

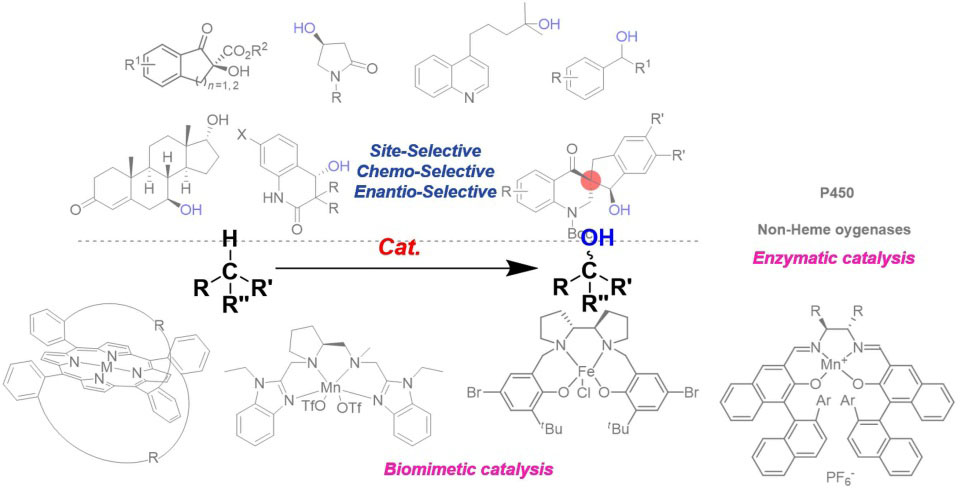
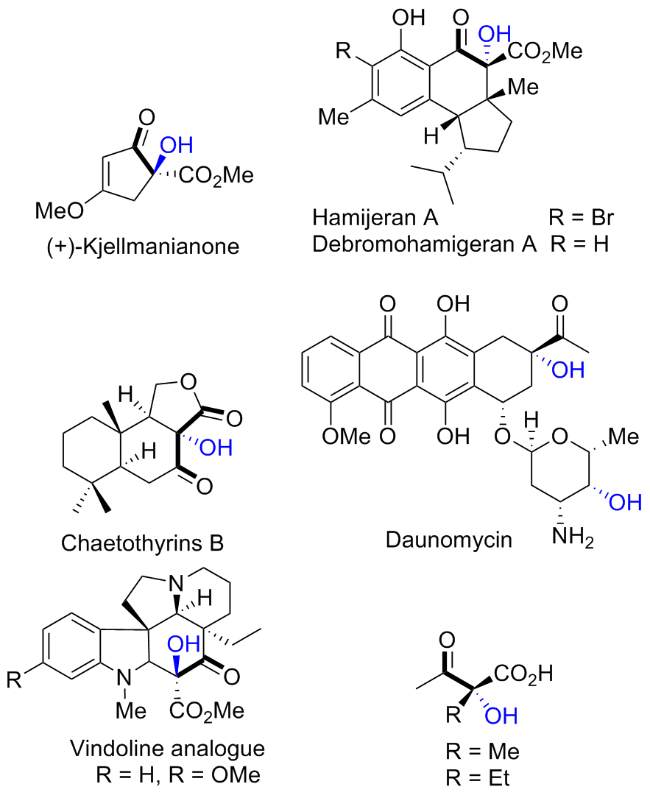
手性化合物作为生物活性分子与天然产物的关键结构单元, 其立体选择性精准构建始终是合成化学领域的核心挑战和研究前沿. 在当代有机合成方法学中, C—H键直接官能化技术因其卓越的原子经济性和合成步骤简洁性而备受瞩目, 其中过渡金属催化的不对称C—H活化策略更是在配体设计与反应机理层面取得突破性进展[1]. 这一创新性策略不仅实现了复杂手性骨架的高效构建, 更在药物先导化合物修饰、天然产物全合成等应用场景中展现出显著的革新潜力, 为现代绿色合成化学的发展提供了具有范式意义的解决方案. C—H键直接羟基化属于C—H键官能化的范畴, 是有机合成化学领域的研究热点之一[2]. 具有光学活性醇结构的化合物广泛地存在于生物活性物质和天然产物中, 它们可作为中间体广泛应用于合成功能有机化合物, 包括抗肿瘤药、抗生素、信息素和糖类等. 例如从马尾藻中提取出来的一种抗生素(+)-Kjellmanianone, 对革兰氏阳性菌具有较强的抗菌活性[3]; Hamigeran A和Debromo-hamigeran A提自一种海绵生物, 其结构特点是有1-氧代-2-羟基四氢萘骨架[4]; Chaetothyrins B是一种地衣内生菌次级代谢物, 具有广谱的抗菌活性和细胞毒性[5]; 从放线菌中获得的Daunorubicin可以用于治疗急性淋巴细胞白血病[6], 该结构还是文朵灵和11-去甲氧基文朵灵全合成的关键中间体. 文朵灵是白坚木属生物碱, 具有抗肿瘤活性; α-乙酰羟基丁酸/乳酸是异亮氨酸与缬氨酸的生物合成前体[7](图1). 具有光学活性的羟基结构单元在生物活性化合物中的重要性以及它们作为手性合成中间体和手性辅助试剂等多样性功能, 促进了诸多不对称氧化合成方法的发展[8].
在过去的几十年里, 生物或化学合成方法为不对称氧化提供了高效和可持续的方法, 包括使用金属催
化[9]、有机小分子催化[10]、酶催化[11]、仿生催化[12]以及光催化[13]等. 在这一过程中, 大量的工作已经试图解决不对称氧化过程中的许多缺点与局限性, 但在反应过程中仍存在多重科学挑战: 首先, 不同位点C—H键的键离解能(BDE)及电子云分布特征存在显著差异, 导致化学选择性的精准调控成为关键瓶颈; 其次, 在复杂分子体系中难以同时实现高区域选择性与立体选择性控制; 再次, 催化体系中普遍存在配体-金属中心动态平衡问题, 配体解离常引发活性物种失活, 显著降低催化循环效率; 特别值得注意的是, 在不对称羟基化构建手性醇的过程中普遍存在过度氧化倾向, 转化为酮类副产物而失去手性中心[14]. 因此, 本文将基于C—H键不对称氧化反应体系的理论突破与合成需求, 从反应机制创新与催化系统进化双重视角对C—H键不对称羟基化反应方法进行综述, 并对可持续发展不对称羟基化反应面临的契机和挑战进行了展望.
1 生物催化C—H键不对称羟基化反应
1.1 P450酶催化不对称羟基化反应
有机化合物中的C—H键常因键能高和极性小而具有化学反应惰性, 在温和条件下将C—H键选择性催化活化以进行不对称羟基化存在热力学和动力学的双重挑战, 因此迫切需要探索新颖的活化策略以实现基于C—H键官能化反应在有机物合成上的突破[15]. 以P450单加氧酶为代表的生物催化方法在不对称羟基化反应方面取得了令人印象深刻的突破性进展.
细胞色素P450单加氧酶(cytochrome P450或CYP450)是一个较大的超家族酶, 可催化多种底物发生氧化反应. 所有的P450都含有血红素辅因子, 当还原的辅因子与CO结合时会在450 nm处观察到强吸收峰, P450酶因此而得名. P450通常可催化底物中的C—H键氧化为羟基, 同时也能进行一系列其他氧化反应[16].
1.1.1 P450酶催化脂肪族C—H键不对称羟基化反应
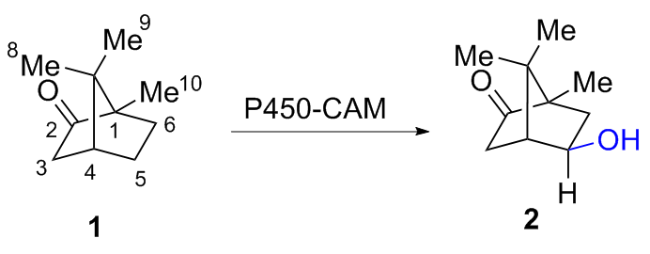
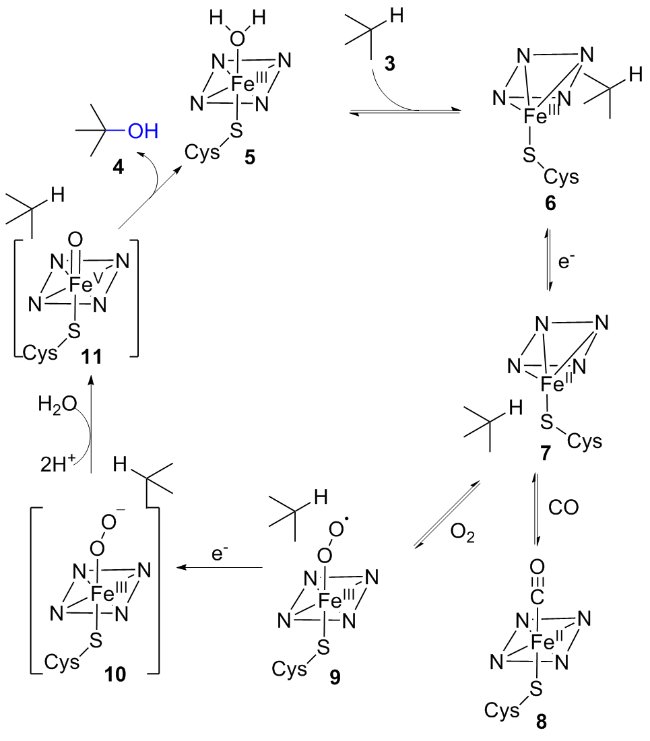
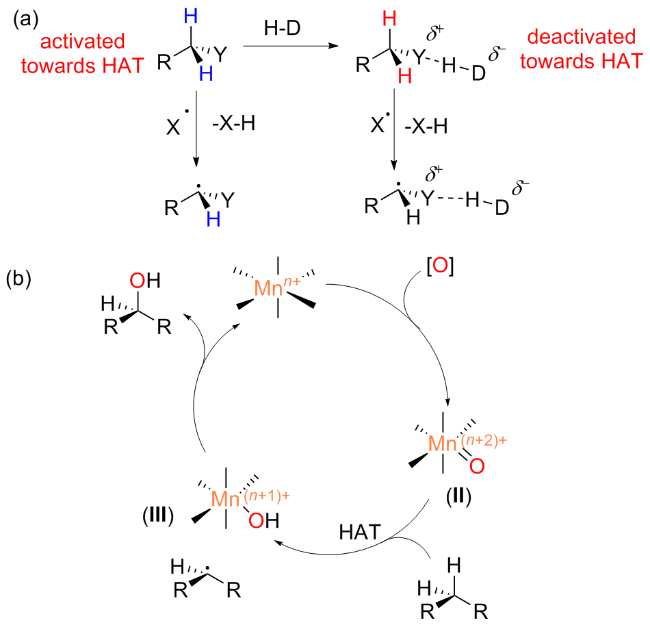
Maryniak提出的P450催化3的C—H键不对称羟基化的机理正是Groves[18]提出的, 被研究者普遍接受的“Oxygen rebound”机制[19]: 低自旋三价铁配合物5在烷烃3存在条件下失去水分子配体后转化为高自旋三价铁6. 6得电子后被还原成二价铁中间体7, 7结合羰基配体后生成羰基化亚铁配合物8, 还可以被氧气氧化成三价的过氧化自由基物种9. 9获得第二个电子后生成过氧负离子10, 随后10在质子作用下通过断裂O—O键脱水得到五价Fe(V)=O物种11, 11为真正的氧化中间体, 随后烷烃被氧化成醇产物4并再生铁酶催化剂5, 进入下一次催化循环(Scheme 2)[14].
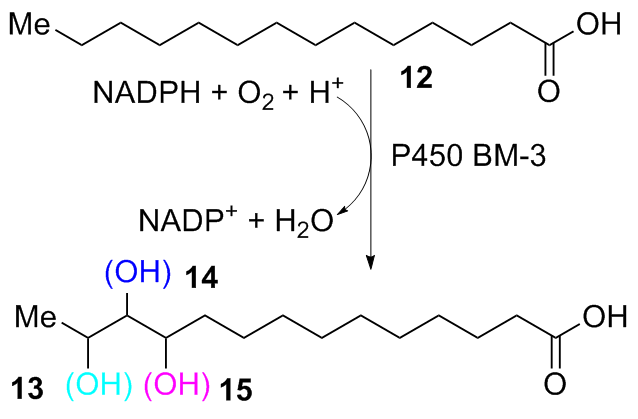
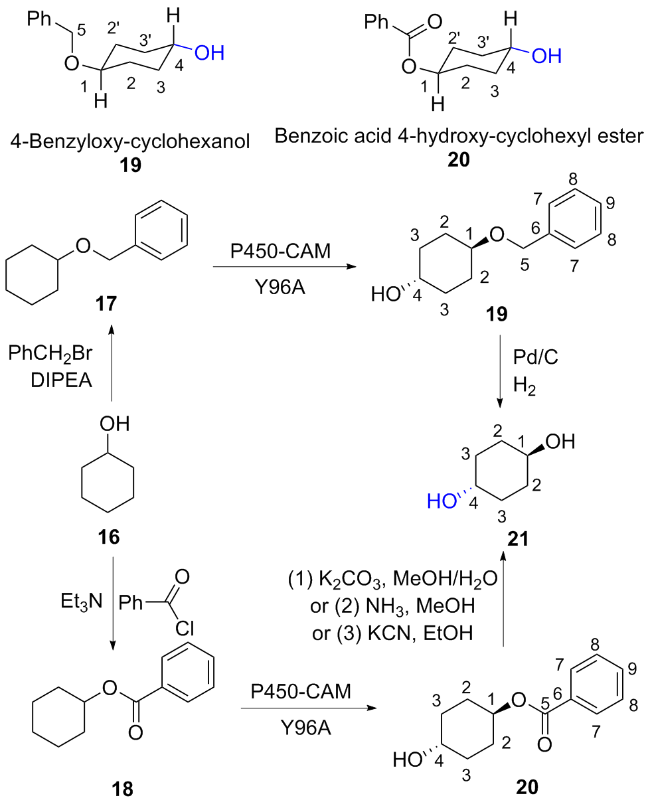
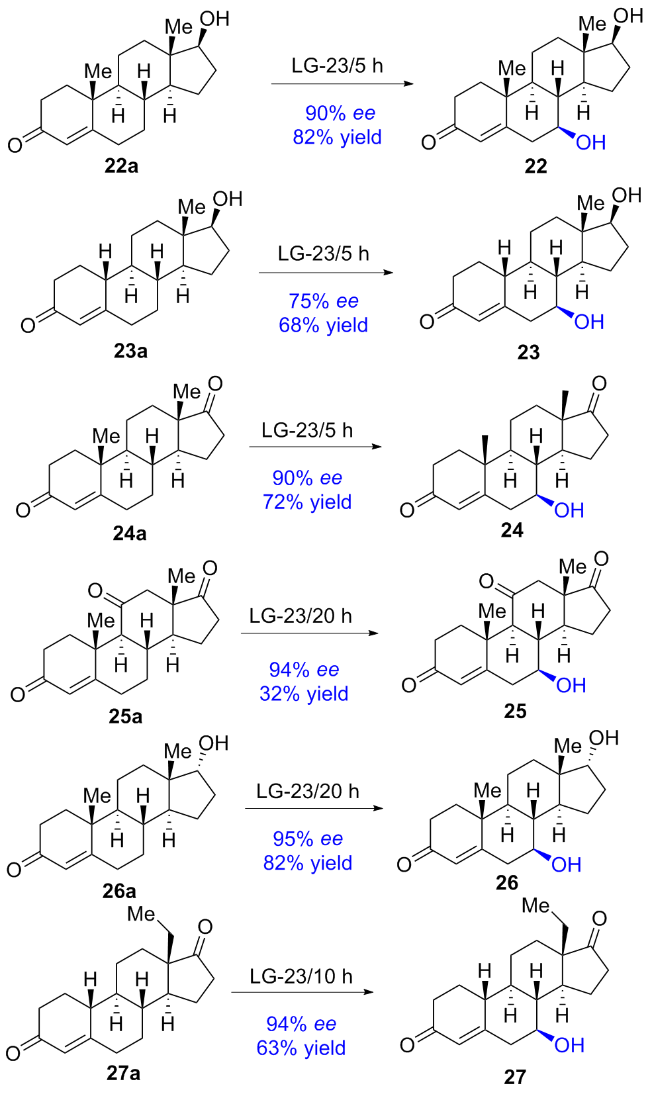
1.1.2 P450酶催化苄位C—H键不对称羟基化反应
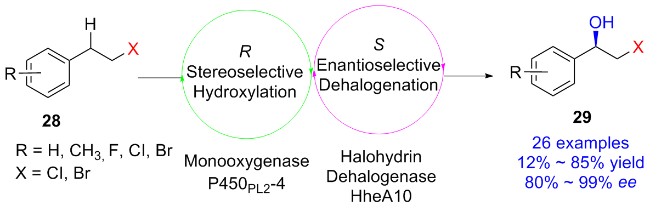
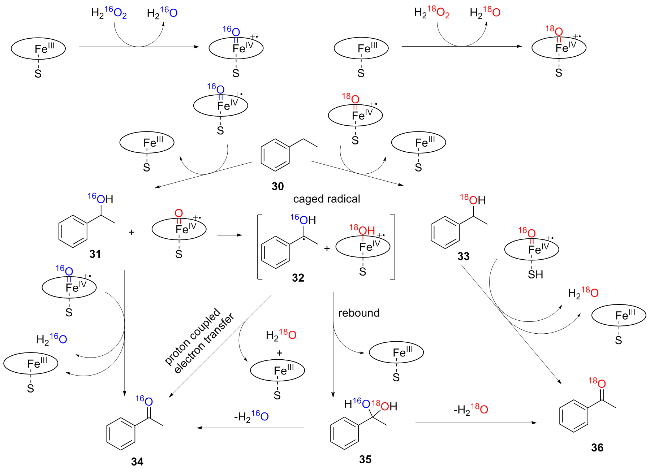
真菌Agrocybe aegerita (AaeAPO)的过氧酶是一种有效的单加氧酶, 可以将过氧化物中的氧原子转移到不同的有机底物上. 2012年Kluge等[25]利用真菌AaeAPO中的血红素-硫醇盐过氧酶对烷基苯进行立体选择性的苄基羟基化反应. 苄位C—H键羟基化反应只生成了 (R)-1-苯基烷基醇产物. (R)-1-苯基乙醇、(R)-1-苯基丙醇和(R)-1-苯基丁醇产物的ee值可达到99%以上. 研究中也发现, 当底物碳链较长时, ee和酶的总转换数会降低, 同时过度氧化副产物(如1-苯基酮)的数量会增加. 作者提出了AaeAPO在H216O2或H218O2存在下对乙苯和1-苯基乙醇进行氧化的双模式机制(Scheme 7).
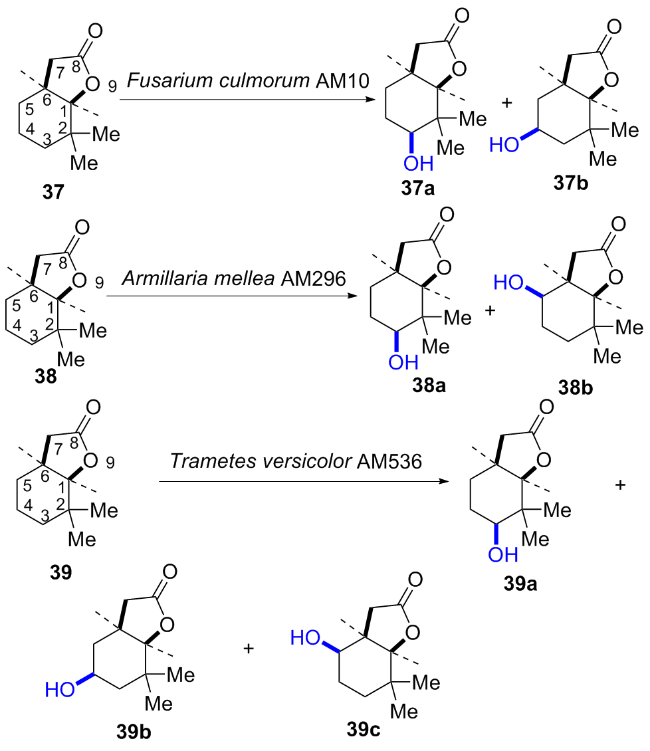
2019年, Mazur等[26]利用丝状真菌的酶系统对多环内酯底物进行C—H键氧化转化研究, 从15个菌株中筛选出镰刀菌AM10、蜜环菌AM296、云芝分枝杆菌AM536 (Fusarium culmorum AM10, Armillaria mellea AM296, Trametes versicolor AM536) 3种菌株可作为有效的生物催化剂(Scheme 8). 其中镰刀菌AM10催化3-C或4-C位羟基官能化, 蜜环菌AM296催化3-C或5-C位的氧化反应, 而云芝分枝杆菌AM536催化3-C、4-C和5-C三个位点均发生羟基化反应. 以上获得的羟基化内酯产物的ee值在17%~99%范围内. 对羟基化产物进行体外抗增殖活性测试, 研究结果表明无论羟基取代基的位置如何, 所有测试的内酯对选定的癌细胞都表现出相似的抑制活性(IC50在22.8~33.9 µg/mL范围内).
1.2 非血红素加氧酶催化的不对称羟基化反应
酶促氧化模型与反应机理研究的突破性进展, 革命性推进了氧原子转移理论在C—H键活化领域的应用深化. 除了血红素酶催化的不对称羟基化反应, 科学家们对非血红素型酶催化的不对称羟基化也进行了广泛的研究. 非血红素加氧酶(Non-Heme oygenases)是一类广泛存在于生物体内的金属酶, 能够高效、选择性地催化惰性C—H键的氧化反应. 这些酶不依赖血红素辅基, 而是通过单核或多核金属中心(如Fe2+、Fe3+和Mn2+等)活化氧气(O₂)或过氧化物(如H₂O₂)生成高活性的金属氧中间体, 进而实现C—H键的氧化, 在生物代谢、药物合成和环境修复中具有重要应用.
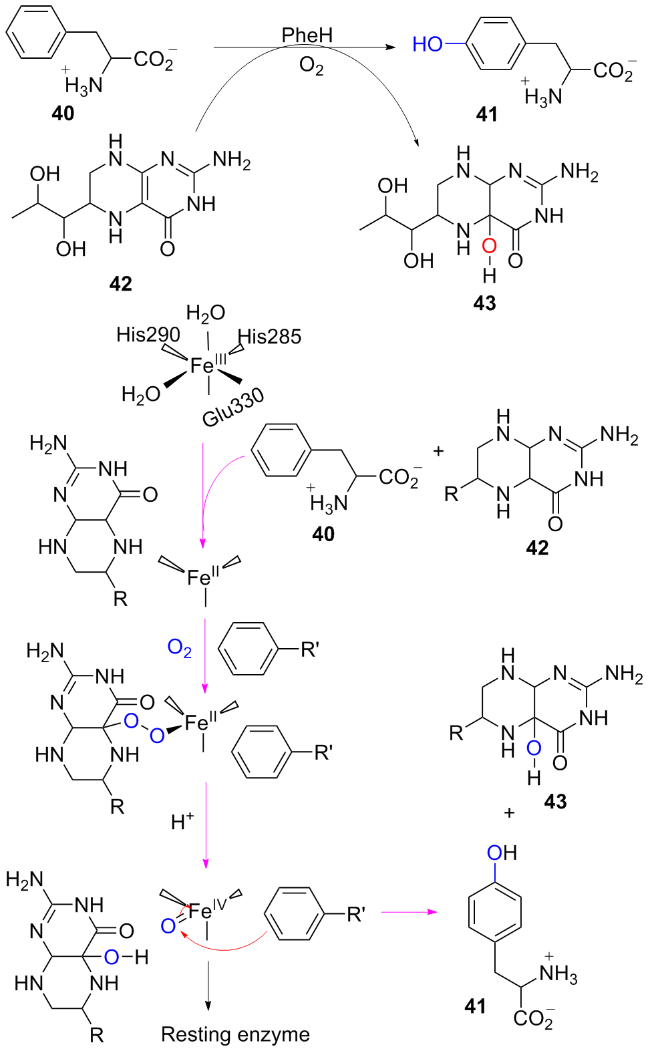
哺乳动物合成神经递质3,4-二羟基苯丙氨酸和血清素以及酪氨酸的反应是由羟化酶催化的, 该酶含有与2-His+Asp/Glu motif结合的Fe(II). 然而, 这些酶催化上述物质的羟基化反应需要有活化的氧[27]. 因此, 该反应主要经历氧激活、与底物结合发生羟基化反应两步. 以酪氨酸的羟基化为例, 首先, 在氧激活阶段, Fe(II)和电子供体四氢生物蝶呤辅因子42之间形成过氧桥键, 4α-过氧蝶呤加合物的稳定性使其发生O—O键异裂, 生成高活性的Fe(IV)=O氧合中间体. 该物种通过亲电芳香取代反应作用于酪氨酸底物, 最终生成相应产物41[28]. 在氧激活阶段, 底物在Fe(II)附近但不与Fe(II)结合, 其起到的作用是促进氧气与Fe(II)的结合(Scheme 9).
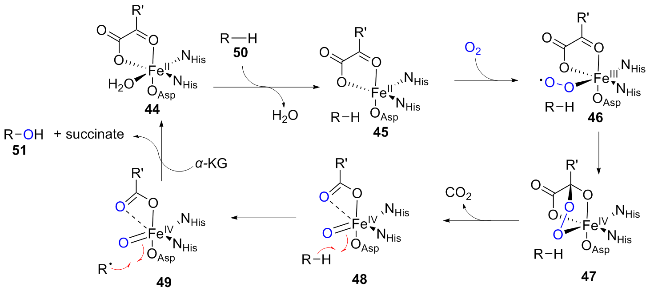
2-His-1-羧酸酯面部三合体(2-His-1-carboxylate facial triad)作为单核非血红素铁酶的核心配位架构, 通过八面体配位几何精确锚定Fe(II)中心, 在分子氧(O2)的活化过程中发挥关键作用. 该结构基序广泛存在于α-酮戊二酸(α-KG)依赖型双加氧酶家族中, 其通过协同电子转移机制实现O2的异裂活化, 生成高反应活性的Fe(IV)=O氧合中间体. 与之协同作用的α-KG不仅是三羧酸循环中琥珀酰-CoA合成的重要前体, 更作为必需共底物参与催化循环: 在氧原子转移反应中, α-KG的1-C羧酸基团通过螯合作用稳定金属中心, 同时其2-C酮基作为电子受体促进O2的还原活化. 这种独特的“代谢物-辅因子”双重功能, 使得α-KG依赖型酶系统在胶原蛋白羟化等关键生物过程中实现精确的氧化修饰. Scheme 10所示为一类依赖α-KG酶共识机制发生的C—H键羟基化反应机理[29], 该过程是基于Hanau-skeabel和Günzler假设的机制而提出的[30].
2 仿生催化不对称羟基化反应
2.1 基于金属-porphyrin体系催化的不对称羟基化反应
受血红素和相关酶非凡的催化能力的启发, 金属卟啉仿生酶已被广泛应用于催化诸多重要的化学转化过程[31]. 卟啉配体可与许多过渡金属形成配合物, 在催化C—H键不对称羟基化反应过程中发挥了重要作用, 以下将依据金属卟啉类催化剂中金属类型的变化来展开详细的介绍.
2.1.1 Mn-porphyrin催化的不对称羟基化反应
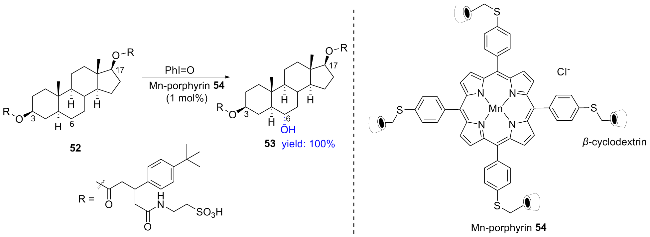
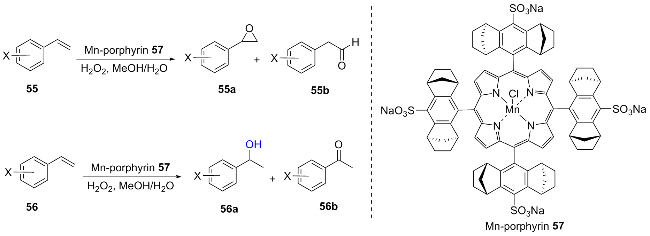
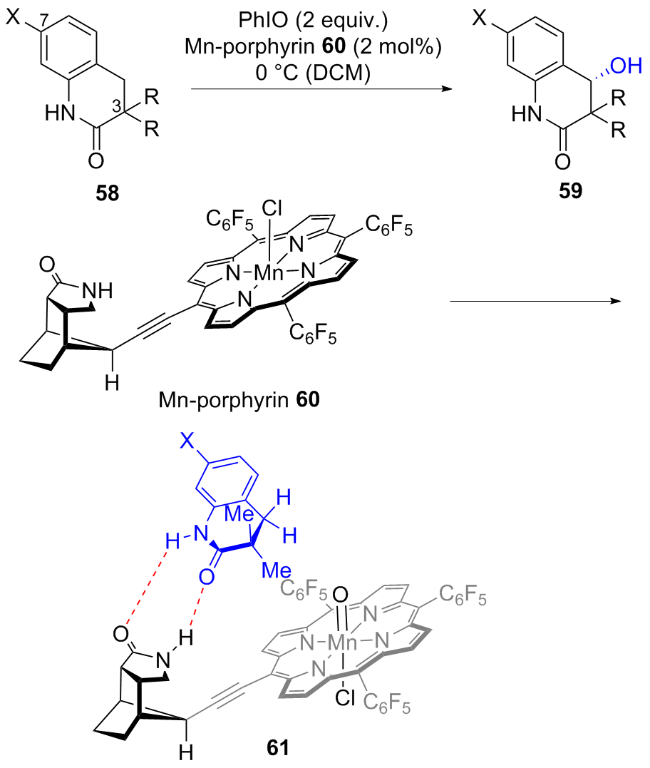
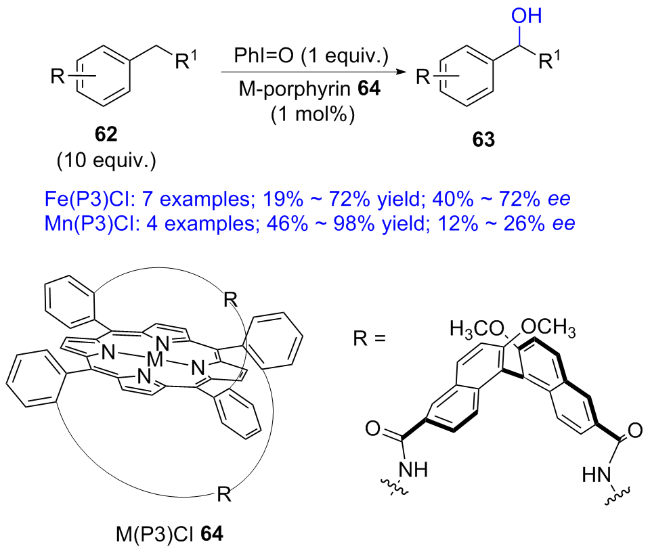
2.1.2 Fe-porphyrin催化的不对称羟基化反应
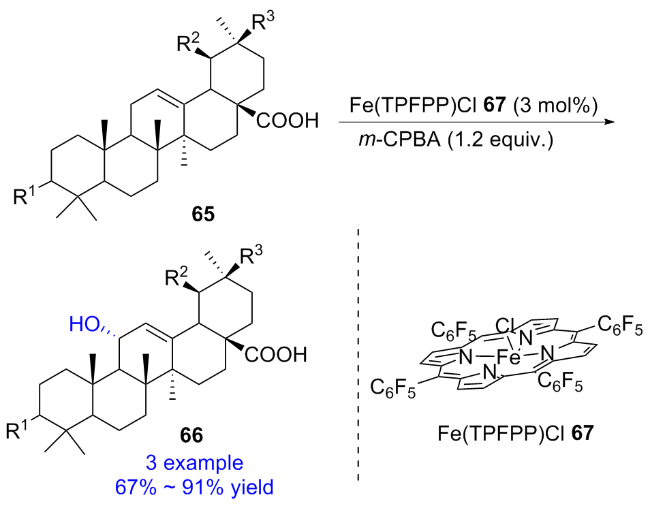
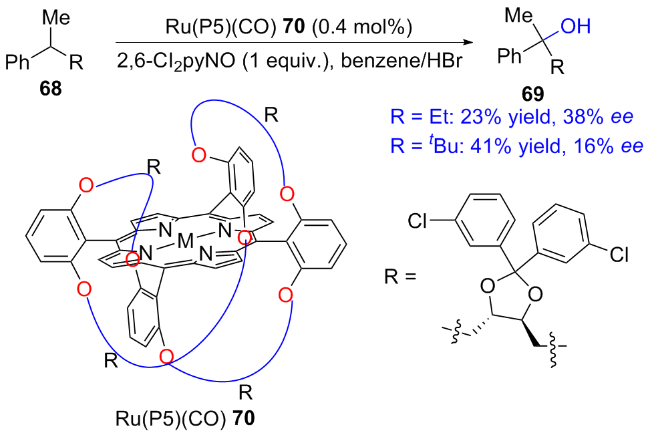
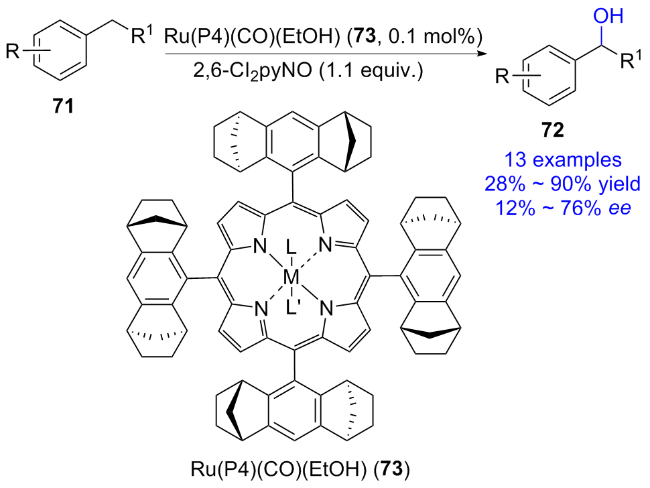
2.1.3 Ru/Os-porphyrin催化的不对称羟基化反应
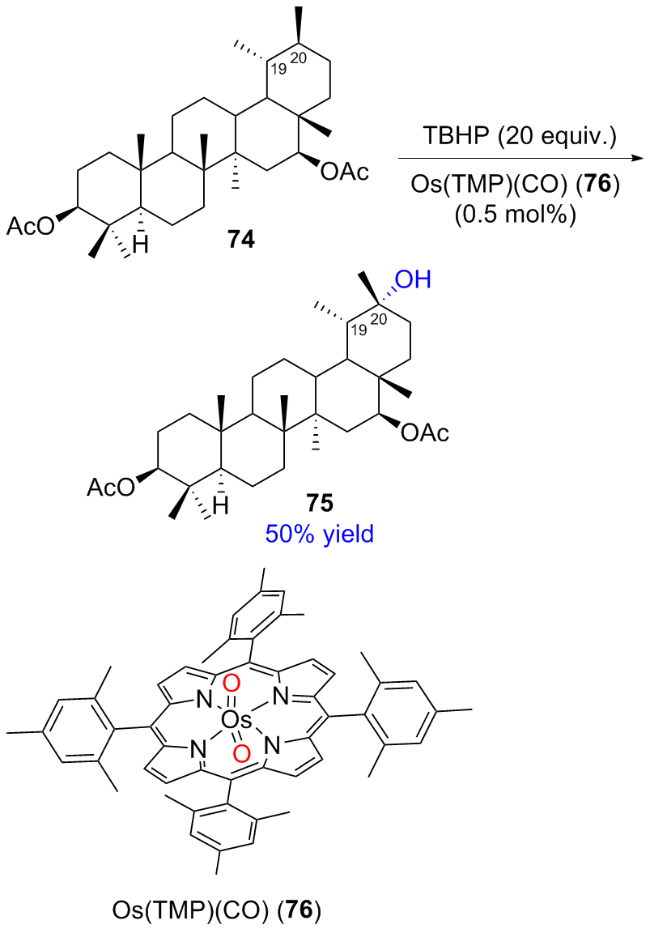
Iida及其同事的研究表明, 叔丁基过氧化氢 (TBHP)与[Os(TMP)(CO)]分别作为氧源和催化剂, 是一种高效、多功能的氧化体系, 可用于各种生物活性分子(包括类固醇[41]、胆汁酸[42]和萜类化合物[43])的功能化, 从而一步直接获得各种新型含氧衍生物. 例如, Os(TMP)(CO) (76)/TBHP氧化官能化系统可以选择性地羟基化二氢法拉醇二乙酸酯74中的一个未活化的叔 C—H键, 以较好的收率形成相应的20a-羟基乌苏酯75[41] (Scheme 18). 值得注意的是, 与分子中的亚甲基碳相比, C-20处的甲基碳更容易被氧化, 这可能是由于其电子密度较高. C-20处的甲基碳更易于羟基化, 这可能是由于其立体结构引起的.
2.2 基于金属-Salen体系催化不对称羟基化反应
席夫碱配合物因其高催化活性在许多领域都广泛应用, 其中一类特殊的席夫碱为Salen, Salen和Salan配体都具有很强的配位能力, 且能与多种金属络合形成手性Lewis酸催化剂. 最近二十多年来, 这类优良的催化剂被广泛应用在不对称催化领域中, 例如烯烃的不对称环氧化[44]等, 以及不对称羟基化反应.
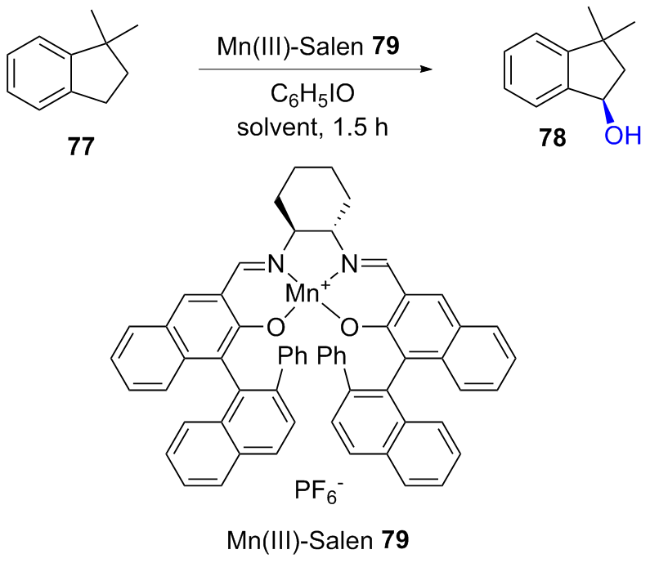
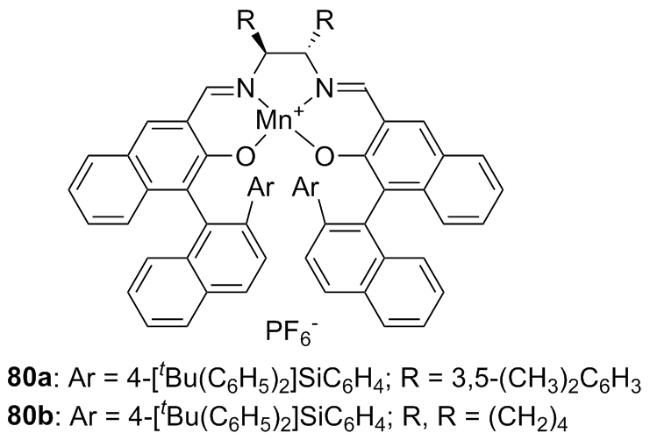
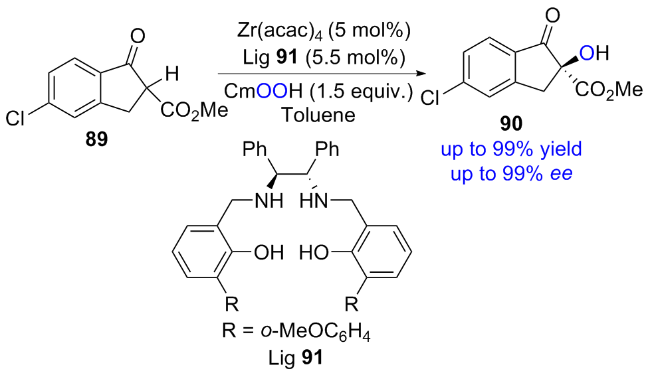
2020年, 王斌课题组[49]对杜邦公司的锆催化体系进行改进, 设计了新型Zr(lV)-Salan催化体系, 对1-茚酮衍生的β-酮酯进行对映选择性和可扩展的α-羟基化反应的研究, 在温和的反应条件下以高产率(高达99%)和优异的对映选择性(高达99% ee) 获得了具有合成价值的羟基化产物(Scheme 23). 机理研究证明: (1)该反应在原位生成了Zr(lV)-Salan复合物, 其作为手性诱导的活性催化剂; (2)亲电片段从氢过氧化枯烯转移到与Zr(lV)结合的烯醇时, 伴随着异溶性O—O键的裂解; (3) Salan配体N上的H原子与氢过氧化枯烯的羟基之间形成氢键, 这对稳定过渡态和提高对映选择性至关重要(Scheme 24).
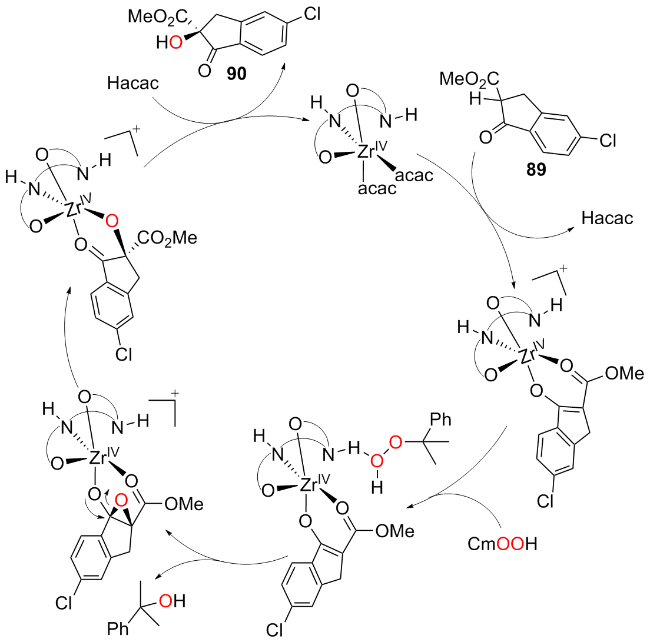
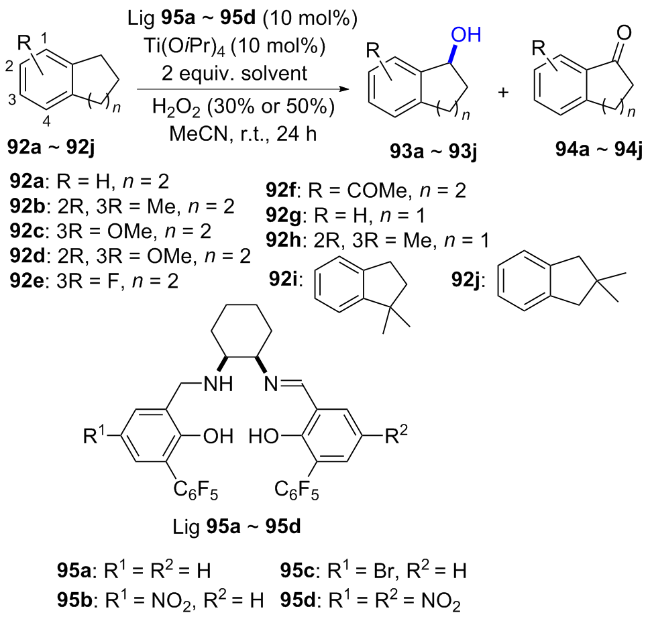
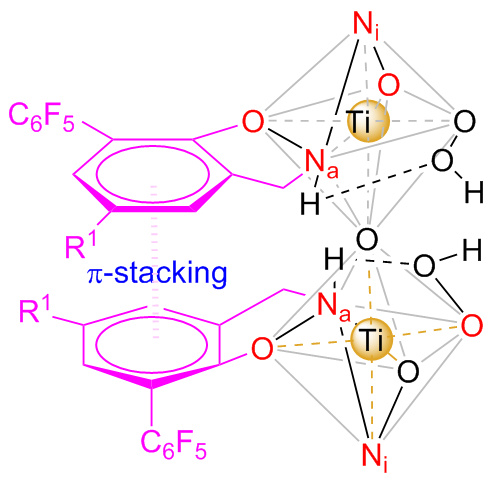
反应机理研究表明, Ti-Salalen催化剂在H2O2氧化剂作用下形成Ti-过氧络合物二聚体, 其侧链上的芳环之间存在着的π-π堆积作用, 有效提高了反应的对映选择性(图2).
2020年, 刘磊课题组[51]开创性开发了Mn-Salen配合物介导的C(sp³)—H键定向氧化新范式, 成功实现了饱和环醚类化合物中惰性C(sp³)—H键的高效对映选择性羟基化. 该催化体系通过精确调控金属中心的配位微环境与氧化电势, 在温和条件下实现C—O键构建, 其对映选择性达到98% ee, 突破了传统自由基机制对立体选择性的限制, 为手性环醚药物中间体的合成提供了革新性方案. 针对无环体系叔炔丙基C(sp³)—H键立体选择性官能化的核心挑战, 研究团队于2024年取得突破性进展[52]. 基于前期环状底物氧化的配体设计经验, 通过引入轴向手性联萘酚骨架修饰Salen配体, 构建了具有动态立体识别功能的催化体系. 该策略创新性结合过渡态构象锁定与动力学拆分双重机制: (1)锰氧活性物种(MnV=O)通过氢原子转移形成碳中心自由基; (2)手性空腔诱导自由基中间体的构象重组; (3)利用底物消旋化速率差异实现动态动力学拆分, 最终获得对映体过量值>96%的叔炔丙醇产物(Scheme 26). 该方法的成功开发标志着无环体系C(sp³)—H键立体控制羟基化技术的突破, 为手性炔丙基叔醇的模块化合成开辟了新途径.
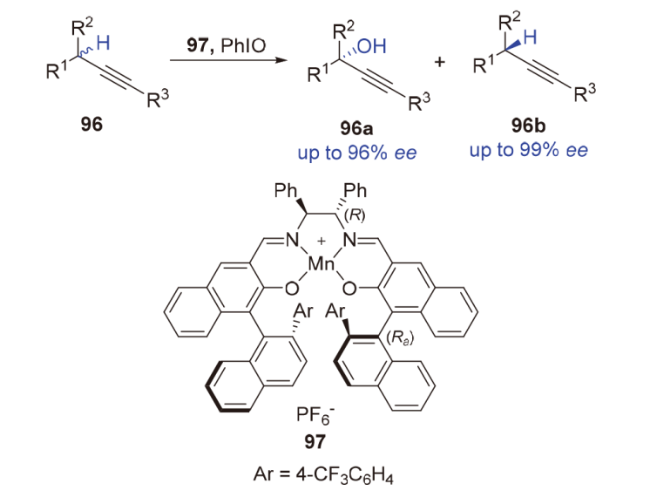
2.3 金属-氨基吡啶体系催化的不对称羟基化反应
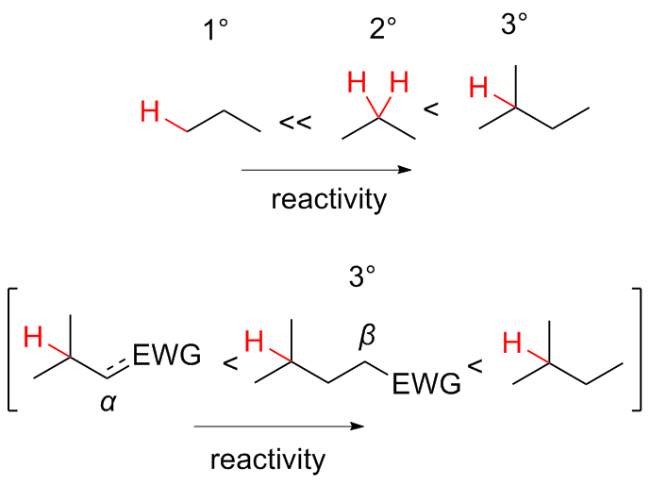
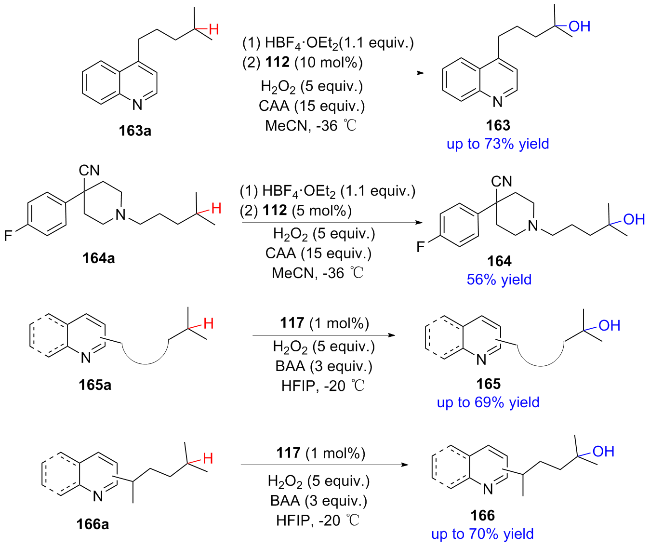
不对称氧化反应的实现常借鉴于由Hamachi等使用的Salen-Mn催化体系. Salen-Mn体系的主要缺点是催化效率低, 转化率通常为20%~50%, 需要使用环境不友好的氧化剂, 如次氯酸盐或间氯过氧苯甲酸等. 在过去的十年里, 以氨基吡啶为配体的过渡金属催化在该领域中得到了广泛的应用, 如使用“绿色”的H2O2作为氧化剂, 即可实现烯烃的不对称环氧化过程, 表现出优异的化学和立体选择性, 其催化效率是Salen-Mn催化剂的100倍以上[53]. 此外, 研究发现氨基吡啶-Mn络合物还能够催化未活化的脂肪族[54]C—H与H2O2发生选择性的氧化反应. 接下来, 将重点介绍氨基吡啶-金属催化体系在C—H不对羟基化反应中的研究进展(Scheme 27).
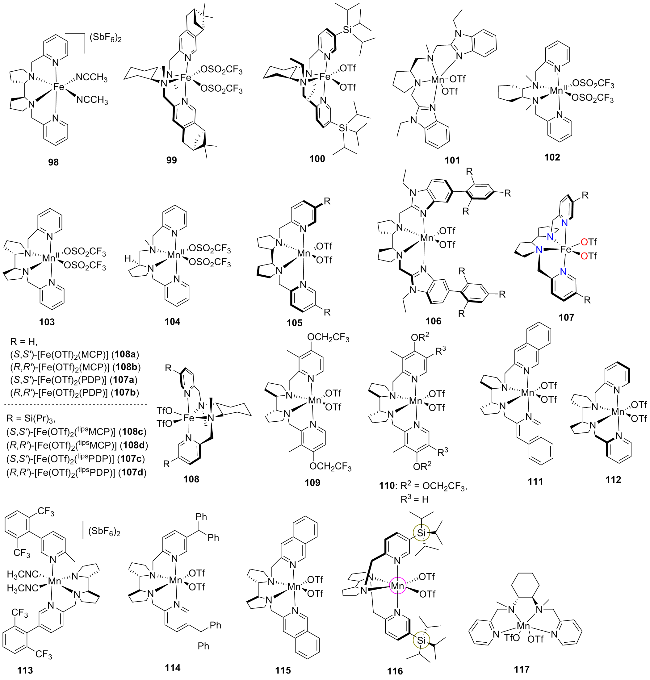
受非血红素-铁催化剂的启发, Que团队[55]设计并应用了氨基吡啶基配位的Fe(II)-TPA络合物作为催化剂, 成功实现了烷烃的立体选择性羟基化, 这一发现填补了铁催化剂在该领域应用的空白, 突破了传统依赖高价金属或贵金属(如Pd、Rh)的局限, 标志着铁催化氧化反应从“非选择性”向“精准合成”的重要跨越.
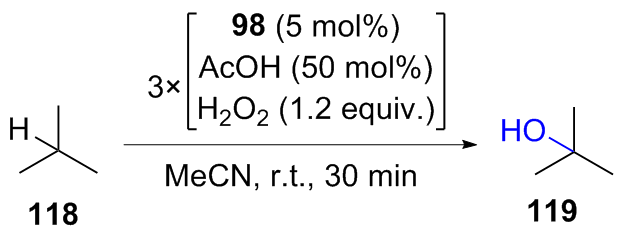
表1 催化剂98对不同类型底物的适用范围Table 1 Range of 98 for different types of substrates |
| Entry | Product | Isolated yield/% (rsma) |
|---|---|---|
| 1 |  | 46 (26) |
| 2 | 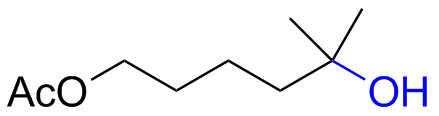 | 53 (43) |
| 3 | 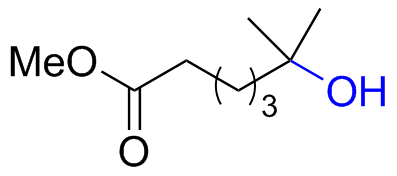 | 60 (18) |
| 4 | 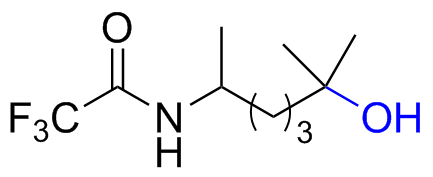 | 43 (33) |
| 5 | 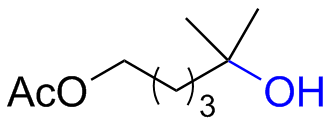 | 52 (21) |
| 6 | 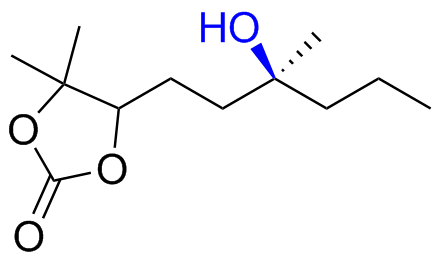 | 57 (27) |
| 7 | 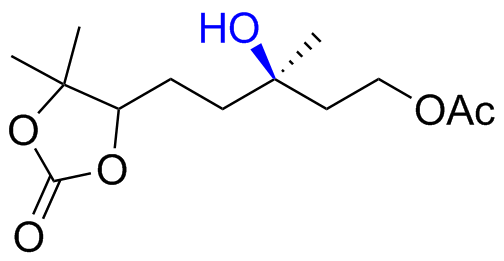 | 43 (42) |
| 8 | 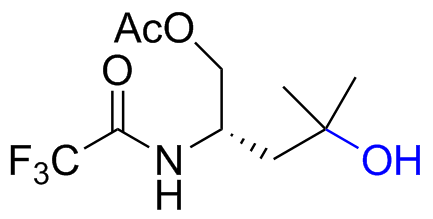 | 33 (67) 90*,b (8) |
| 9 | 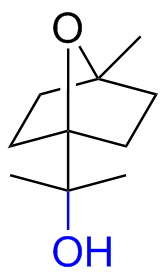 | 52 (20) |
| 10 |  | 92* |
a Recovered starting material. b GC yield. |
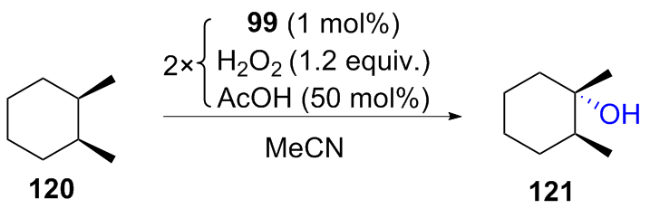
Costas课题组[57]在前人研究工作基础上, 改进了氨基吡啶配体的结构并合成了新型非血红素-Fe配合物99, 配体上的吡啶氮原子处于反式位置, 脂环上的氮原子位于顺式, 可有效阻止氧代二聚体非催化物种的形成, 该催化剂仍遵循血红素复合物催化氧化的机制[58]. 作者首先以顺-1,2-二甲基环己烷120为底物, H2O2为绿色氧化剂, 在1 mol%负载量的99催化作用下, 底物转化率可达到79%, 获得了57%产率的立体结构保持的顺-1,2-二甲基叔醇(121)(Scheme 30). 通过分批次等量加入催化剂和添加剂的方法, 反应产率可提高至70%. 研究结果表明, 该催化剂对三级C—H键具有良好的化学选择性.
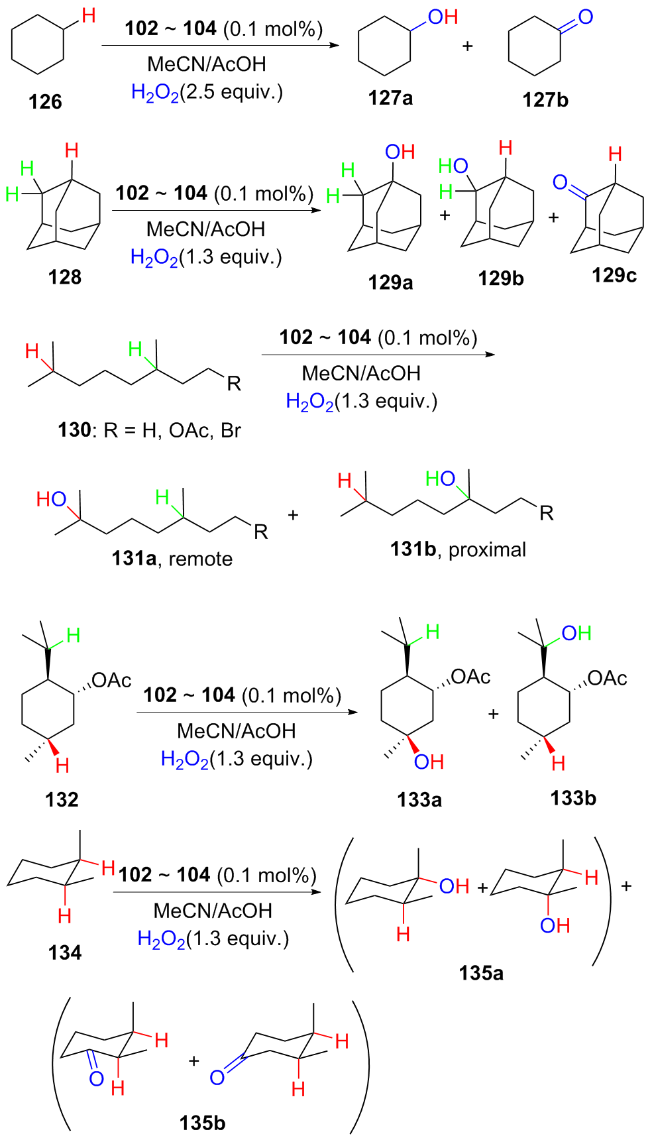
Ottenbacher等[54a]报道了三种手性Mn配合物102~104催化的C—H键氧化反应(Scheme 32), 三种催化剂都表现出了高选择性和高效率, 转化数可达到970. 使用H2O2作为绿色氧化剂, 催化剂负载量仅需0.1 mol%就能快速完成反应(2~4 h). 相较于铁催化体系, Mn催化剂的合成效率高得多, 使得催化剂的用量可由15 mol%降低到千分之一. 例如, 环己烷126在催化剂102~104作用下, 产率高达84%, 醇/酮产物比例约为5∶1; 金刚烷底物128的氧化反应主要发生在富电子的3° C—H上, 对于底物130、132、134的氧化反应, 产率在44%~97%范围之内, 其中底物130氧化的主要产物为三级醇130b, 甲基处于顺式关系的产率占比高于99%, 过度氧化生成的酮为副产物. 需要指出的是, 催化剂103对反应中的每类底物都起到最好的催化效果(Scheme 32).
2016年, Font等[60]设计并合成了一类三烷基硅烷基取代的氨基吡啶-Fe催化剂, 氧化反应的产率得到了显著的提高, 此外, 大空间位阻配体使得催化反应具有良好的区域选择性, 可对萜类和甾体底物的亚甲基位点进行区域选择性的氧化, 即甾体底物中6-C和12-C的C—H键优先被氧化. 以萜类和甾体底物为代表, 作者研究了107和108两类催化剂对复杂分子C—H键选择性氧化的能力. 以(+)-新薄荷脑新戊酸酯(136)为底物, 108a将其氧化成三种类型的产物137a~137c (Scheme 33, 总产率为87%, 酮产物占比71%). 在此基础上, 作者以类固醇为底物进行氧化反应研究(Scheme 34), 结果表明, 当反式雄酮醋酸酯被107c~107d和108c~108d分别催化氧化时(反应选择性结果见表2). 107c和107d选择性氧化位点主要在6-C上(72%~82% ee), 产生相应的酮产物139a和139b, 分离收率约为50%; 相反, 当使用108c和108d时, 优先反应位点为12-C[61].
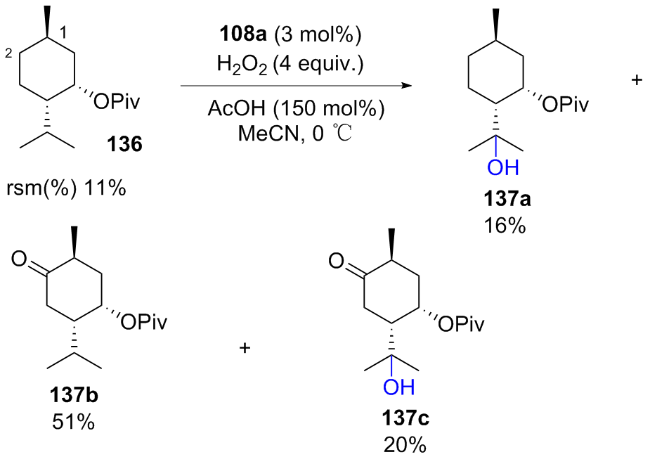
图式33 (+)-新薄荷脑新戊酸酯136选择性氧化反应Scheme 33 Selective oxidation of (+)-neomenthol neopentanoate 136 |
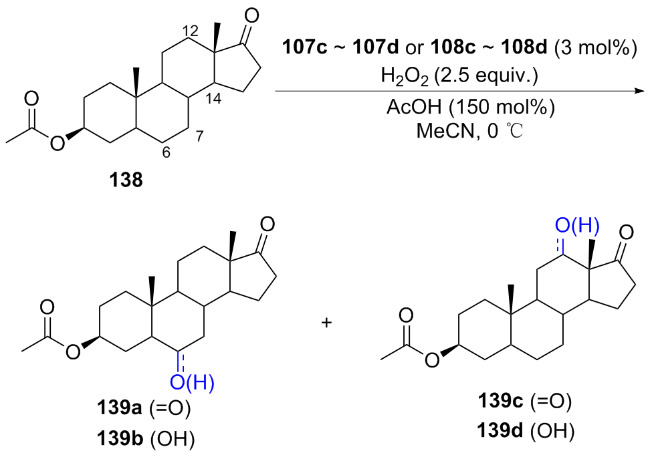
图式34 反式雄酮醋酸酯138的催化氧化Scheme 34 Selective oxidation of (+)-neomenthol neopentanoate 138 |
表2 反式雄酮醋酸酯的氧化Table 2 Oxidation of trans-androsterone acetate |
| Entry | Cat. | Conv/% | Total yield (isolated yield)/% | C6/C7/C12/C14 |
|---|---|---|---|---|
| 1 | 107c | 85 | 70 (50) | 75/5/23/— |
| 2 | 107d | 86 | 80 (71) | 11/1/88/— |
| 3 | 108c | 83 | 60 (49) | 82/12/3/3 |
| 4 | 108d | 50 | 48 (33) | 22/6/69/3 |
基于前人的研究工作, Costas课题组[62]从研究反应溶剂对底物的活化作用出发, 以极性反转为策略实现了脂肪族C—H键选择性氧化反应过程(Scheme 35). 当底物中带有氢键受体(HBA)官能团时, 溶剂通过与底物HBA中心发生氢键作用, 增加了官能团上正电荷密度而逆转直接相连的C—H键极性, 从而使得HAT过程失活, Scheme 35(a)中的Y表示HBA基团, H-D为溶剂, X•为CumO•[63]. 能与C—H键发生氧化反应的活性物种通常会表现出很强的亲电性, 与胺、酰胺、醚和醇等给电子基团接近的C—H键对氧化剂表现出更高的反应活性. 作者通过氟代醇溶剂诱导HBA基团发生极性反转, 使得与HBA基团邻近的C—H键失活, 防止其发生过度氧化反应, 为C—H键选择性氧化反应开辟了一种新的途径.
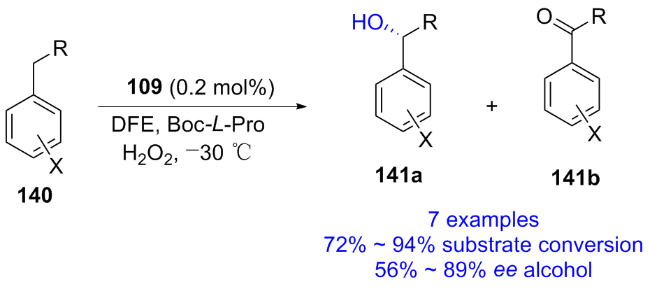
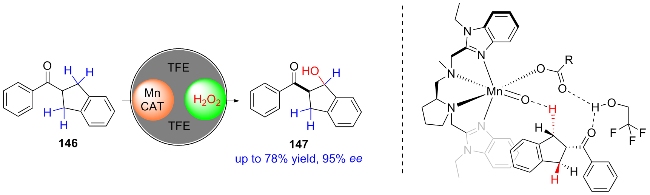
2018年, Ottenbacher及同事[64]通过使用改进的双吡咯烷衍生的锰络合物, 实现了H2O2在氟化乙醇溶液中对苄位C—H键的不对称氧化, 手性苄醇高达89% ee值, 产率接近45%. 作者讨论了溶剂和添加剂对羟基化产率和对映选择性的影响, 结果表明, 用氟化乙醇替代乙腈可极大提高目标产物的收率, 从5%提高到45%, 产率随溶剂不同而增加的次序为: 乙腈<2-氟乙醇<2,2-二氟乙醇<2,2,2-三氟乙醇, 在2,2-二氟乙醇中获得最高的不对称诱导, ee值高达89%. 除了催化剂及溶剂的性质外, 反应的对映选择性还取决于羧酸添加剂的性质, N-Boc-L-脯氨酸表现效果最佳(Scheme 36).
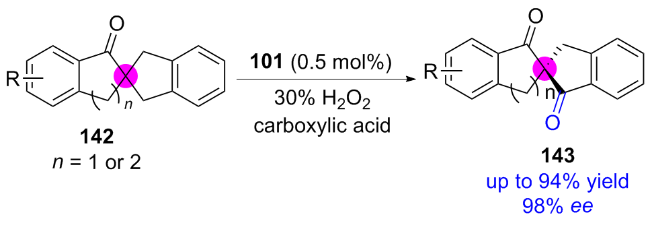
螺环烃凭借其刚性稠环架构与多手性中心特征, 在抗抑郁药物和手性发光材料领域展现出独特价值. 然而, 其惰性C(sp³)—H键的立体选择性氧化面临挑战, 现有策略多依赖铑(III)/钌(II)等贵金属催化剂或高价碘试剂, 普遍存在原子经济性低、ee值波动大及氧化剂环境毒性等问题. 针对这一领域瓶颈, 孙伟团队[65]受细胞色素P450酶氧活化机制启发, 构建了仿生锰催化平台, 利用过氧化氢作为绿色氧化剂, 以92%产率和97% ee合成螺环氧化物中间体, 实现螺环烃C—H键的高对映选择性氧化(Scheme 37). 该工作建立非活化螺环体系 C—H键定向氧化的普适性模型, 为稠环药物分子的后期功能化提供了全新策略.
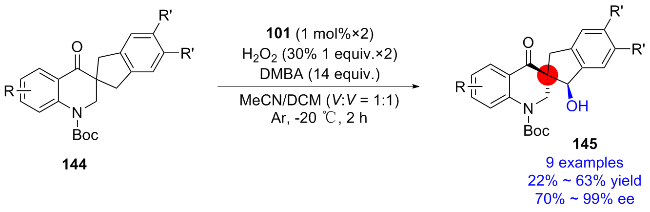
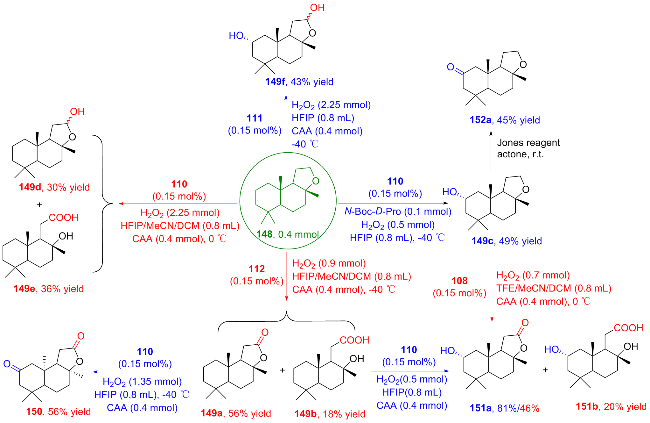
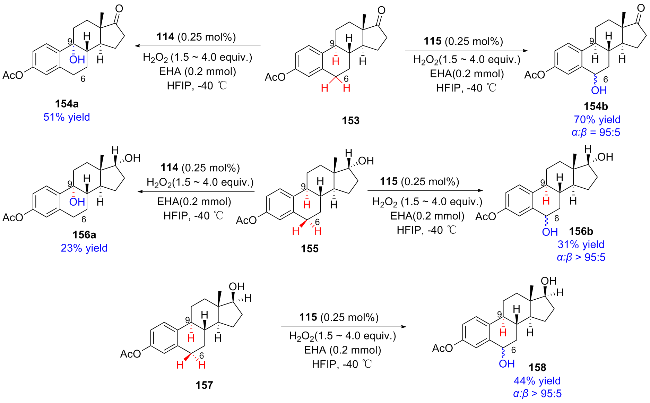
2022年, Ottenbacher等[69]系统研究了Mn-氨基吡啶配合物催化雌酮-3-醋酸酯、17α和17β-雌二醇-3-醋酸酯等底物的C—H键氧化反应. 结果表明, 雌酮-3-醋酸酯底物153在不同的催化剂114和115条件下, 氧化反应的区选择性也发生了变化, 从9-C α-羟基化产物154a转变为6-C α-羟基化产物154b, 而且反应具有良好的非对映选择性和较高的分离产率(Scheme 41). 17α-雌二醇-3-醋酸酯底物155在不同的催化剂114和115催化下, 氧化反应的9-C α-羟基化产物156a与6-C α-羟基化产物156b的产率相对较低. 17β-雌二醇-3-醋酸酯157在催化剂115催化下得到6-C α-羟基化产物158, 产率44%.
关于C—H键的不对称羟基化, 除了C—H键直接氧化, C—H键的不对称内酯化本质也是羟基化, 因为其立体化学在羟基引入时已确定, 而内酯环的形成仅是对羟基酸中间体的后续处理. 这一观点强调了羟基化步骤在构建手性中心中的核心作用, 体现了不对称催化中“关键步骤决定产物性质”的原则.
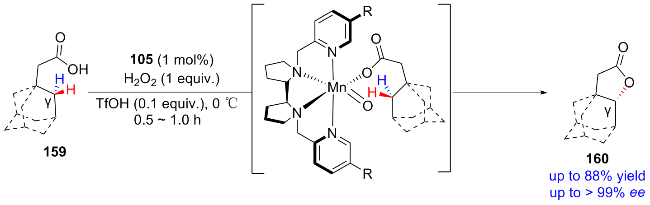
Costas小组[70]利用氨基吡啶-金属配合物催化在活化C—H键不对称内酯化领域内有多项重要研究成果, 实现了脂肪族C—H键的高对映选择性氧化, 未活化的C—H键不对称内酯化[71]等. 2020年, Costas小组[71b]中的Cianfanelli等[71b]针对非酶促体系中未活化亚甲基C—H键不对称氧化难题, 尤其是在面对如何区别同一亚甲基中两个对映异构的C—H键高对映选择性和γ位点选择性的挑战, 采用Mn配合物105作为催化剂, 羧酸基团作为导向基团, 通过羧酸配位作用与锰催化剂结合形成刚性中间体, 从而实现对底物反应位点和对映选择性的精准控制. 该反应在温和的条件下实现了高效转化, γ-内酯产物160的ee值高达99%. 当底物含非等价γ-亚甲基时, 邻近取代基的C—H活化/失活效应可预测内酯化的位点选择性. 此外, 催化剂设计还可调控非对映体C—H键的选择性. 该反应通过创新的锰催化体系, 解决了未活化亚甲基C—H键的不对称内酯化难题, 为复杂手性分子的高效合成提供了重要工具(Scheme 42).
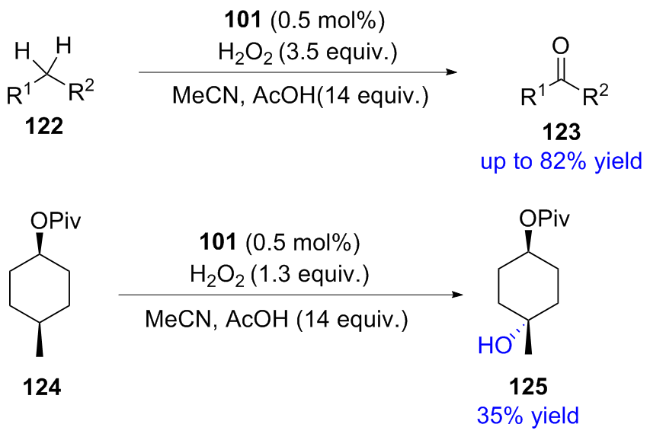
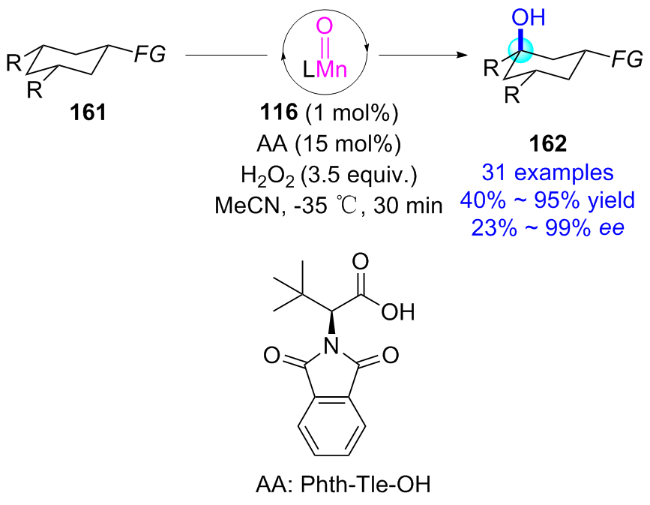
2023年, Palone及其同事[72]首次探究了未活化的叔C(sp3)—H键的非导向和非酶催化对映选择性羟基化, 并产生了多个立体中心. 通过改进Mn催化剂结构获得了MnTips(pdp) 116, 催化体系由氧化锰中心和邻苯二甲酰亚胺保护的氨基酸配体组成, 以H2O2为氧化剂, 二甲基环己烷衍生物161为底物, 作者实现了三级C(sp3)—H键不对称羟基化过程[73]. 不对称羟基化反应可在短时间内发生, 在一步反应中产生了多达四个稳定的手性中心[74], 对映体选择性通过环己烷骨架精确结合到催化活性位点而得到调控, 所得到的手性醇产物162可广泛用于手性环己烯、萜类、二醇、内酯、氨基酸和大环内酯前体的合成(Scheme 43).
目前还需要关注的一个实际重要问题是解决锰介导的氧化反应对底物结构中的杂原子(如N)的耐受性问题[75]. 事实上, 含氮杂环在天然产物和生物活性化合物中普遍存在, 但它们的存在可能通过氮的配位作用使催化剂失活, 或导致含氮底物氧化为相应的氧化产物. 另外, 对于超共轭电子效应活化的C—H官能化反应, 通常需要对氨基进行保护, 这会明显降低合成的步骤经济性, 使整个合成过程复杂化.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
鉴于含氮分子在药物化学中的重要作用, 以及锰介导的C(sp3)—H氧化反应中的官能团耐受性在很大程度上未被充分探索, 期望在不久的将来能看到该领域内更多的研究工作.
3 总结与展望
综上所述, 本篇综述总结了近几十年来C—H键不对称羟基化反应进展. 第一类反应涉及生物催化, 这类酶催化大多数受到酶自身因素的限制, 稳定性有限, 反应速率较慢. 受到酶催化的启发, 研究者们制备了一系列仿生酶催化剂(如金属卟啉催化剂、带有Salan类配体的催化剂以及带有氨基吡啶类配体的新型催化剂)用于不对称羟基化反应中. 其中, 卟啉类配体由于其具有较多的共轭结构, 使得它在反应过程中有较多的位点选择性, 但由于结构刚性较强, 转化率中等, 对映选择性较低, 对于底物的位点选择要求比较苛刻. Salan类配体的催化剂则是增强了配体的柔性, 在较大的配体内形成空腔以供底物发生选择性氧化, 因此这一类配体提高了羟基化反应的对映选择性, 但也存在底物适用范围狭窄、配体与金属易解离导致催化效率低等缺点. 氨基吡啶结构作为一类新型配体, 由其所形成的金属催化剂具有对映选择性优异、催化周转数更高、条件温和、大部分使用绿色氧化剂且底物适用范围广等优点. 上述催化方法极大地丰富了不对称C—H键直接羟基化反应研究领域, 为手性醇、生物活性分子的高效合成提供了强大的技术支持.
手性醇物质是一类重要的合成砌块, 在临床药物、农药、材料以及其他精细化学品的合成中具有重要的应用. 通过C—H键的直接不对称羟基化反应合成手性醇的方法具有高原子经济性和步骤经济性, 但该合成方法学仍面临着诸多挑战: (1)复杂底物的区域选择性与立体选择性调控: 在含共轭双键、多羰基或烯酮的底物中, 如何精准控制羟基化的位点(如区分双键与羰基的还原顺序)仍具挑战. 当反应位点与前手性中心距离较远时 (如自由基反应中的远程立体控制), 如何通过催化剂设计实现高效对映选择性仍不明确. (2)催化体系的设计与优化: 现有高效体系多依赖贵金属(如Ru、Rh), 而基于地球丰产金属(如Mn、Cu)的催化剂在活性和选择性上仍有差距. 多组分催化体系中, 各组分的作用机制(如氢键网络、π-π相互作用) 尚未完全明晰, 影响进一步优化. (3)反应机理的深入解析: 许多反应涉及高活性中间体(如金属卡宾、自由基物种), 其生成与转化路径难以通过实验直接观测, 需依赖理论计算(如DFT)辅助推测等. 因此, 生物及仿生催化C—H键的不对称羟基化反应未来仍有广阔的研究前景, 例如在仿生催化脂肪族非活化C—H键选择氧化的催化效率和对映选择性方面. 可以采用先进的技术手段监测, 根据监测结果, 及时调整反应条件, 实现反应的精准控制; 还可以运用量子化学计算、分子模拟等方法, 对催化剂的结构、反应机理和对映选择性进行深入研究. 通过计算预测不同结构和反应条件下的催化性能, 为实验设计提供理论指导, 加速催化剂的优化和反应条件的筛选. C—H键不对称羟基化反应在药物合成、天然产物合成和绿色化学等领域具有广泛应用前景. 随着催化剂、机理研究和多功能反应的深入, 该领域将持续发展, 为有机合成提供更多高效、环保的解决方案.
(Lu, Y.)