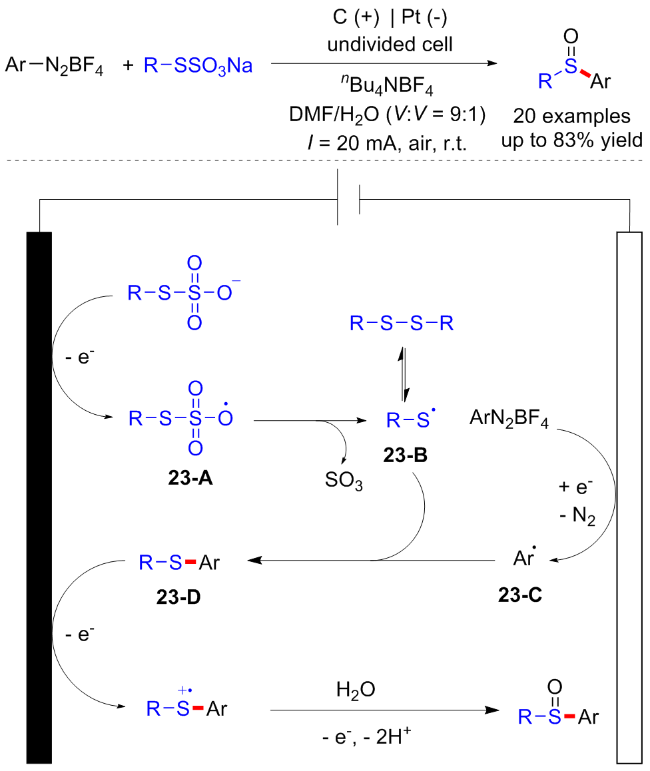
2024年, Singh课题组[33]报道了一例在室温恒电流电解条件下, 以Bunte盐和芳基重氮四氟硼酸盐为原料合成不对称亚砜的电化学方法. 这一合成手段不使用金属或氧化剂, 在温和条件下以相当高的产率合成了带有不同取代基的不对称亚砜衍生物, 并具有良好的官能团耐受性. 机理研究表明, Bunte盐首先发生阳极氧化形成硫代硫酸根自由基23-A, 随后失去一分子SO3得到硫自由基23-B, 硫自由基容易发生二聚得到二硫化物. 芳基重氮盐还原产生的芳基自由基23-C与硫自由基发生自由基偶联得到硫化物中间体23-D. 硫化物23-D进一步阳极氧化再与水的反应, 接着氧化去质子化得到目标化合物(Scheme 23).
3 电化学氧化磺酰化构建砜类化合物
3.1 亚磺酸起始的C—S键构建合成砜类化合物
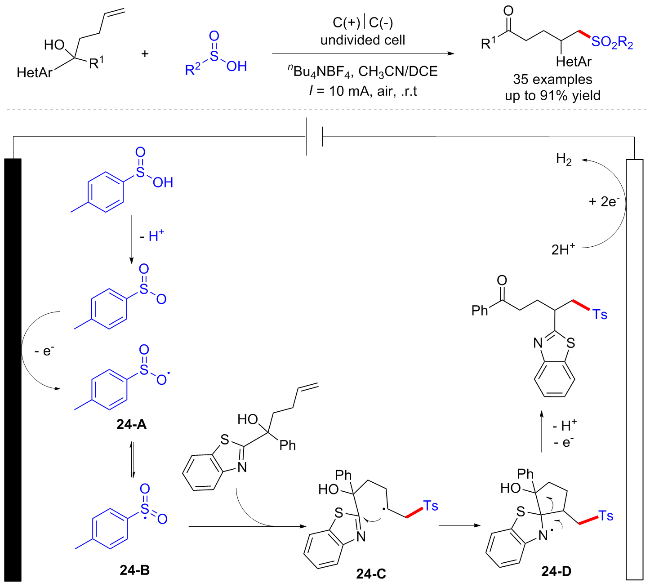
2018年, 郭凯课题组[36]报道了一例烯烃与亚磺酸的直接电化学氧化磺酰化/杂芳基化反应, 该反应通过远端杂芳基的迁移以及C—S和C—C键的形成进行(Scheme 24). 该方法为室温条件下制备各种磺酰化的杂芳烃提供了高效且环境友好的合成新策略, 避免使用任何金属催化剂、添加剂和氧化剂. 初步的机制研究表明, 磺酰基负离子中间体首先在阳极通过单电子转移过程被氧化为新的以氧为中心的自由基中间体24-A同时共振为磺酰基自由基24-B, 并与苯并噻唑取代的叔醇发生分子间自由基加成反应, 生成中间体24-C, 后者通过五元环状过渡态被分子内杂芳基捕获, 生成螺环N-自由基24-D. 24-D最后开环, 并在阳极发生氧化去质子化得到产物.
3.2 亚磺酸钠起始的C—S键构建合成砜类化合物
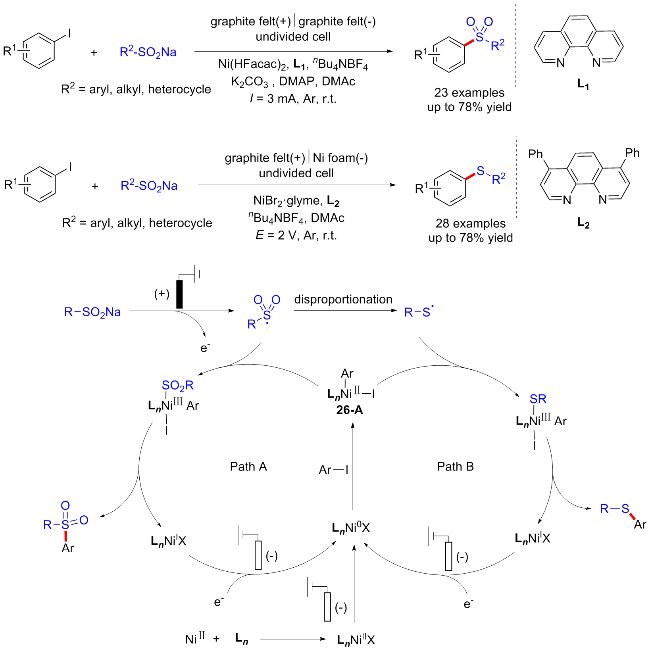
2022年, 潘毅课题组[38]报道了一种电化学条件下镍催化的C—S键偶联反应, 用于制备芳基硫化物和砜类化合物. 通过改变镍催化剂与电极的种类, 能够实现化学选择性的构建C—S键, 且该反应具有良好的可扩展性和可持续性. 此外, 作者还对这种电化学交叉偶联反应的机制进行了研究. 首先, Ni(II)在阴极还原产生Ni(0), 并氧化加成到芳基碘化物中产生Ni(II)中间体26-A. 同时, 亚磺酸钠在阳极氧化产生磺酰基自由基, 并可能发生歧化生成硫自由基中间体. 由此, 该反应可能经历两个途径. 在路径A中, 磺酰基自由基与Ni(II)中间体26-A发生氧化加成随后还原消除以产生砜类目标产物. 在路径B中, 硫自由基与Ni(II)中间体26-A发生氧化加成随后还原消除, 最终生成硫醚产物. 这一反应的化学选择性主要由镍催化剂决定. 此外, 所用的电极和碱可能对磺酰基自由基的还原过程有一定影响(Scheme 26).


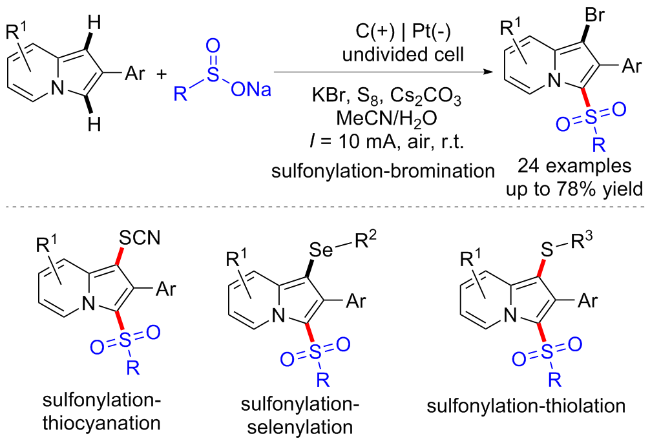
3.3 磺酰肼起始的C—S键构建合成砜类化合物
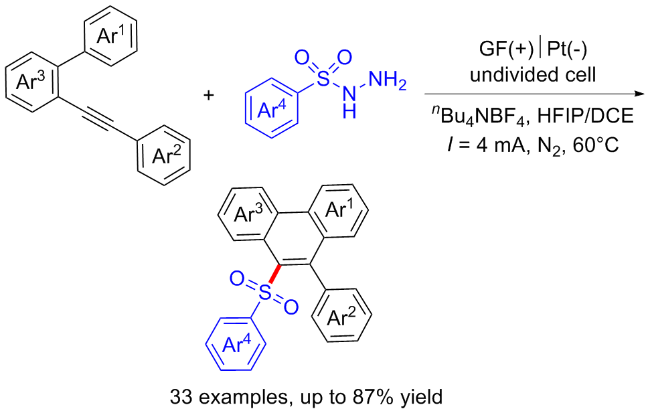
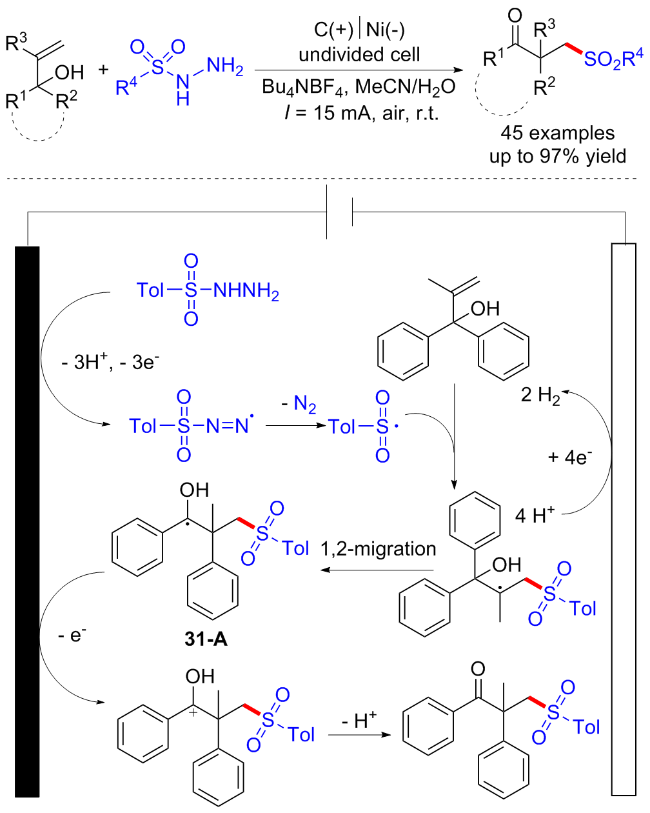
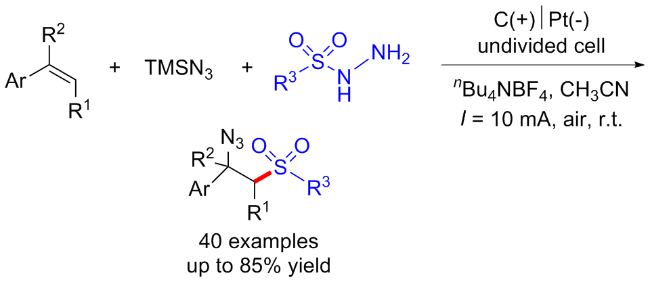
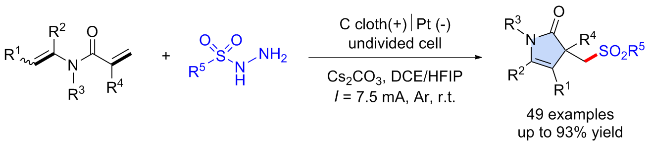
3.4 焦亚硫酸盐起始的C—S键构建合成砜类化合物
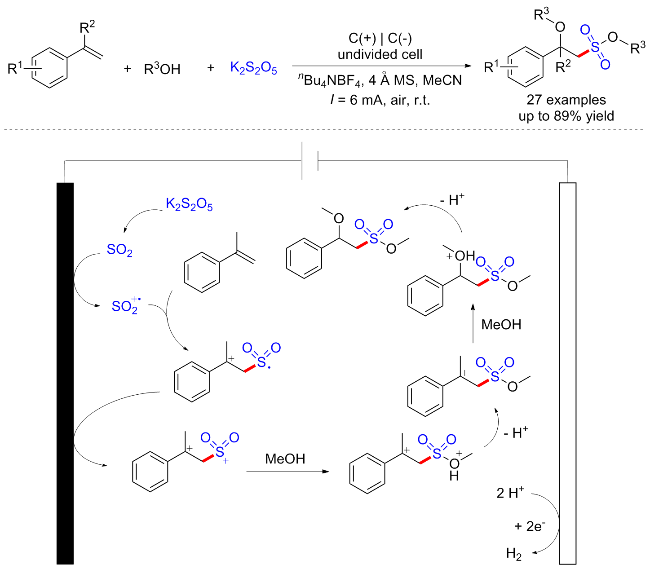

4 电化学氧化硫氰化构建硫氰化合物
有机硫氰酸酯是存在于生物活性分子中的常见官能团, 也是合成含有芳基硫键的生物活性化合物的重要中间体[48]. 硫氰根负离子容易在电化学条件下发生阳极氧化, 生成硫氰根自由基, 随后被一系列化合物捕获, 构建新的C—S键, 合成多样化的硫氰化合物.
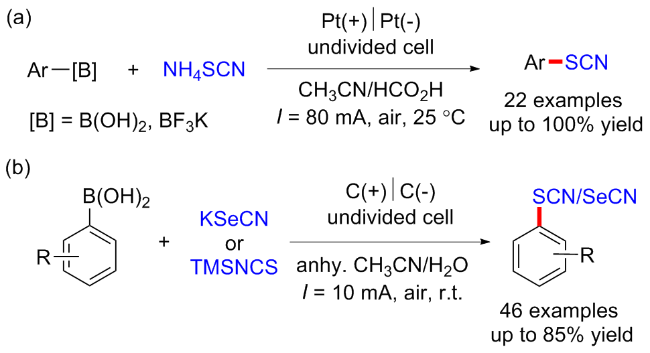
2019年, Gooßen课题组[49]发展了一种无过渡金属催化下芳基硼酸和芳基三氟硼酸酯的硫氰化电化学方法. 该反应通过阳极氧化硫氰酸根阴离子原位生成SCN亲电试剂, 从而避免了化学计量氧化剂的使用以及盐废弃物的产生(Scheme 36, a). 2020年, 蔡琥课题组[50]报道了一例芳基硼酸的电化学条件下脱硼硒/硫氰酸化反应, 在室温条件下合成了相应的芳基硒/硫氰酸酯, 该反应具有良好的官能团耐受性, 同时能够扩大到克级规模(Scheme 36, b).
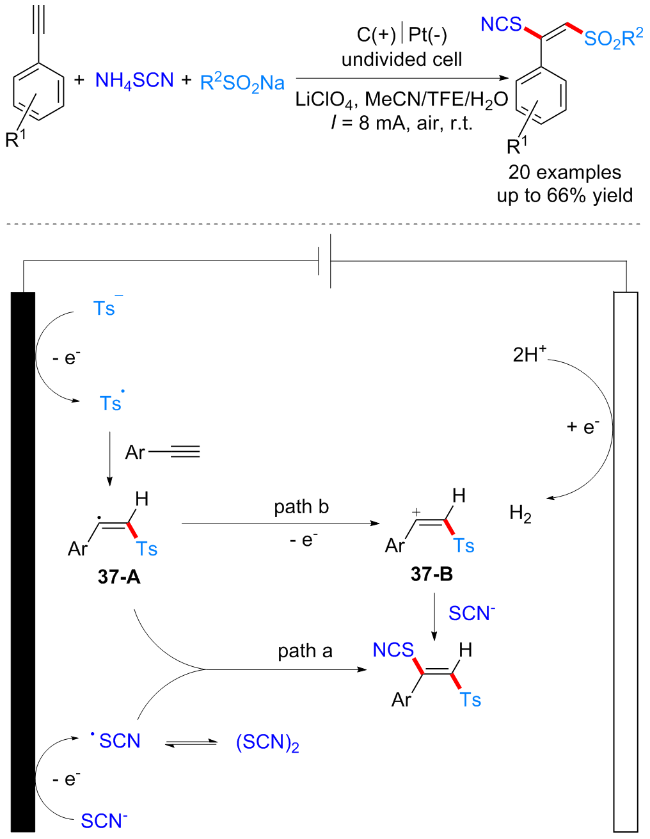
2022年, 黄申林课题组[51]报道了一例简单的电化学条件下芳基端炔、亚磺酸钠和硫氰酸铵的三组分反应, 成功实现了炔烃的C(sp)—H双官能团化, 合成了一类硫氰代烯基砜类化合物. 该反应原料廉价易得, 并无需添加外源氧化剂, 反应条件温和且具有高度的立体选择性. 机理研究表明, 对甲苯亚磺酸根阴离子首先在阳极被优先氧化, 得到砜自由基, 随后与炔烃加成得到乙烯基砜自由基中间体37-A. 同时, 阳极处形成硫氰基自由基并与自由基中间体37-A发生自由基偶联得到目标化合物. 此外, 在另一条可能的反应途径中, 乙烯基自由基37-A可能被进一步氧化成乙烯基阳离子37-B, 随后与硫氰基负离子发生亲核加成反应得到目标化合物(Scheme 37).
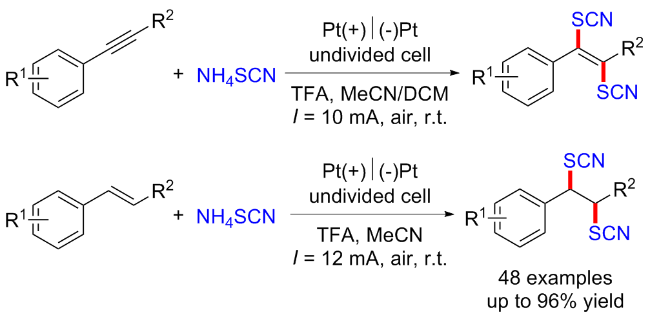

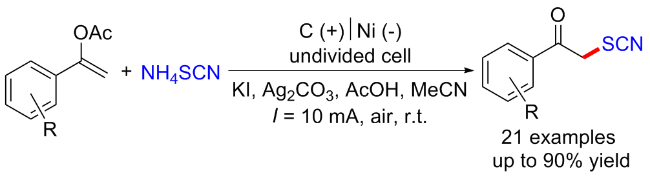
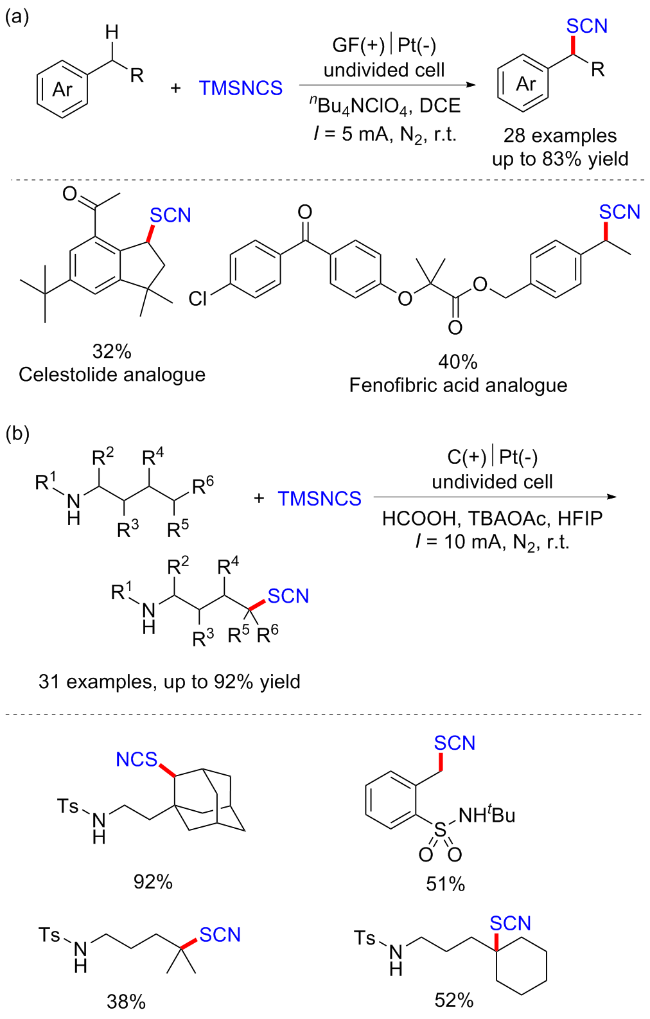
2022年, 郭维斯课题组[55]报道了一例电化学条件下苄基C(sp3)—H键直接硫氰化反应. 与现有方法相比, 该反应显示出广泛的底物范围和独特的苄基C—H位点选择性(2°>3°>1°). 初步的机理研究表明, 该反应可能经历了一个自由基-极性交换过程. 该方法也被成功地用于后续衍生化及生物活性分子的后期硫氰化反应(Scheme 41, a). 2024年, 该课题组[56]通过使用三甲基氰基硅烷(TMSNCS)捕获原位产生的碳正离子, 成功地在电化学条件下实现了远端C(sp3)—H键的硫氰酸化. 控制实验和密度泛函理论(DFT)计算表明, 该反应可能涉及自由基极性交叉机制. 初级、二级和三级底物和生物活性分子均能兼容于该反应, 表现出广泛的底物范围(Scheme 41, b).
5 电化学氧化环化构建含硫杂环化合物


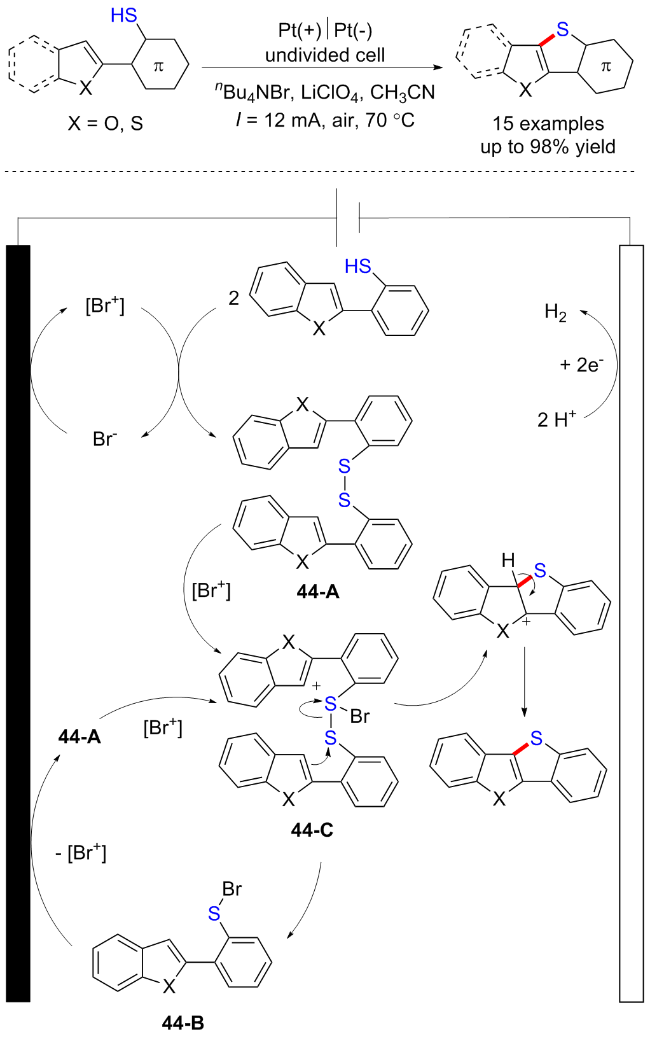
2020年, Suga课题组[61]报道了一例[Br+]促进的电化学脱氢C—S键形成的反应. 该方法通过C—H/ S—H偶联反应合成了几种π-扩展的噻吩并苯衍生物. 其中使用nBu4NBr作为卤素介体催化促进对于这一反应来说是必不可少的. 其可能的反应机理是nBu4NBr的 Br首先被氧化得到[Br+], 随后底物被[Br+]氧化得到二硫化物44-A和Br, 后者将在阳极上再次氧化为[Br+]; 接下来, 二硫化物44-A将与[Br+]反应, 随后分子内环化去质子化得到目标化合物. 芳基硫代溴化物44-B将通过阳极氧化或[Br+]氧化得到二硫化物44-A, 其与[Br+]反应能够得到44-C, 继续参与催化循环. 阴极反应则是将H+还原生成氢气(Scheme 44).
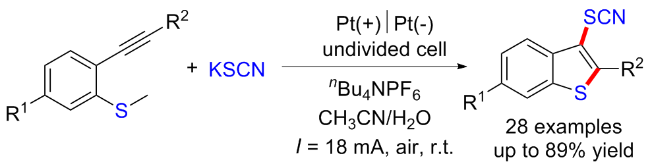
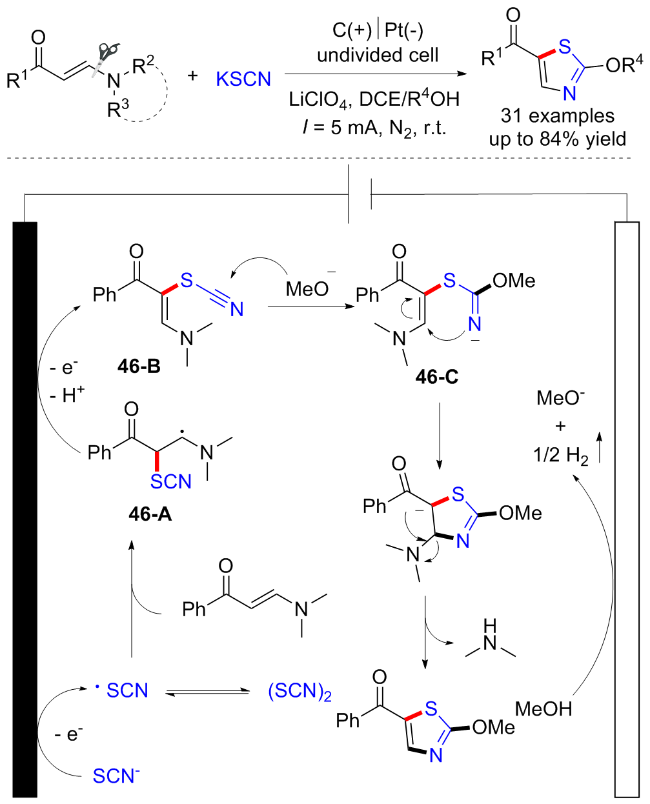
2023年, 李丹丹课题组[63]在无外部氧化剂的条件下, 开发了一例烯胺酮与硫氰化钾和醇的三组分串联环化反应, 得到2-烷氧基噻唑衍生物. 该反应经历电化学氧化C—H硫醇化、亲核串联环化构建C—O、C—S和C—N键以及C—N键的断裂. 这一电化学串级环化反应表现出广泛的底物兼容性和优异的官能团耐受性. 在该反应中, SCN−通过阳极氧化转化为硫氰基自由基, 随后加成到烯胺酮上得到中间体46-A, 其进一步氧化和脱氢成中间体46-B. 同时, 甲醇的阴极还原提供甲氧基阴离子并进攻中间体46-B得到中间体46-C, 最后发生分子内亲核加成以及N,N-二甲胺消除得到2-甲氧基噻唑(Scheme 46).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 结论与展望
综述了近年来电化学条件下C—S键的交叉偶联反应构建有机含硫化合物的新方法. 通过电化学有机合成这一新兴的绿色合成手段, 以无质量的电子作为氧化剂或还原剂, 能够避免额外的化学氧化剂或还原剂的添加, 以高度的原子经济性和化学选择性构建C—S键. 这一电化学合成方法能够通过调节电极、电流以及电解质种类, 有效地向目标分子骨架中引入各种含硫的官能团, 如硫烷基/硫芳基、亚磺酰基、磺酰基或硫氰基等, 且具有良好的官能团兼容性和潜在的应用性, 具有充足的发展潜力和应用前景. 未来, 进一步扩大这一类反应在工业生产方面的应用也将是化学家们所面临的新挑战.
(Zhao, C.)