


1 结果与讨论
1.1 菌株鉴定

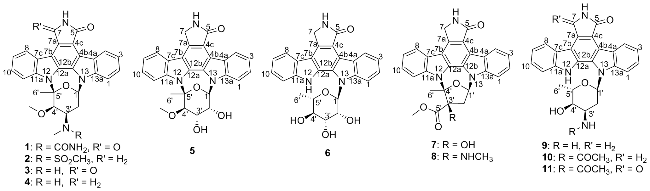
1.2 化合物的结构鉴定
表1 化合物1的1H NMR (600 MHz)和13C NMR (150 MHz)数据(DMSO-d6)Table 1 1H NMR (600 MHz) and 13C NMR (150 MHz) sata for compound 1 (DMSO-d6) |
| Position | δC (type) | δH (mult. J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 109.9 (CH) | 7.75 (d, 8.2) | 2 | 3, 4a | |
| 2 | 127.2 (CH) | 7.61 (t, 7.4) | 1, 3 | 4, 13a | |
| 3 | 120.7 (CH) | 7.43 (t, 7.8) | 2, 4 | 1, 4a | |
| 4 | 124.9 (CH) | 9.07 (d, 8.1) | 3 | 2, 4b, 13a | |
| 4a | 121.4 (C) | ||||
| 4b | 115.2 (C) | ||||
| 4c | 119.5 (C) | ||||
| 5 | 171.2 (C) | ||||
| 6 | — | 11.1 (s) | 4c | ||
| 7 | 170.9 (C) | ||||
| 7a | 120.9 (C) | ||||
| 7b | 116.1 (C) | ||||
| 7c | 122.8 (C) | ||||
| 8 | 124.9 (CH) | 9.25 (d, 7.9) | 9 | 10, 7b, 11a | 10 |
| 9 | 120.6 (CH) | 7.39 (t, 7.3) | 8, 10 | 11, 7c | |
| 10 | 126.9 (CH) | 7.56 (t, 7.8) | 9, 11 | 8, 11a | 8 |
| 11 | 113.7 (CH) | 7.71 (d, 8.1) | 10 | 9, 7c | |
| 11a | 139.9 (C) | ||||
| 12a | 130.5 (C) | ||||
| 12b | 128.8 (C) | ||||
| 13a | 137.7 (C) | ||||
| 1' | 82.6 (CH) | 7.03 (td, 5.8, 2.1) | 2' | 12b 13a, 5' | 3' |
| 2' | 27.4 (CH2) | 2.68~2.71 (m); 2.22 (td, 5.8, 12.8) | 1', 3' | 3', 4' | |
| 3' | 48.3 (CH) | 4.85 (d, 12.8) | 2', 4' | 4' | 1', 6' |
| 4' | 84.0 (CH) | 4.22 (d, 3.0) | 3' | 2', 6', 4'-OCH3 | 11 |
| 5' | 95.2 (C) | ||||
| 6' | 29.2 (CH3) | 2.35 (s) | 4' | 3' | |
| 3'-NCH3 | 30.2 (CH3) | 2.63 (s) | 3', 3'-NCO | ||
| 4'-OCH3 | 60.2 (CH3) | 2.68 (d, 3.0) | 4' | ||
| 3'-NCO | 158.9 (C) | 3'-NCH3 | |||
| NH2 | 6.02 (s) | 3', 3'-NCH3 |
表2 化合物3和4的1H NMR (400 MHz)和13C NMR (100 MHz)数据(DMSO-d6)Table 2 1H NMR (400 MHz) and 13C NMR (100 MHz) data for compounds 3 and 4 (DMSO-d6) |
| Position | 3 | 4 | ||||
|---|---|---|---|---|---|---|
| δC (type) | δH (mult. J in Hz) | δC (type) | δH (mult. J in Hz) | |||
| 1 | 110.0 (CH) | 7.60 (d, 8.0) | 108.4 (CH) | 7.57 (d, 8.2) | ||
| 2 | 125.2 (CH) | 7.45 (dd, 8.0, 7.4) | 124.9 (CH) | 7.42 (dd, 8.2, 7.4) | ||
| 3 | 119.3 (CH) | 7.63 (dd, 8.2, 7.4) | 118.8 (CH) | 7.27 (dd, 7.9, 7.4) | ||
| 4 | 125.0 (CH) | 9.25 (d, 8.2) | 125.6 (CH) | 9.29 (d, 7.9) | ||
| 4a | 122.9 (C) | 122.5 (C) | ||||
| 4b | 115.0 (C) | 113.5 (C) | ||||
| 4c | 121.0 (C) | 119.0 (C) | ||||
| 5 | 171.1 (C) | 172.3 (C) | ||||
| 6 | 11.1 (s) | 8.54 (s) | ||||
| 7 | 170.9 (C) | 45.4 (CH2) | 4.95 (s) | |||
| 7a | 131.0 (C) | 132.1 (C) | ||||
| 7b | 114.9 (C) | 114.1 (C) | ||||
| 7c | 121.0 (C) | 123.9 (C) | ||||
| 8 | 121.5 (CH) | 9.05 (d, 8.0) | 120.9 (CH) | 7.97 (d, 8.6) | ||
| 9 | 121.1 (CH) | 7.45 (dd, 8.0, 7.4) | 119.8 (CH) | 7.27 (dd, 8.6, 8.2) | ||
| 10 | 127.2 (CH) | 7.63 (dd, 8.5, 7.4) | 124.4 (CH) | 7.45 (dd, 7.9, 8.2) | ||
| 11 | 112.7 (CH) | 8.05 (d, 8.5) | 115.3 (CH) | 7.96 (d, 7.9) | ||
| 11a | 138.9 (C) | 139.5 (C) | ||||
| 12a | 129.8 (C) | 130.0 (C) | ||||
| 12b | 127.3 (C) | 126.7 (C) | ||||
| 13a | 137.6 (C) | 136.4 (C) | ||||
| 1' | 81.0 (CH) | 6.95 (dd, 2.7, 9.3) | 80.0 (CH) | 6.69 (t, 3.7) | ||
| 2' | 27.0 (CH2) | 2.13 (td, 1.8, 2.7); 3.33~3.35 (m) | 29.4 (CH2) | 3.17~3.20 (m); 2.49~2.51 (m) | ||
| 3' | 53.2 (CH) | 4.04~4.06 (m) | 50.1 (CH) | 3.22~3.25 (m) | ||
| 4' | 79.3 (CH) | 4.46 (d, 2.0) | 82.8 (CH) | 4.04 (d, 3.4) | ||
| 5' | 93.3 (C) | 91.1 (C) | ||||
| 6' | 27.8 (CH3) | 2.48 (s) | 29.8 (CH3) | 2.30 (s) | ||
| 3'-NH | 9.01~9.04 (m) | |||||
| 3'-NCH3 | 30.7 (CH3) | 2.20 (s) | 33.4 (CH3) | 1.44 (s) | ||
| 4'-OCH3 | 59.9 (CH3) | 2.71 (s) | 57.3 (CH3) | 3.32 (s) | ||
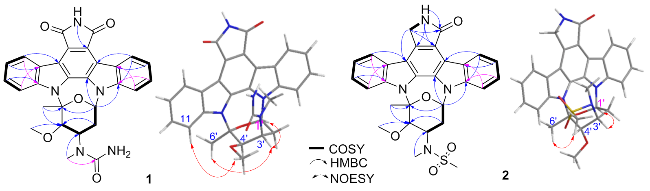
图3 化合物1和2的1H-1H COSY、HMBC和NOESY相关Figure 3 1H-1H COSY, HMBC and NOESY correlations of compounds 1 and 2 |
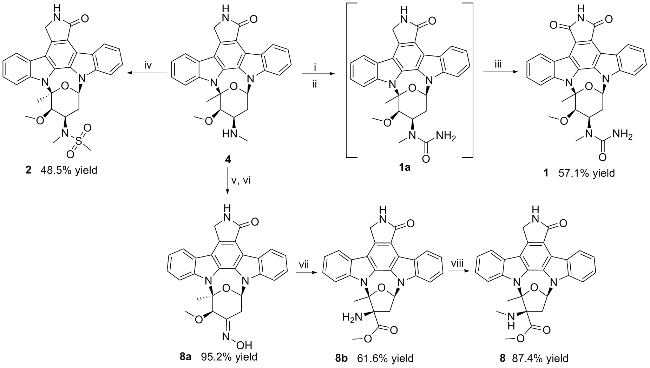
图式1 化合物1、2和8的化学合成Scheme 1 Synthesis of compounds 1, 2 and 8 Condition and reagents: (i) 1.5 equiv. BTC, 5 equiv. DMAP, 6 equiv. DIEA, CH2Cl2, r.t., 12 h; (ii) 50 equiv. NH4HCO3, 40 °C, 2 h; (iii) 10 equiv. t-BuOK, DMSO, air, r.t., 12 h; (iv) 10 equiv. DMAP, 3 equiv. Ms2O, CH2Cl2, r.t., 1 h; (v) 4 equiv. Na2WO4, 30 equiv. H2O2, MeOH/CH2Cl2 (V∶V = 1∶1), r.t., 2 h; (vi) Py, NH2OH, r.t., 0.5 h; (vii) 10 equiv. H2SO4, 1,4-dioxane, air, 100 °C,1 h; (viii) 10 equiv. NaBH3CN, 6 equiv. HCHO, MeOH, r.t., 0.5 h. |
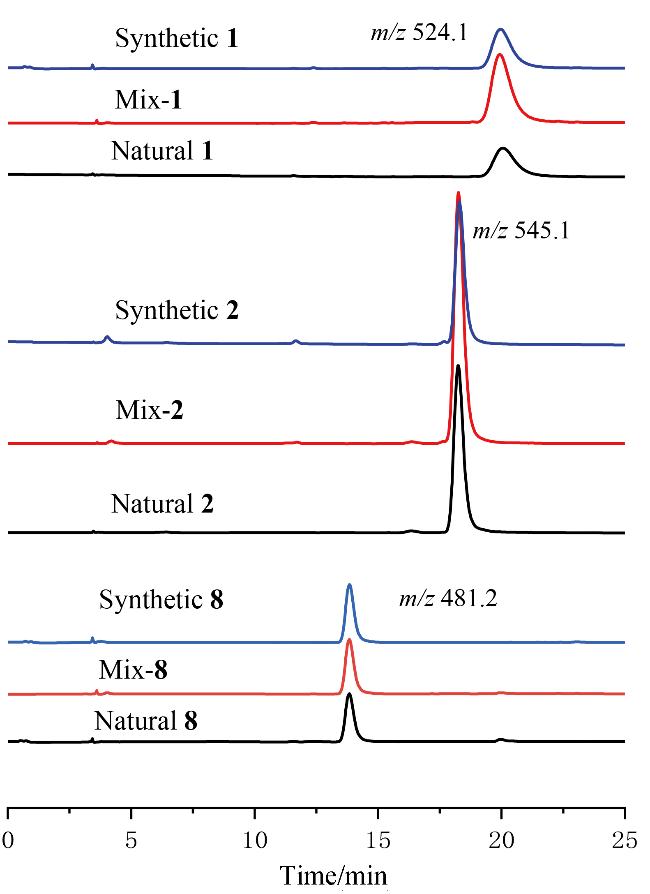
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
表3 化合物2的1H NMR (600 MHz)和13C NMR (150 MHz)数据(DMSO-d6)Table 3 1H NMR (600 MHz) and 13C NMR (150 MHz) data for compound 2 (DMSO-d6) |
| Position | δC (type) | δH (mult. J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 109.0 (CH) | 7.61 (d, 8.4) | 2 | 3, 4a | 1' |
| 2 | 125.1 (CH) | 7.50 (dd, 8.4, 7.5) | 1, 3 | 4, 13a | |
| 3 | 119.6 (CH) | 7.31 (dd, 8.0, 7.5) | 2, 4 | 1, 4a | |
| 4 | 125.8 (CH) | 9.29 (d, 8.0) | 3 | 2, 4b, 13a | |
| 4a | 122.7 (C) | ||||
| 4b | 115.2 (C) | ||||
| 4c | 119.5 (C) | ||||
| 5 | 172.0 (C) | ||||
| 6 | 8.60 (s) | 5, 7, 7a, 4c | |||
| 7 | 45.5 (CH2) | 5.00 (s) | 5, 4c, 7a, 7b | 8 | |
| 7a | 132.7 (C) | ||||
| 7b | 114.2 (C) | ||||
| 7c | 123.8 (C) | ||||
| 8 | 121.6 (CH) | 8.04 (d,7.6) | 9 | 10, 7a, 11a | 7, 10 |
| 9 | 120.5 (CH) | 7.37 (dd, 7.6, 7.5) | 8, 10 | 11, 7c | |
| 10 | 125.1 (CH) | 7.50 (dd, 8.3, 7.5) | 9, 11 | 8, 11a | 8 |
| 11 | 113.7 (CH) | 8.06 (d, 8.3) | 10 | 9, 7c | |
| 11a | 138.8 (C) | ||||
| 12a | 129.2 (C) | ||||
| 12b | 125.4 (C) | ||||
| 13a | 136.3 (C) | ||||
| 1' | 82.3 (CH) | 7.04 (dd, 8.5, 5.9) | 2' | 12b, 2', 5' | 1, 3' |
| 2' | 26.9 (CH2) | 2.22 (dt, 12.7, 5.9), 2.81~2.83 (m) | 1', 3' | 3' | 4'-CH3 |
| 3' | 51.4 (CH) | 4.40 (dt, 12.7, 3.5) | 2' | 3'-NCH3 | 1' |
| 4' | 85.2 (CH) | 4.29 (brs) | 2', 4'-OCH3 | 4'-OCH3 | |
| 5' | 94.9 (C) | 6' | |||
| 6' | 29.2 (CH3) | 2.36 (s) | 5' | 4' | |
| 3'-NCH3 | 30.4 (CH3) | 2.67 (s) | 3' | ||
| 4'-OCH3 | 60.5 (CH3) | 2.78 (s) | 4' | ||
| 3'-NSO2CH3 | 36.8 (CH3) | 3.12 (s) |
表4 化合物5和6的1H NMR (400 MHz)和13C NMR (100 MHz)核磁数据(DMSO-d6)Table 4 1H NMR (400 MHz) and 13C NMR (100 MHz) data for compounds 5 and 6 (DMSO-d6) |
| Position | 5a | 6 | |||
|---|---|---|---|---|---|
| δC (type) | δH (mult. J in Hz) | δC (type) | δH (mult. J in Hz) | ||
| 1 | 108.8 (CH) | 7.65 (d, 8.2) | 109.9 (CH) | 7.69 (d, 8.5) | |
| 2 | 125.5 (CH) | 7.54 (dd, 8.2, 7.0) | 125.2 (CH) | 7.49 (dd, 8.5, 7.5) | |
| 3 | 119.9 (CH) | 7.33 (dd, 7.9, 7.0) | 119.3 (CH) | 7.27 (dd, 7.9, 7.5) | |
| 4 | 125.8 (CH) | 9.31 (d, 7.9) | 125.6 (CH) | 9.47 (d, 7.9) | |
| 4a | 122.8 (C) | 122.3 (C) | |||
| 4b | 114.9 (C) | 117.6 (C) | |||
| 4c | 119.3 (C) | 118.6 (C) | |||
| 5 | 171.9 (C) | 172.3 (C) | |||
| 6 | 8.61 (s) | 8.55 (s) | |||
| 7 | 45.5 (CH2) | 4.98 (dd, 17.6, 24.0) | 45.2 (CH2) | 5.00 (dd, 18.8, 22.1) | |
| 7a | 132.7 (C) | 133.9 (C) | |||
| 7b | 114.4 (C) | 114.9 (C) | |||
| 7c | 123.7 (C) | 121.9 (C) | |||
| 8 | 121.0 (CH) | 8.00 (d, 7.8) | 121.2 (CH) | 8.07 (d, 7.7) | |
| 9 | 120.3 (CH) | 7.33 (dd, 7.8, 7.0) | 119.9 (CH) | 7.31 (dd, 7.7, 7.5) | |
| 10 | 125.0 (CH) | 7.46 (dd, 7.0, 8.5) | 125.2 (CH) | 7.49 (dd, 7.5, 8.0) | |
| 11 | 115.5 (CH) | 7.98 (d, 8.5) | 77.2 (CH) | 7.60 (d, 8.0) | |
| 11a | 140.3 (C) | 139.0 (C) | |||
| 12 | 11.69 (s) | ||||
| 12a | 127.9 (C) | 127.6 (C) | |||
| 12b | 124.7 (C) | 124.6 (C) | |||
| 13a | 136.5 (C) | 140.3 (C) | |||
| 1' | 87.3 (CH) | 6.60 (d, 1.8) | 77.2 (CH) | 6.39 (dd, 2.7, 9.5) | |
| 2' | 71.8 (CH) | 4.13 (dd, 3.1, 1.8) | 67.0 (CH) | 4.49 (dt, 2.5, 9.5) | |
| 3' | 65.7 (CH) | 3.57 (dd, 3.1, 10.5) | 71.7 (CH) | 4.18 (q, 3.4) | |
| 4' | 83.1 (CH) | 4.13 (d, 10.5) | 71.5 (CH) | 4.05 (t, 3.5) | |
| 5' | 95.6 (C) | 76.5 (CH) | 4.48 (q, 6.1) | ||
| 6' | 29.1 (CH3) | 2.39 (s) | 15.4 (CH3) | 1.70 (dd, 2.6, 7.1) | |
a The NMR data for 2'-OH, 3'-OH and 4'-OCH3 were δH 6.21 (d, 3.7, 1H), 5.10 (d,5.7, 1H), and δC/H 61.7 (CH3)/3.63 (s, 3H), respectively. |
表5 化合物7和8的1H NMR (400 MHz)和13C NMR (100 MHz)数据(DMSO-d6)aTable 5 1H NMR (400 MHz) and 13C NMR (100 MHz) data for compounds 7 and 8 (DMSO-d6) |
| Position | 7 | 8 | |||
|---|---|---|---|---|---|
| δC (type) | δH (mult. J in Hz) | δC (type) | δH (mult. J in Hz) | ||
| 1 | 109.2 (CH) | 7.94 (d, 8.5) | 109.2 (CH) | 7.92 (d, 8.3) | |
| 2 | 125.5 (CH) | 7.50 (dd, 8.5, 7.4) | 125.4 (CH) | 7.48 (dd, 8.3, 7.2) | |
| 3 | 119.6 (CH) | 7.29 (dd, 7.4, 8.0) | 119.6 (CH) | 7.29 (dd, 7.2, 8.0) | |
| 4 | 125.7 (CH) | 9.22 (d, 8.0) | 125.6 (CH) | 9.21 (d, 8.0) | |
| 4a | 122.6 (C) | 122.6 (C) | |||
| 4b | 115.8 (C) | 115.9 (C) | |||
| 4c | 120.5 (C) | 119.7 (C) | |||
| 5 | 171.8 (C) | 171.8 (C) | |||
| 6 | 8.65 (s) | 8.66 (s) | |||
| 7 | 45.5 (CH2) | 5.01 (dd, 17.8, 23.0) | 45.6 (CH2) | 5.00 (dd, 16.0, 20.0) | |
| 7a | 133.0 (C) | 133.0 (C) | |||
| 7b | 114.9 (C) | 114.7 (C) | |||
| 7c | 124.0 (C) | 124.0 (C) | |||
| 8 | 121.3 (CH) | 8.07 (d, 7.6) | 121.4 (CH) | 8.05 (d, 7.8) | |
| 9 | 119.6 (CH) | 7.37 (dd, 7.4, 7.6) | 120.6 (CH) | 7.36 (dd, 7.4, 7.8) | |
| 10 | 125.1 (CH) | 7.50 (dd, 8.5, 7.4) | 125.7 (CH) | 7.49 (dd, 8.5, 7.4) | |
| 11 | 114.6 (CH) | 7.91 (d, 8.5) | 114.9 (CH) | 8.09 (d, 8.5) | |
| 11a | 140.0 (C) | 139.7 (C) | |||
| 12a | 128.4 (C) | 128.5 (C) | |||
| 12b | 124.2 (C) | 124.2 (C) | |||
| 13a | 136.9 (C) | 136.8 (C) | |||
| 1' | 85.1 (CH) | 7.15 (dd, 5.0, 7.1) | 85.9 (CH) | 7.20 (dd, 5.1, 7.3) | |
| 2' | 42.6 (CH2) | 2.02 (dd, 5.0, 14); 3.40 (t, 7.1) | 40.1 (CH2) | 2.03 (dd, 5.1, 13.8); 3.43 (dd, 7.3, 13.8) | |
| 3' | 85.0 (C) | 78.1 (C) | |||
| 4' | 99.4 (C) | 100.5 (C) | |||
| 5' | 172.9 (C) | 173.0 (C) | |||
| 6' | 22.9 (CH3) | 2.16 (s) | 23.9 (CH3) | 2.17 (s) | |
| 5'-OCH3 | 52.8 (CH3) | 3.94 (s) | 53.0 (CH3) | 3.94 (s) | |
a The NMR data for 3'-OH (7) and 3'-NHCH3 (8) was δH 6.37 (s, 1H), and δC/H 32.4 (CH3)/1.94 (d, 5.9, 3H), respectively. |
表6 化合物9~11的1H NMR (400 MHz)和13C NMR (100 MHz)数据(DMSO-d6)Table 6 1H NMR (400 MHz) and 13C NMR (100 MHz) data for compounds 9~11 (DMSO-d6) |
| Position | 9 | 10a | 11a | |||||
|---|---|---|---|---|---|---|---|---|
| δC (type) | δH (mult. J in Hz) | δC (type) | δH (mult. J in Hz) | δC (type) | δH (mult. J in Hz) | |||
| 1 | 109.5 (CH) | 7.95 (d, 8.5) | 109.7 (CH) | 8.02 (d, 8.4) | 110.5 (CH) | 8.14 (d, 8.5) | ||
| 2 | 125.5 (CH) | 7.52 (dd, 8.5, 7.3) | 125.4 (CH) | 7.50 (dd, 8.4, 7.3) | 126.9 (CH) | 7.60 (dd, 7.6, 8.5) | ||
| 3 | 120.0 (CH) | 7.32 (dd, 7.9, 7.3) | 119.7 (CH) | 7.29 (dd, 7.3, 7.9) | 120.2 (CH) | 7.37 (dd, 7.6, 8.0) | ||
| 4 | 125.9 (CH) | 9.47 (d,7.9) | 125.7 (CH) | 9.46 (d, 7.9) | 124.6 (CH) | 9.10 (d, 8.0) | ||
| 4a | 122.3 (C) | 122.2 (C) | 121.1 (C) | |||||
| 4b | 117.5 (C) | 117.3 (C) | 117.3 (C) | |||||
| 4c | 118.6 (C) | 118.5 (C) | 119.2 (C) | |||||
| 5 | 172.2 (C) | 172.3 (C) | 171.1 (C) | |||||
| 6 | 8.58 (s) | 8.55 (s) | 11.11 (s) | |||||
| 7 | 45.2 (CH2) | 5.00 (dd, 16.9, 20.3) | 45.2 (CH2) | 5.00 (dd, 17.4, 20.2) | 171.2 (C) | |||
| 7a | 134.3 (C) | 134.1 (C) | 120.8 (C) | |||||
| 7b | 115.1 (C) | 114.9 (C) | 116.5 (C) | |||||
| 7c | 121.8 (C) | 121.8 (C) | 121.0 (C) | |||||
| 8 | 121.2 (CH) | 8.08 (d, 7.7) | 121.2 (CH) | 8.07 (d, 7.9) | 124.7 (CH) | 9.20 (d, 8.1) | ||
| 9 | 120.0 (CH) | 7.32 (dd, 7.7, 7.5) | 119.8 (CH) | 7.32 (dd, 7.9, 7.4) | 120.9 (CH) | 7.42 (dd, 7.4, 8.1)) | ||
| 10 | 125.1 (CH) | 7.49 (dd, 7.2, 8.8) | 125.0 (CH) | 7.50 (dd, 7.4, 8.1) | 127.1 (CH) | 7.61 (dd, 7.4, 8.3) | ||
| 11 | 113.6 (CH) | 8.01 (d, 8.5) | 111.7 (CH) | 7.71 (d, 8.1) | 111.8 (CH) | 7.73 (d, 8.3) | ||
| 11a | 139.2 (C) | 139.3 (C) | 139.8 (C) | |||||
| 12 | 11.79 (s) | 12.13 (s) | 12.34 (s) | |||||
| 12a | 127.2 (C) | 127.4 (C) | 127.6 (C) | |||||
| 12b | 124.0 (C) | 124.2 (C) | 128.7 (C) | |||||
| 13a | 138.5 (C) | 138.6 (C) | 140.4 (C) | |||||
| 1' | 75.1 (CH) | 6.71 (dd, 11.2, 4.1) | 75.9 (CH) | 6.73 (dd, 4.3, 11.3) | 76.1 (CH) | 6.77 (dd (3.3, 11.5) | ||
| 2' | 30.7 (CH2) | 2.02 (dt, 4.1, 12.7); 2.55 (dd, 11.2, 12.7) | 32.2 (CH2) | 1.80 (dt, 4.3, 12.9); 2.52 (d, 12.9) | 32.0 (CH2) | 1.85 (dt, 3.3, 12.4); 2.50 (m) | ||
| 3' | 45.8 (CH) | 4.12~4.15 (m) | 44.3 (CH) | 4.70 (dd, 3.3, 7.7) | 44.1 (CH) | 4.71 (dd, 5.7, 7.8) | ||
| 4' | 66.1 (CH) | 4.08~4.10 (m) | 68.2 (CH) | 3.92 (d, 3.3) | 68.2 (CH) | 3.92 (brs) | ||
| 5' | 76.5 (CH) | 4.62 (q, 6.9) | 76.7 (CH) | 4.52 (q, 6.7) | 77.0 (CH) | 4.53 (q, 7.1) | ||
| 6' | 14.0 (CH3) | 1.60 (d, 7.2) | 14.4 (CH3) | 1.61 (d, 7.1) | 14.3 (CH3) | 1.61 (d, 7.1) | ||
| 3'-NH | 8.19~8.21 (m, 2H, NH2) | 7.92 (d, 7.7) | 7.93 (d, 7.8) | |||||
| 4'-OH | 7.67 (m) | 6.96 (s) | 7.02 (s) | |||||
a The NMR data of N3'-acetyl for both 10 and 11 were δC 168.8 (C) and δH/C 1.82 (s, 3H)/22.7 (CH3), respectively |
1.3 化合物活性测试结果
2 结论
3 实验部分
3.1 仪器与试剂
3.2 菌株的分离鉴定
表7 培养基配方一览表Table 7 List of Culture Medium Formulations |
| 培养基名称 | 配方 |
|---|---|
| A1 | 可溶性淀粉10 g, 酵母浸膏4 g, 蛋白胨2 g, Fe2(SO4)3•4H2O 0.04 g, KBr 0.1 g, 海水1 L, 自然pH |
| Kazuo | 可溶性淀粉25 g, 酵母浸膏2 g, 豆粉15 g, CaCO3 2 g, Amberlite XAD-16N 10 g, 海水1 L, 自然pH |
| LB | 胰蛋白胨10 g, NaCl 5 g, 酵母浸膏5 g, 自来水1 L, 自然pH |
| YPD | 酵母浸膏10 g, 蛋白胨20 g, 葡萄糖20 g, 海水1 L, 自然pH |
| 分离培养基 | 可溶性淀粉10 g, 干酪素0.3 g, 硝酸钾2 g, 七水合硫酸镁0.05 g, 氯化钠2.0 g, 磷酸氢二钾2.0 g, 碳酸钙0.02 g, 七水合硫酸亚铁0.01 g, 海水500 L, 水500 L, 琼脂20 g, 重铬酸钾50 mg, pH 7.2~7.4 |
| 纯化培养基 | 葡萄糖4.0 g, 酵母膏4.0 g, 麦芽浸粉10.0 g, 海盐27.0 g, 水1000 mL, pH 7.0 |