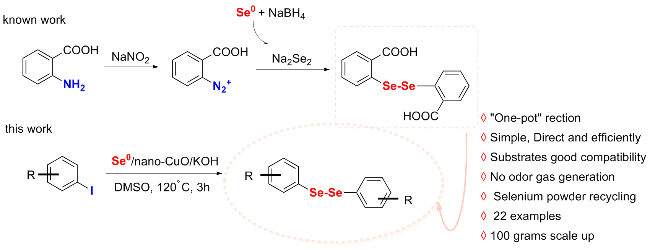
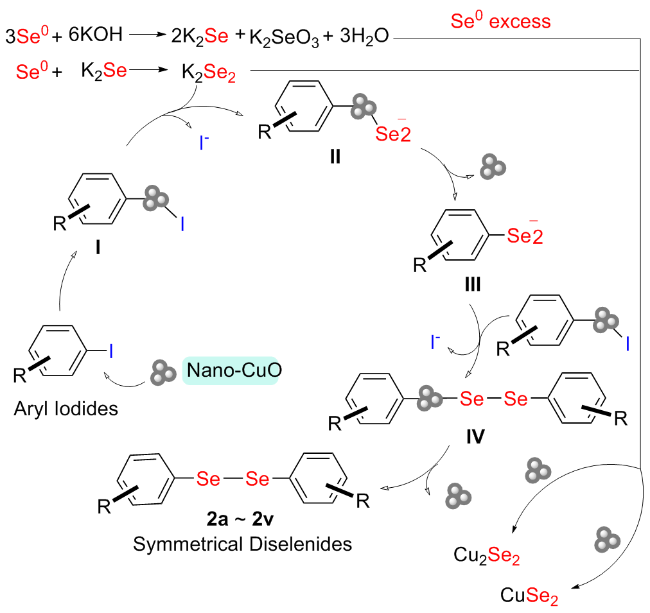
将2-碘苯甲酸(248.02 mg, 1 mmol)、硒粉(157.92 mg, 2 mmol)和纳米氧化铜(3.97 mg, 0.05 mmol)分别加入到装有干燥DMSO (3 mL)的反应瓶中, 氮气置换三次, 升温至120 ℃后, 加入氢氧化钾(126.22 mg, 2.25 mmol), 搅拌反应3 h. TLC检测反应完全后, 将反应液冷却到室温, 并用蒸馏水稀释至30 mL, 转移至烧杯中, 加入3 mL的浓盐酸并于85~90 ℃度下搅拌30 min. 放入冰箱冷藏1.5~2 h, 过滤. 滤饼用少量稀盐酸洗涤, 溶于15 mL的饱和碳酸氢钠溶液, 加入活性炭脱色, 趁热过滤, 滤液加入浓盐酸调节pH至1, 将滤液放入0~5 ℃冰箱静置1.5 h, 过滤, 滤饼用水洗涤3次, 得到化合物2, 为黄色或白色固体粉末. 精制采用乙醇/水(V∶V=1∶1.2)进行重结晶.
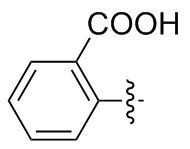
2,2'-二苯甲酸二硒醚(2a): 186 mg, 白色固体, 产率94%. Rf=0.67(石油醚/乙酸乙酯, V∶V=3∶1); m.p. 284~286 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 13.72 (s, 2H), 8.04 (dd, J=7.8, 1.6 Hz, 2H), 7.68 (dd, J=8.1, 1.2 Hz, 2H), 7.50 (td, J=7.6, 1.6 Hz, 2H), 7.37 (td, J=7.5, 1.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 169.04, 134.09, 133.95, 132.06, 129.98, 129.23, 127.09; HRMS (ESI) calcd for C14H9NaO4Se2 (M-2H+Na+) 423.3274, found 423.3269.
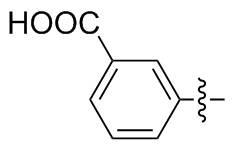
3,3'-二苯甲酸二硒醚(2b): 175 mg, 黄色固体, 产率87.42%. Rf=0.67(石油醚/乙酸乙酯, V∶V=3∶1); m.p. 228~230 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 13.16 (s, 2H), 8.18 (t, J=1.8 Hz, 2H), 7.84 (td, J=7.7, 1.6 Hz, 4H), 7.46 (t, J=7.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 166.94, 135.38, 132.42, 131.82, 130.85, 130.22, 129.16; HRMS (ESI) calcd for C14H9NaO4Se2 (M-2H+Na+) 423.3274, found 423.3279.
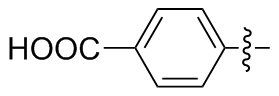
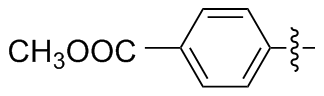
4,4'-二苯甲酸二硒醚(2c): 155 mg, 黄色固体, 产率77.22%. Rf=0.67(石油醚/乙酸乙酯, V∶V=3∶1); m.p. 296~298 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 13.04 (s, 2H), 7.87 (d, J=8.2 Hz, 4H), 7.74 (d, J=8.1 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ: 169.43, 138.20, 132.49, 131.88, 101.41; HRMS (ESI) calcd for C14H9NaO4Se2 (M-2H+Na+) 423.3274, found 423.3278.
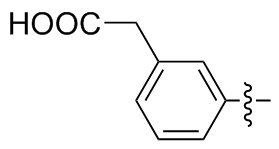
3,3'-二乙羧基苯二硒醚(2d): 184 mg, 黄色油状物, 产率85.43%. Rf=0.45(石油醚/乙酸乙酯, V∶V=20∶1); 1H NMR (400 MHz, CDCl3) δ: 7.26 (dt, J=7.7, 1.5 Hz, 2H), 7.09 (t, J=7.6 Hz, 2H), 7.03 (dt, J=7.6, 1.5 Hz, 2H), 6.78 (t, J=1.8 Hz, 2H), 3.83 (s, 4H); 13C NMR (101 MHz, CDCl3) δ: 185.95, 140.44, 135.30, 133.38, 128.55, 128.52, 128.12, 31.62; HRMS (ESI) calcd for C16H13O4Se2 (M- H+) 428.9144, found428.9132.
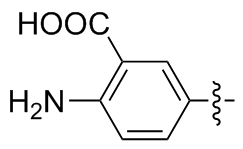
4,4'-二氨基-3,3'-二苯甲酸二硒醚(2e): 188 mg, 黄色固体, 产率87.36%. Rf=0.53 (DCM/MeOH, V∶V=20∶1); m.p. 210~212 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.05 (d, J=27.1 Hz, 2H), 7.38~7.17 (m, 2H), 6.65 (d, J=8.6 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 152.21, 138.78, 117.44, 113.92; HRMS (ESI) calcd for C14H12Cl- N2O4Se2 (M+Cl-) 466.8821, found 466.8844.
将不同取代的碘代底物(1 mmol)、硒粉(0.1579g, 2 mmol)和纳米氧化铜(3.97 mg, 0.05 mmol)分别加入装有干燥DMSO (3 mL)的反应瓶中, 氮气置换三次, 加热至120 ℃, 加入氢氧化钾(0.1262 g, 2.25 mmol), 搅拌反应3 h. TLC检测反应完全后, 将反应液冷却到室温, 过滤, 滤饼分别用水和二氯甲烷洗涤, 滤液用二氯甲烷萃取, 蒸馏水洗涤, 无水硫酸镁干燥, 过滤后减压蒸馏得到粗产物, 纯化通过柱层析分离得到对称二硒化物产物2.
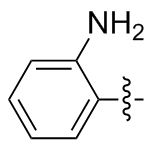
2,2'-二氨基二苯二硒醚(2g): 109 mg, 橙色固体, 产率63.85%. Rf=0.42(石油醚/乙酸乙酯, V∶V=3∶1); m.p. 72~74 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.36~7.33 (m, 2H), 7.21~7.10 (m, 2H), 6.75~6.69 (m, 2H), 6.59~6.52 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 138.31, 131.53, 118.43, 114.75; HRMS (ESI) calcd for C12H13N2Se2 (M+H+) 344.9404, found 344.9379.
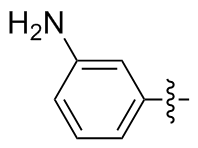
3,3'-二氨基二苯二硒醚(2h): 124 mg, 黄色油状物, 产率72.35%. Rf=0.45(石油醚/乙酸乙酯, V∶V=3∶1); 1H NMR (400 MHz, CDCl3) δ: 7.12~7.02 (m, 4H), 6.92~6.78 (m, 2H), 6.66~6.57 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 155.70, 136.06, 129.05, 127.09, 118.38, 100.47; HRMS (ESI) calcd for C12H13N2Se2 (M+H+) 344.9404, found 344.9387.
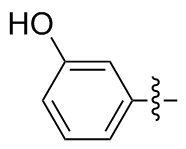
3,3'-二羟基二苯二硒醚(2i): 130 mg, 棕色油状物, 产率75.47%. Rf=0.51(石油醚/乙酸乙酯, V∶V=3∶1); 1H NMR (400 MHz, CDCl3) δ: 7.28~7.10 (m, 6H), 6.78~6.68 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 156.03, 131.89, 130.20, 123.38, 117.99, 115.00; HRMS (ESI) calcd for C12H9O2Se2 (M-H+) 344.8933, found 344.8925.
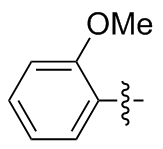
2,2'-二甲氧基二苯二硒醚(2j): 117 mg, 黄色固体, 产率62.78%. Rf=0.73(石油醚/乙酸乙酯, V∶V=5∶1); m.p. 85~86 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.56 (dd, J=7.8, 1.6 Hz, 2H), 7.23~7.18 (m, 2H), 6.88~6.82 (m, 2H), 3.91 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 154.69, 130.64, 128.18, 121.95, 121.92, 110.20, 55.96, 55.90; HRMS (ESI) calcd for C14H14O2Se2 373.9324, found 373.9314.
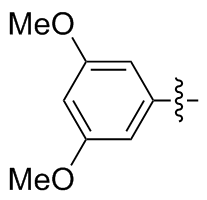
3,3',5,5'-四甲氧基二苯二硒醚(2k): 181 mg, 橙色固体, 产率83.84%. Rf=0.23(石油醚/乙酸乙酯, V∶V=6∶1); m.p. 156~157 ℃; 1H NMR (400 MHz, CDCl3) δ: 6.78 (dd, J=4.8, 2.2 Hz, 4H), 6.32 (dt, J=4.1, 2.2 Hz, 2H), 3.74 (d, J=9.5 Hz, 12H); 13C NMR (101 MHz, CDCl3) δ: 160.85, 132.34, 109.08, 100.43, 55.54, 55.44, 55.40; HRMS (ESI) calcd for C16H17O4Se2 (M-H-) 432.9467, found 432.9494.
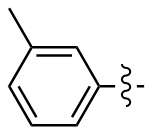
3,3'-二甲基二苯二硒醚(2l): 159 mg, 黄色油状物, 产率92.93%. Rf=0.88(石油醚/乙酸乙酯, V∶V=30∶1); 1H NMR (400 MHz, CDCl3) δ: 7.46~7.38 (m, 4H), 7.16 (t, J=7.6 Hz, 2H), 7.09~7.02 (m, 2H), 2.32 (d, J=2.4 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ: 139.00, 132.15, 128.98, 128.89, 128.66, 128.61, 21.31, 21.21; HRMS (ESI) calcd for C14H14Se2 341.9426, found 341.9418.
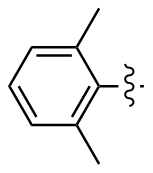
2,2',6,6'-四甲基二苯二硒醚(2m): 162 mg, 棕色固体, 产率87.52%. Rf=0.64(石油醚/乙酸乙酯, V∶V=10∶1); m.p. 102~104 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.09~6.98 (m, 6H), 2.26 (s, 12H); 13C NMR (101 MHz, CDCl3) δ: 149.42, 143.83, 129.24, 127.43, 24.21; HRMS (ESI) calcd for C16H18Se2 369.9739, found 369.9728.
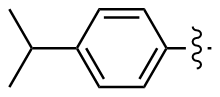
4,4'-二异丙基二苯二硒醚(2n): 148 mg, 黄色油状物, 产率74.56%. Rf=0.85(石油醚/乙酸乙酯, V∶V=30∶1); 1H NMR (400 MHz, CDCl3) δ: 7.71~7.35 (m, 4H), 7.25~7.08 (m, 4H), 2.99~2.82 (m, J=6.8 Hz, 2H), 1.27 (dd, J=6.9, 3.0 Hz, 12H); 13C NMR (101 MHz, CDCl3) δ: 148.87, 132.07, 130.91, 127.38, 33.80, 23.96, 23.94; HRMS (ESI) calcd for C18H22Se2 398.0052, found 398.0043.
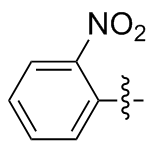
2,2'-二硝基二苯二硒醚(2o): 161 mg, 黄色固体, 产率79.98%. Rf=0.32(石油醚/乙酸乙酯, V∶V=4∶1); m.p. 132~134 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.34 (dd, J=8.0, 1.7 Hz, 2H), 7.58 (dd, J=7.6, 1.6 Hz, 2H), 7.02 (dd, J=7.9, 1.6 Hz, 2H), 6.87 (dd, J=8.1, 1.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 139.24, 133.91, 132.33, 129.70, 119.30, 115.20; HRMS (ESI) calcd for C12H9N2O2Se2 (M+H+) 404.8888, found 404.8893.
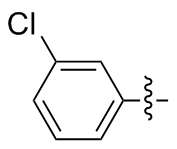
3,3'-二氯基二苯二硒醚(2p): 146 mg, 黄色固体, 产率76.45%. Rf=0.50(石油醚/乙酸乙酯, V∶V=8∶1); m.p. 75~76 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.90~7.78 (m, 2H), 7.82~7.65 (m, 2H), 7.65~7.50 (m, 2H), 7.47~7.33 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 135.18, 134.23, 131.54, 129.96, 118.88, 113.75; HRMS (ESI) calcd for C12H8Cl2Se2 381.8333, found 381.8322.
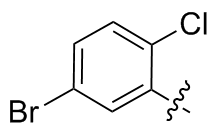
2,2'-二氯-5,5'-二溴二苯二硒醚(2q): 144 mg, 黄色固体, 产率53.58%. Rf=0.45(石油醚/乙酸乙酯, V∶V=30∶1); m.p. 133~135 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.73 (d, J=8.4 Hz, 2H), 7.32 (dd, J=8.4, 2.3 Hz, 2H), 7.21 (d, J=8.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ: 133.51, 131.82, 130.67, 121.86; HRMS (ESI) calcd for C12H5F4Se2 (M- H+) 538.6460, found 538.6488
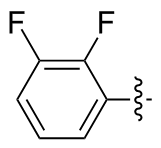
3,3',4,4'-四氟二苯二硒醚(2r): 86 mg, 橙色油状物, 产率44.68%. Rf=0.57(石油醚/乙酸乙酯, V∶V=30∶1); 1H NMR (400 MHz, CDCl3) δ: 7.45~7.24 (m, 4H), 7.16~6.99 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 128.49, 128.46, 128.43, 128.39, 121.53, 121.35, 118.14, 117.96; HRMS (ESI) calcd for C12H6ClF4Se2 (M+Cl-) 420.8430, found 420.8454.
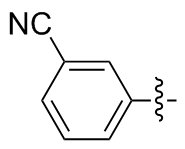
3,3'-二氰基二苯二硒醚(2s): 137 mg, 黄色油状物, 产率75.45%. Rf=0.83(石油醚/乙酸乙酯, V∶V=25∶1); 1H NMR (400 MHz, CDCl3) δ: 7.64 (t, J=1.8 Hz, 2H), 7.53~7.47 (m, 2H), 7.29~7.22 (m, 4H); 13C NMR (101 MHz, CDCl3) δ: 130.92, 130.91, 130.04, 129.17, 127.92, 127.76; HRMS (ESI) calcd for C14H8ClN2Se2 (M+Cl-) 398.8711, found 398.8724.
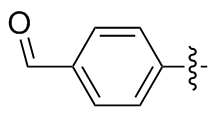
4,4'-二苯甲醛二硒醚(2t): 138 mg, 黄色油状物, 产率74.37%. Rf=0.35(石油醚/乙酸乙酯, V∶V=7∶1); 1H NMR(400 MHz, CDCl3) δ: 9.96 (s, 2H), 7.79~7.73 (m, 8H); 13C NMR (101 MHz, CDCl3) δ: 191.19, 138.42, 135.53, 130.34, 130.32; HRMS (ESI) calcd for C14H9O2Se2 (M-H+) 368.8538, found 368.8554.
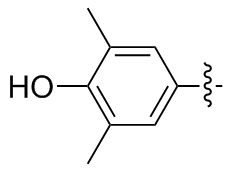
3,3',5,5'-四甲基-4,4-二羟基二苯二硒醚(2u): 160 mg, 黄色油状物, 产率79.47%. Rf=0.25(石油醚/乙酸乙酯, V∶V=8∶1); 1H NMR (400 MHz, CDCl3) δ: 7.15 (t, J=0.7 Hz, 4H), 2.29 (d, J=0.7 Hz, 12H); 13C NMR (101 MHz, CDCl3) δ: 151.25, 127.25, 126.99, 123.13, 16.06; HRMS (ESI) calcd for C16H17O2Se2 (M-H+) 400.9564, found 400.9543
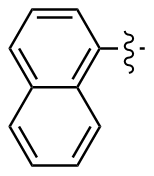
1,1'-二萘二硒醚(2v): 135 mg, 红色油状物, 产率65.47%. Rf=0.54(石油醚/乙酸乙酯, V∶V=30∶1); 1H NMR (400 MHz, CDCl3) δ: 8.42~8.13 (m, 2H), 7.94~7.72 (m, 6H), 7.61~7.13 (m, 6H); 13C NMR (101 MHz, CDCl3) δ: 134.09, 134.05, 129.86, 129.82, 128.59, 128.55, 127.99, 126.35, 126.32, 125.64; HRMS (ESI) calcd for C20H14Se2 (M-H+) 412.9353, found 412.9353.



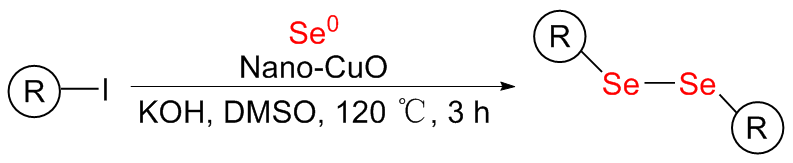

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}