


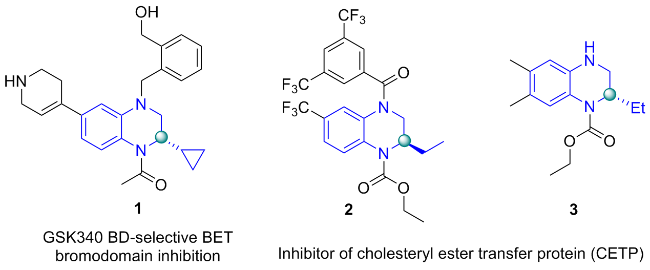
近几十年来, 手性N-杂芳烃化合物作为许多生物活性分子的核心结构受到了广泛地关注[1-4], 通常, 手性四氢喹喔啉(THQ)片段是胆固醇酯转移蛋白(CETP)抑制剂和BD-2选择性BET抑制剂的关键结构单位, 它们在治疗动脉粥样硬化和肥胖中起着关键作用或在体内表现出抗炎活性(图1)[5-7]. 不对称催化主要包括手性有机催化、手性过渡金属催化和酶催化三大体系, 是高效获取对映体纯分子的重要策略[8-12]. 其中, 不对称氢化因其出色的原子经济性和无副产物等优点, 成为合成手性四氢喹喔啉最直接高效的方法之一[13-15].
近年来, 过渡金属催化的不对称氢化技术已成为构建手性杂环化合物的重要策略. 2009年, de Vries等[16]报道了一种高对映选择性的铱催化不对称氢化方法, 采用双膦氧配体(R)-H8-binapo和单齿膦酰胺配体(S)-PipPhos实现了烷基取代喹喔啉的高效对映选择性氢化(Scheme 1A). 随后, Liu等[17]发展的新型锰催化体系实现了立体多向不对称氢化, 用于二取代喹喔啉的氢化反应, 能够以高产率和良好的立体选择性获得顺式(cis)或反式(trans)手性四氢喹喔啉(THQs) (Scheme 1B). 2023年, 张俊良和刘媛媛等[18]开发了一类具有碳手性中心、构型可翻转的氮手性中心的新型氮膦配体Rong-Phos. 通过调控催化剂非对映异构体的构型翻转, 实现了α,β-不饱和内酰胺的立体发散性氢化. 除手性催化剂外, 溶剂效应在立体控制中的独特作用逐渐引起关注. 张绪穆和董秀琴等[19]报道了一种Ir/N-Me-ZhaoPhos催化体系, 在Brønsted酸活化下, 通过溶剂极性调控成功实现了四氢喹喔啉对映异构体的选择性制备, 然而其溶剂作用的催化机制有待深入的研究. 尽管催化体系不断革新, 越来越多的对映催化体系已相继被报道, 但现有研究对立体选择性的调控机制仍缺乏深入的理论阐释[20-21]. 结合上述喹喔啉对映选择性氢化的研究, 我们针对2-芳基喹喔啉氢化体系进行优化设计. 通过采用溶剂控制的对映选择性氢化方案(辅助材料, 表S1和表S2), 以邻苯二胺和2-溴苯乙酮衍生物为起始原料, 在无添加剂条件下通过溶剂切换即可高效制备了两种对映异构体. 实验表明, 在甲苯/二氧六环溶剂中可获得R-构型产物(产率93%, ee值98%); 而在乙醇溶剂中则生成S-构型产物(产率83%, ee值93%). 为阐明溶剂效应的调控机制, 本文使用密度泛函理论(DFT)计算对反应机理进行了全面探索, 在甲苯/二氧六环混合溶剂与乙醇溶剂中建立了不同的反应路径模型(Scheme 1C), 为溶剂调控的立体选择性机制提供了理论依据[22].
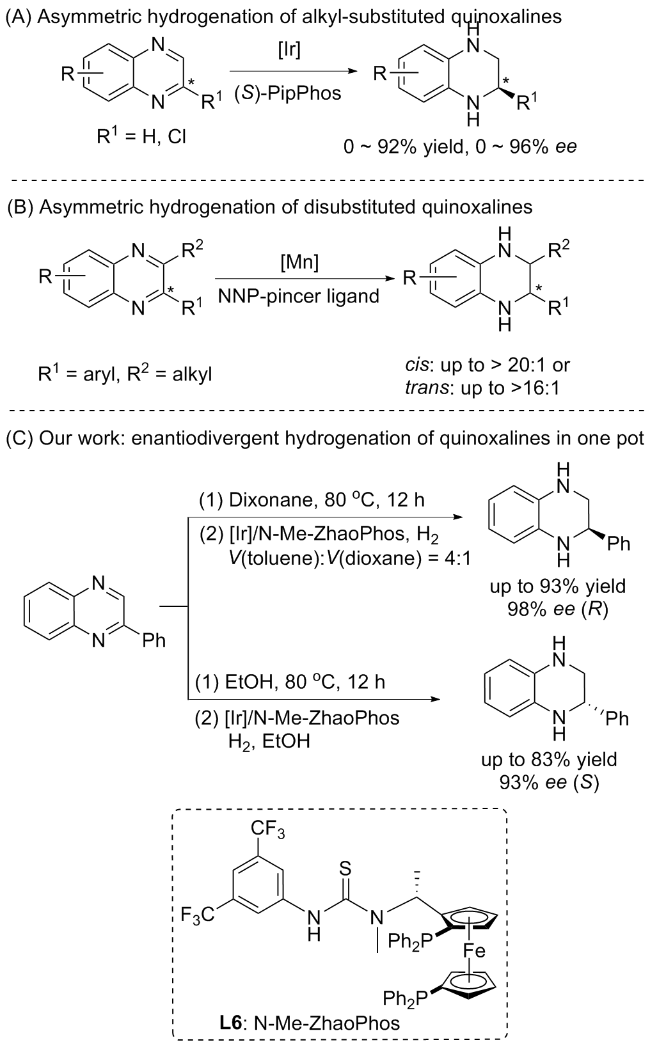
图式1 不对称催化体系: (A)不对称氢化烷基取代喹喔啉; (B)不对称氢化二取代喹喔啉; (C)溶剂控制的喹喔啉对映氢化Scheme 1 Asymmetric catalytic systems: (A) Asymmetric hydrogenation of alkyl-substituted quinoxalines; (B) Asymmetric hydrogenation of disubstituted quinoxalines; (C) Solvent-contro- lled enantiodivergent hydrogenation of quinoxaline |
1 结果与讨论
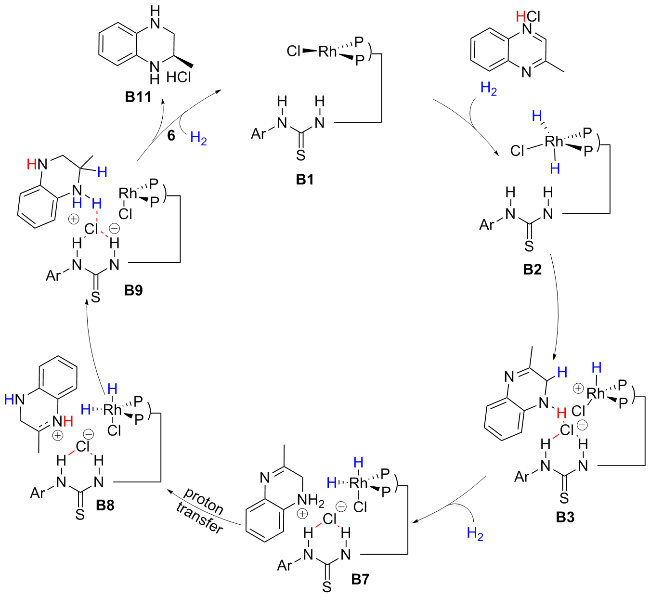
在前期的工作中, 我们[23]对喹喔啉和喹喔啉酮底物的不对称氢化反应进行了研究, 提出了一条可能的催化循环途径(Scheme 2), 进一步结合DFT计算揭示了该转化过程为经典的外球转化机制. 2-甲基喹喔啉被盐酸(HCl)质子化形成催化剂-底物复合物. 同时, 氯离子与催化剂的硫脲和质子化的2-甲基喹喔啉上的NH基团形成氢键. 然后将活性铑络合物的氢化物转移到底物上. 第一次氢化生成部分还原中间体B3. 随后, 另一个H2分子与催化剂配位, 氯离子促进二氢的异质裂解, 生成活性二氢化物. 经过质子转移过程以产生更稳定的中间体B8. 接着, 活性二氢化物通过底物与配体之间的阴离子结合对底物进行第二次识别, 氢再次转移到底物的NH上, 同时. 另一个氢转移至质子化喹喔啉的C2位, 完成氢化, 生成R构型产物. 最后HCl离去得到催化剂, 完成催化循环.
本研究参考之前提出的反应机理, 对2-苯基喹喔啉对映选择性氢化机制进行了研究. 由氘代实验(辅助材料, Scheme S2)可知, 在甲苯/二氧六环催化体系中, 氢化产物中的氢原子主要来自氢气, 而在乙醇催化体系中氢原子主要来源于乙醇. 再根据铱催化2-苯基喹喔啉不对称氢化的转化途径, 进行了相关的对照实验(辅助材料, Scheme S3). 实验结果表明, 半还原中间体Ⅳ-5a在两种催化体系(辅助材料, Scheme S1)下均顺利被氢化, 产物的ee值分别为90%和87%. 此反应结果与直接使用2-苯基喹喔啉的氢化结果一致, 说明铱催化的喹喔啉不对称氢化反应可能是一个分步氢化的过程, 涉及3,4-和1,2-氢化加成反应, 从而产生中间产物Ⅳ-5a. 为了进一步确定关键中间体Ⅳ-5a的存在, 本研究还比较了2-苯基喹喔啉中两种不同C=N双键的氢化反应活性(辅助材料, 图S1). 进一步说明在2-取代喹喔啉底物的不对称氢化反应中, 不含取代基的C=N双键比含有取代基的C=N双键更容易被氢化, 进而产生半还原中间体Ⅳ-5a. 为了研究两种对映异构体产物的动力学特性, 本研究分别对底物在甲苯/二氧六环和乙醇催化体系中的不对称氢化反应过程进行了监测(辅助材料, 图S2、表S3、表S4). 实验结果表明在甲苯/二氧六环催化体系中, 反应在前10 h内进行得较为缓慢, 并伴随着少量中间体Ⅳ-5a的产生, 在12 h后反应进行得较为迅速, 此时已监测不到中间体Ⅳ-5a的含量(辅助材料, 图S2, 表S3). 中间体Ⅳ-5a的存在进一步证实了该催化体系为分步氢化过程, 而非取代的C=N双键更容易被氢化的猜想. 相比之下, 在乙醇体系中底物的转化率要明显快于甲苯/二氧六环体系, 在11 h内就实现了完全转化(辅助材料, 图S2, 表S4). 但是, 在反应过程中几乎没有观察到中间体Ⅳ-5a的形成, 这可能是由于中间体Ⅳ-5a在乙醇体系中的氢化反应速度较快, 从而无法监察, 该结果进一步表明乙醇溶剂能够激活底物, 从而加速氢化反应的进程.
1.1 在甲苯/二氧六环混合溶剂中选择性生成R构型的手性喹喔啉的反应机理
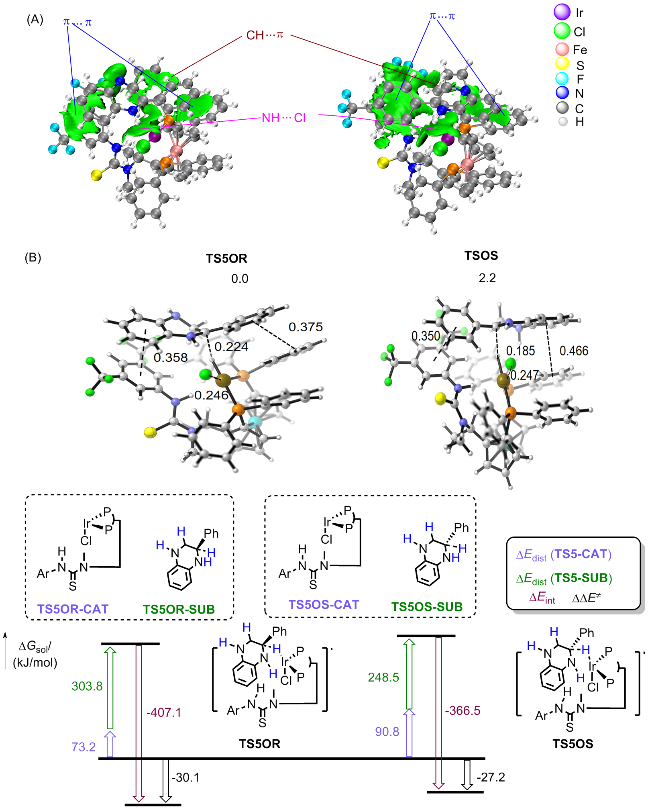
根据机理实验, 本文对2-苯基喹喔啉不对称氢化过程提出了机理猜想, 进一步结合DFT计算验证了该机理的合理性. 2-苯基喹喔啉在甲苯/二氧六环反应体系中的势能面计算结果如图2. 首先, 催化剂A-1物理吸附一分子氢气后得到中间体A-TS2, 再通过无势垒过渡态A-TS1与氢气发生氧化加成, 生成稳定的Ir(III)中间体A-3 (-23.8 kJ•mol-1), 此中间体作为后续氢转移步骤中活性氢的主要来源. 通过对照实验(辅助材料, Scheme S3)表明杂原子N4在第一步氢转移中发挥了重要的作用. 结合两个C=N双键不同的反应活性, 确定了第一个氢转移可能发生的四个活性位点N1、C2、C3和N4, 在其相应的过渡态中底物通过N4—Ir键与催化剂配位. 结果表明, 第一个氢通过过渡态A-TS2-C3转移到C3位最为有利, 能垒最低为46.0 kJ/mol. 随后, 生成的中间体A-4通过A-TS3向N4位进行第二个氢转移, 其活化自由能为67.4 kJ/mol, 从而形成中间体A-5. 接下来, 通过复合物A-5的快速解离得到半还原中间体Ⅳ-5a. 计算结果与对照实验(辅助材料, Scheme S3)和动力学实验结论一致(辅助材料, 图S2), 即非取代的 C=N双键氢化活性更高, 中间体Ⅳ-5a为该反应的关键中间体. 随后, 另一分子氢气与催化剂A-6配位, 第二次氢化反应启动. 如图4, 第三个氢转移涉及三条相互竞争的途径, 最有利的途径是通过过渡态A-TS4生成中间体A-8, 其自由能为64.0 kJ/mol, 明显低于竞争过渡态A-TS4-1和A-TS4-2. 然后, 通过N1和N4之间的质子转移生成较为稳定的中间体A-9. 通过对照实验表明, 在第二步氢化过程中不需要N4原子参与辅助配位, 可以直接识别底物中不饱和C=N双键. 氢化物通过A-TS5OR或A-TS5OS从底物的Re或Si面向C2位转移, 该过程为对映选择性决定步. 计算结果表明, A- TS5OR过渡态的能垒为78.7 kJ/mol, 明显低于A- TS5OS过渡态, 该计算结果与实验结果一致, 氢化产物为R-构型.
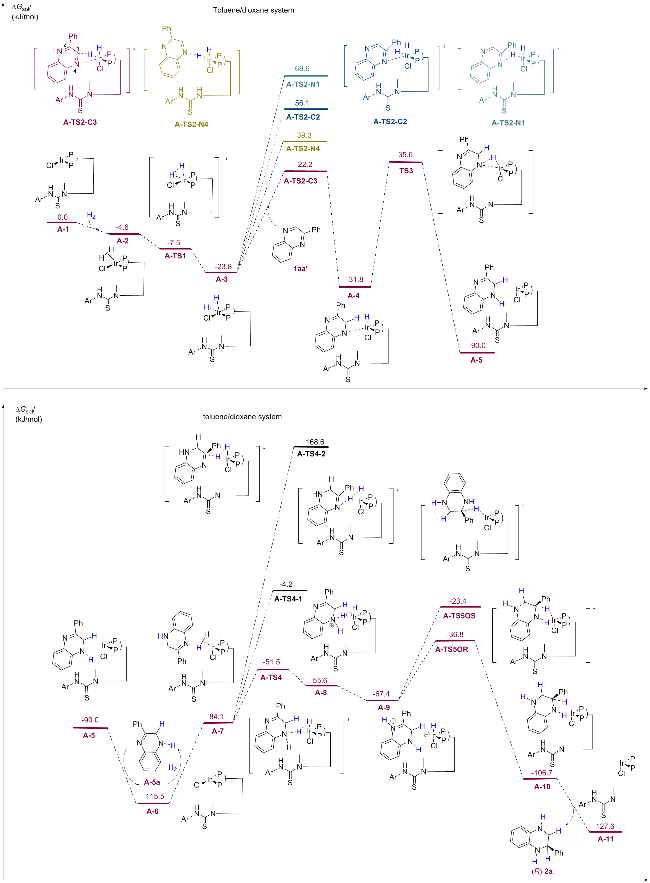
此外, 还对过渡态A-TS5OR、A-TS5OS进行了基于Hirshfeld分区的独立梯度模型(IGMH)分析和distortion-interaction分析(图3). 该复合物分为2-苯基喹喔啉和催化剂片段. 分析表明, 催化剂和底物之间的相互作用能控制对映体选择性. A-TS5OR的相互作用能为30.1 kJ/mol高于A-TS5OS, 这很大一部分归因于底物和配体之间良好的π-π相互作用(A-TS5OR和A-TS5OS中两个苯环之间的距离分别为0.375和0.466 nm). 两过渡态自由能差异即为甲苯/二氧六环烷系统中对映选择性的来源.
1.2 在乙醇溶剂中选择性生成S构型的手性喹喔啉的反应机理
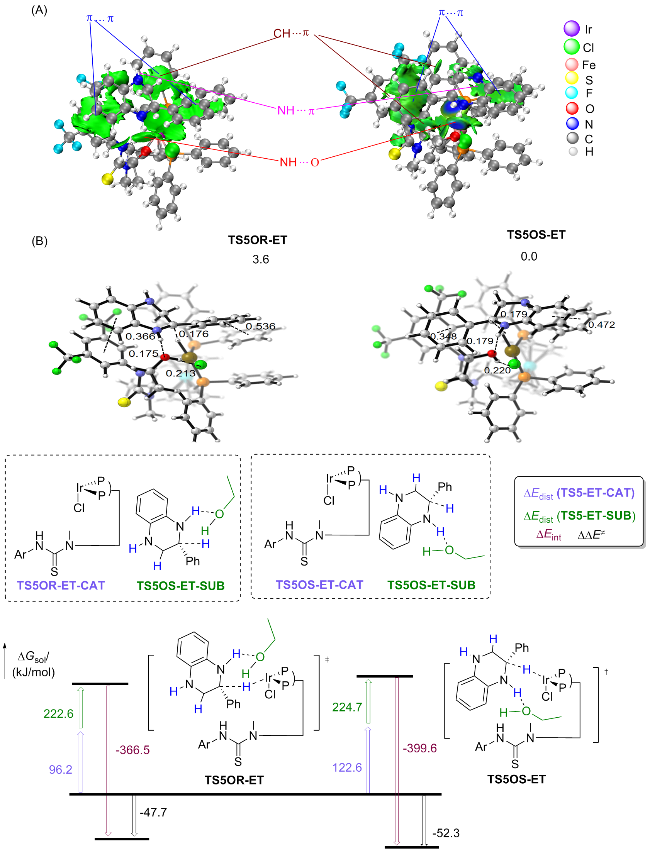
2-苯基喹喔啉在乙醇反应体系中势能面计算结果如图4. 首先, 催化剂C-1经过18.0 kJ/mol过渡态C-TS1与氢气发生氧化加成, 生成稳定的Ir(III)中间体C-3(-33.5 kJ/mol). 在第一次氢转移过程中, 通过两个C=N双键不同的反应活性结合对照实验(辅助材料, Scheme S3)确定第一次氢转移发生的两个活性位点N1、C2, 金属Ir配位的负氢与乙醇分子羟基中的活泼氢同时参与反应, 通过协同转移的方式经历六元环过渡态(C-TS2, 能量为24.7 kJ/mol)分别转移到底物分子的N1和C2位点上, 生成中间体C-4. 然后, 为保证催化剂Ir的六配位结构另一分子氢气再通过σ电子与Ir空轨道配位形成中间体C-6(能量为19.7 kJ/mol). 催化剂上活化的氢原子首先从Ir转移至底物分子的N1位点, 并促进N1上已有的一个氢原子向乙醇氧发生转移. 这一过程通过六元环过渡态C-TS3(能量为41.8 kJ/mol)实现, 并释放大量自由能生成稳定的中间体C-7(能量为-47.7 kJ/mol). 随后, Ir中心上的另一个氢原子继续转移至N1位点, 伴随着乙醇分子中OH基团的氢与N1形成氢键, 最终生成产物中间体C-8(能量为4.6 kJ/mol). 此后, 底物中N1位上的氢通过质子转移过程转移至 N4 位点, 进一步形成产物C-9(能量为10.0 kJ/mol). 最后, Ir配位中心的最后一个氢原子转移到底物的C3位点, 完成最终的产物生成. 氢化物通过C-TS5OR-ET或C- TS5OS-ET从底物的Re或Si面向C2位转移, 该过程为对映选择性决定步. 计算结果表明, C-TS5OS-ET过渡态的能垒为93.7 kJ/mol, 明显低于C-TS5OR-ET过渡态, 该计算结果与实验结果一致, 氢化产物为S-构型. 我们同样对过渡态C-TS5OR-ET、C-TS5OS-ET进行了IGMH分析和distortion-interaction分析(图5). 图中复合物分为2-苯基喹喔啉和催化剂片段. 分析表明, C-TS5OR-ET中四氢喹喔啉底物和催化剂的相互作用能为52.3 kJ/mol, 高于A-TS5OS, 这很大一部分归因于C-TS5OR-ET内底物和配体之间较强的π-π相互作用(A-TS5OR和A-TS5OS中两个苯环之间的距离分别为0.366和0.348 nm). 两个过渡态自由能差异便是乙醇系统中对映选择性的来源. 整个反应过程体现了Ir催化剂的高效活化作用、底物和催化剂的相互作用, 以及乙醇溶剂通过氢键对过渡态的稳定作用, 最终实现对映选择性的不对称氢化. 手性Ir催化剂在氢气分子活化与选择性氢转移过程中起到了核心作用, 并通过多步协同反应实现底物的高效转化.
2 结论
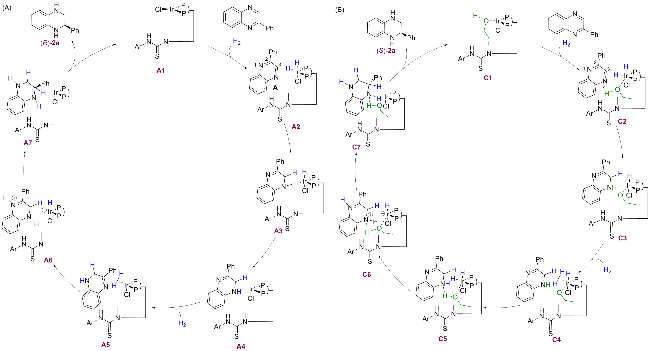
通过一系列计算研究深入探讨了两种不同溶剂中生成不同构型手性四氢喹喔啉的反应机理. 在甲苯/二氧六环体系中, 手性四氢喹喔啉中的氢主要来源于实验中通入的氢气(H₂), Ir(I)催化剂A1与H2作用后形成Ir-H物种, 实现氢气的活化. 随后, 底物配位到铱中心, 并在Ir-H作用下发生逐步氢转移. 第一步氢转移到C=N上生成中间体A4, 继续与第二分子氢气反应生成中间体A6. 最后, 产物2-四氢喹喔啉(R)-2a离去, 催化剂经历配位解离或重排后回到初始状态, 完成催化循环. 底物与催化剂之间的相互作用共同决定了氢化过程的选择性, 确保产物具有高对映选择性, 同时催化剂可循环使用(Scheme 3A). 相比之下, 在乙醇体系中, 手性四氢喹喔啉中的氢则来源于乙醇溶剂中羟基上的活性氢和实验中通入的氢气. 乙醇与Ir(I)催化剂配位形成复合物C2, 直接参与氢转移生成中间体C3. 随后, 第二分子氢气结合到金属中心, Ir-H上的氢转移到N上, 同时NH上的氢转移到CH3O-上生成乙醇分子, 得到中间体C5. 然后, 再次发生分步氢转移到另一个C=N键上, 生成中间体C7. 最后, 氢化产物逐步释放形成目标化合物(S)-2a, Ir催化剂通过配位交换或氢转移完成再生, 进入新的催化循环(Scheme 3B). 该循环的关键在于乙醇分子和底物通过氢键相互作用, 同时底物和配体之间的π-π相互作用, 二者的协同效应实现了氢化反应立体选择性反转.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
研究结果表明, 手性催化剂、溶剂及底物之间的非共价相互作用(包括π-π相互作用和氢键相互作用)对控制反应的对映选择性至关重要. 机理研究进一步揭示了溶剂在反应中的关键作用, 为开发其它溶剂控制的对映催化体系提供了重要的理论指导和参考价值.
3 计算方法
所有量子化学计算均在中国国家超级计算深圳中心使用Gaussian 16软件包进行, 采用密度泛函理论(DFT)方法. 几何结构优化使用了B3LYP-D3(BJ)泛函[24-25], Ir和Fe的基组采用SDD基组[26], 其他元素则使用6-31g*基组[24,27]. 然后, 在优化的几何结构上进行了频率计算(在298.15 K下), 以确认局部极小值(无虚频)和过渡态(有一个虚频)[28]. 为了获得更精确的能量数据, 在B3LYP-D3(BJ)/def2-tzvp理论级别下计算了单点溶剂化能, 采用SMD溶剂化模型, 混合溶剂[solvent=generic, ε=2.34, ε∞=2.05, 溶剂混合物为V(甲苯)∶V(二氧六环)=4∶1]用于混合体系, 乙醇作为溶剂则在EtOH体系中使用SMD溶剂化模型. 优化后的过渡态的三维结构通过CYLView软件[29]进行可视化. IGMH分析使用Multiwfn 3.3.9程序[30]进行, IGMH图的可视化通过VMD程序[31]生成.
辅助材料(Supporting Information) 实验部分、能量和笛卡尔坐标. 这些材料可以免费从本刊网站(http://sioc- journal.cn/)上下载.
(Cheng, F.)