


1 Introduction
Dydrogesterone is a semi-synthetic progesterone receptor agonist used to treat various gynecological disorders associated with luteinizing hormone deficiency, including infertility,[1] menstrual disorders,[2] threatened miscarriage,[3] endometriosis[4] and dysmenorrhoea.[5] Preclinical studies indicated minimal teratogenic, mutagenic, and carcinogenic risks.[6] Since its market launch, global demand has continuously rising, surpassing $500 million in worldwide sales in 2023.[7]
Structurally, unlike the natural progestin progesterone, dydrogesterone owns a 9β-hydrogen, 10α-methyl, and an additional Δ6,7 double bond (Figure 1). These features enhance oral bioavailability and metabolic stability, but make its preparation from conventional steroidal raw materials challenging.[8] As a result, the synthesis of dydrogesterone has been a hot topic in organic chemistry and pharmaceutical industry, with numerous promising strategies reported.[9]
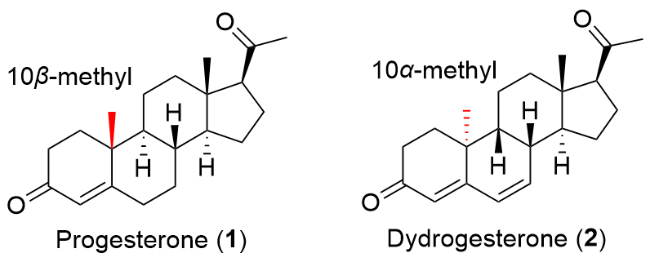
Currently, the industrial production of dydrogesterone relies on progesterone 1 as the raw material, undergoing multiple reactions, including two key photochemical steps, to construct the 10α-methyl skeleton 4.[9] However, this method is costly, energy-intensive, and low yielding, which does not meet the global demand. In 2023, a dearomatization strategy was reported using Hajos-Parrish ketone (5) as the starting material to construct the 10α-methyl tetracyclic skeleton, followed by conventional multi-step reactions to obtain dydrogesterone 2[7] (Scheme 1, b, Tangs’ work). Inspired by this work, we would like to develop an effective and mild synthetic route to gain intermediate 20 from easily available starting materials.
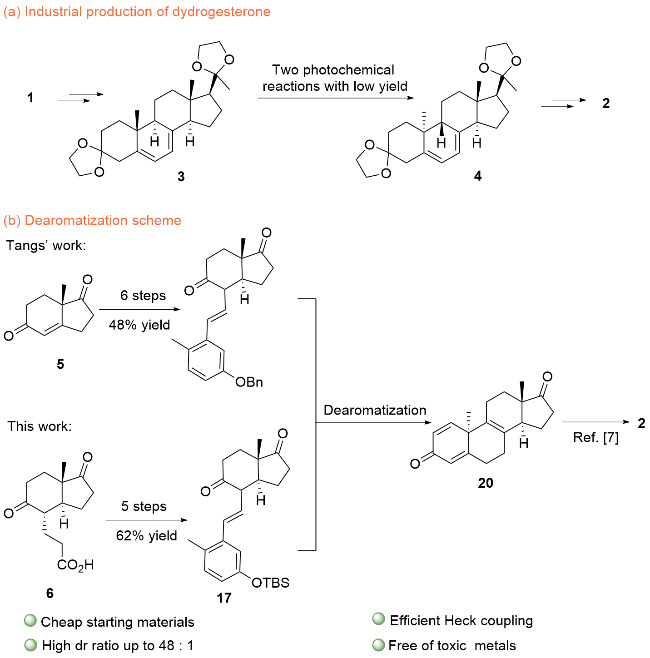
Based on our group’s extensive experience in this field,[10] as well as the availability of a by-product form biodegradation process of sterols,[11] we developed an alternative dearomatization strategy for dydrogesterone’s intermediate. The advantage of this strategy is the use of cheap and commercially available dicetolic acid (6) as a starting material.[12] What’s more, it significantly reduces the use of hazardous chemicals and avoids harsh reaction conditions (Scheme 1, b).
2 Results and discussion
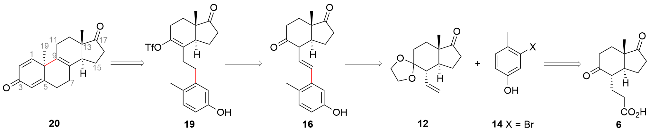
Our retrosynthetic analysis is shown in Scheme 2. Compound 20 was regarded as key intermediate to achieve the formal synthesis of dydrogesterone 2. it can be obtained by dearomatization of triflate 19,[7] which can be accessed by organic transformation from phenol 16. It was proposed that phenol 16 can be synthesized via a Heck coupling reaction between 3-bromo-4-methylphenol (14) and olefin 12, which can be derived from the cost-effective and readily available dicetolic acid (6).
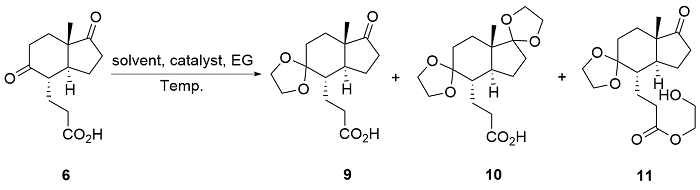
Initially, the direct decarboxylation of 6 was attempted. However, due to the presence of the carbonyl group, the more stable conjugated by-products 7 and 8 were generated, which is similar to the conclusion of Rigby et al.[13] (Scheme 3). To avoid this, the carbonyl groups should be protected with ethylene glycol before proceeding with this decarboxylation. However, when employing triditional p-toluenesulfonic acid as a catalyst,[14] we observed a significant amount of esterification by-product 11 formed probably due to the use of the strong acidic catalyst (Table 1, Entries 1, 2). Moreover, this classical approach requires large amount of glycol and acid, which does not adhere to the principles of green chemistry.[15] In contrast, the approach proposed by Gui et al.,[16] which uses oxalic acid as a catalyst with lower acidity, can yield satisfactory results while preventing the formation of by-product 11. But it still requires a large amount of ethylene glycol (Table 1, Entries 3~5). Based on the previous work in our group,[17] the reaction conditions were further optimized and found the addition of 3.0 equiv. of ethylene glycol and 3 mol% polyvinylpyrrolidone iodine (PVP-I) were enough to obtain the precursors 9 and 10 in yields of 68.3% and 23.4%, respectively, without the formation of by-product 11 (Table 1, Entry 7).
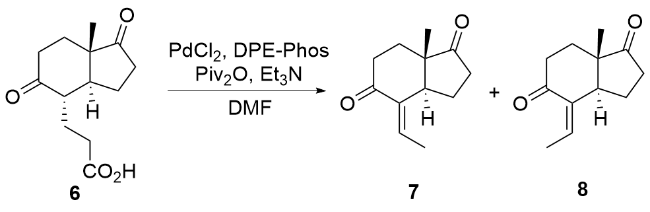
Table 1 Optimization of the carbonyl protectiona |
| Entry | Solvent | Catalyst (Dosage/mol%) | EG/equiv. | T/℃ | Yieldb/% | ||
|---|---|---|---|---|---|---|---|
| 9 | 10 | 11 | |||||
| 1 | — | PTSAc (3) | 60 | r.t. | 35.1 | 29.9 | 24.3 |
| 2 | CH3CN | PTSAc (3) | 3 | r.t. | 37.4 | 19.2 | 32.2 |
| 3 | CH3CN | Oxalic acid (50) | 2 | 80 | 11.4 | 6.7 | N.D. |
| 4 | CH3CN | Oxalic acid (50) | 8 | 80 | 33.2 | 19.3 | N.D. |
| 5 | CH3CN | Oxalic acid (50) | 25 | 80 | 59.6 | 23.5. | Trace |
| 6 | CH3CN | PVP-I (3) | 2 | 60 | 9.8 | N.D. | N.D. |
| 7 | CH3CN | PVP-I (3) | 3 | 60 | 68.3 | 23.4 | N.D. |
| 8 | CH3CN | PVP-I (3) | 8 | 60 | 42.6 | 29.7 | Trace |
| 9 | CH3CN | PVP-I (3) | 30 | 60 | 26.5 | 42.3. | 54.2 |
| 10 | — | PVP-I (3) | 60 | 60 | Trace | N.D. | N.D. |
| 11 | EtOAc | PVP-I (3) | 3 | 60 | 15.8 | Trace | N.D. |
| 12 | THF | PVP-I (3) | 3 | 60 | 25.7 | Trace | N.D. |
| 13 | CH3CN | PVP-I (3) | 3 | r.t. | 39.5 | 9.5 | N.D. |
| 14 | CH3CN | PVP-I (3) | 3 | 80 | 45.3 | 21.3 | 29.4 |
a Unless otherwise specified, the reactions were all carried out in the presence of compound 6 (1.0 mmol), catalyst, ethylene glycol (EG), and solvent (3.0 mL); b GC-MS yield; c PTSA: p-Toluenesulfonic acid. |
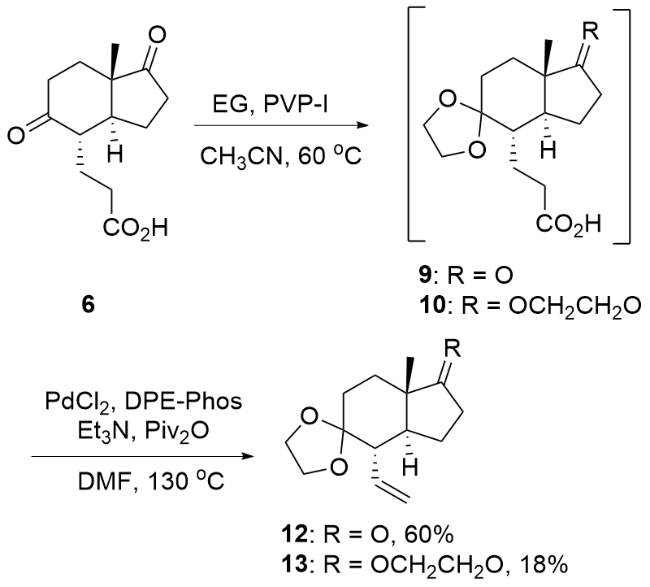
It is not necessary to purify compounds 9 and 10. A mixture of these two crude products can be directly applied to the next decarboxylation after removing the excess ethylene glycol by extraction at the end of the reaction. Next, decarboxylation condition with 1.0 mol% palladium chloride, 3.0 mol% bis(2-diphenylphosphophenyl) ether, 6.0 mol% triethylamine, and 1.5 equiv. of pivalic anhydride in N,N-dimethylacetamide (DMF) was applied to the synthesis of the crude product. By combining these two steps, we obtained compounds 12 and 13 in 60% and 18% yields, respectively (Scheme 4).
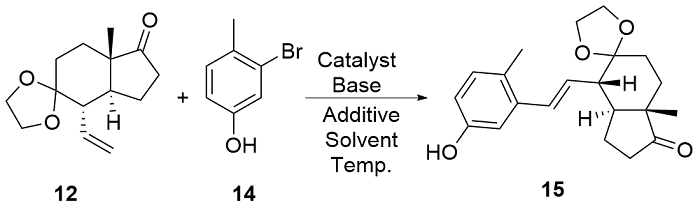
With compounds 12 and 13 in hand, our attention was turned to their arylation, which is a crucial step for providing the precursor necessary for the installation of the A/B rings. The Heck coupling reaction condition was optimized to use compound 12 with compound 14. Initially, the reaction was conducted with Pd(OAc)₂ and K₃PO₄ at 140 ℃ in DMF, giving product 15 in 52% yield (Table 2, Entry 1). After then, other palladium catalysts were screened, such as PdCl₂, PdCl₂(PPh₃), Pd(PPh₃)₄, and Pd/C, in the combination with various bases, including K₂CO₃, NaHCO₃, and Na₂CO₃. Notably, the reaction yield increased to 61% when using Pd(OAc)₂ and Na₂CO₃ (Table 2, Entries 2~9). Furthermore, the addition of phosphorus-based ligands significantly improved the reaction efficiency. When tri(o-tolyl)- phosphine was used as a ligand, the yield increased to 85%, with complete consumption of the starting material and shorter reaction time (Table 2, Entry 11).
Table 2 Optimization of the Heck couplinga |
| Entry | Catalyst | Base | Additive | T/℃ | Yieldb/% |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | K3PO4 | — | 140 | 52 |
| 2 | PdCl2 | K3PO4 | — | 140 | 49 |
| 3 | PdCl2(PPh3)2 | K3PO4 | — | 140 | 43 |
| 4 | Pd(PPh3)4 | K3PO4 | — | 140 | 25 |
| 5 | Pd/C | K3PO4 | — | 140 | 29 |
| 7 | Pd(OAc)2 | K2CO3 | — | 140 | 46 |
| 8 | Pd(OAc)2 | NaHCO3 | — | 140 | 43 |
| 9 | Pd(OAc)2 | Na2CO3 | — | 140 | 61 |
| 10 | Pd(OAc)2 | Na2CO3 | PPh3 | 140 | 69 |
| 11d | Pd(OAc)2 | Na2CO3 | P(o-Tol)3c | 140 | 85 |
| 12d | Pd(OAc)2 | Na2CO3 | P(o-Tol)3 | 150 | 78 |
| 13d | Pd(OAc)2 | Na2CO3 | P(o-Tol)3 | 100 | 67 |
a Unless otherwise stated, reactions were carried out at 140 ℃ for 18 h at 12 (0.5 mmol), 14 (0.5 mmol), catalyst (2.0 mol%), additive (6.0 mol%), base (1.5 mmol), and DMF (5.0 mL); b HPLC yields; c P(o-Tol)3: tri(o-tolyl)phosphine. d Reaction time: 10 h. |
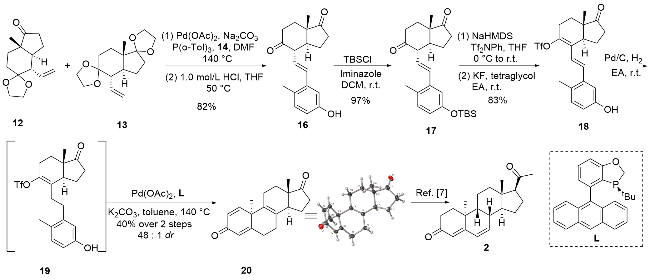
Then, the optimized reaction condition was applied to the mixture of 12 and 13 obtained in the previous steps. Upon the completion of Heck coupling, the residue was treated with 1.0 mol/L HCl to give the deprotected product 16 in 82% isolated yield over two steps. Next, the phenolic hydroxyl group was protected using t-butyldimethylsilyl chloride (TBSCl), giving compound 17 with the yield of 97%. Triflate formation of 17 under sodium bis(trimethyl- silyl) amide/N-phenyl bis(trifluoromethanesulfonyl) imide (NaHMDS/Tf2NPh) afforded compound 18 in 83% yield. Hydrogenation of compound 18 over Pd/C under H₂ atmosphere smoothly afforded the reduction product 19. Since triflate 19 is unstable, we opt not to purify it and proceed directly to the next step. In the final cyclization stage, we observed that using the combination of Pd(OAc)₂, (S)-4-(anthracen-9yl)-3-(tert-butyl)-2,3-dihydrobenzo[d][1,3]-oxaphosphole (L), and K₂CO₃ resulted in compound 20 with improved stereoselectivity (dr=48:1) but with a lower yield (40%) compared to the condition used by Tang et al.[7] (Scheme 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In summary, starting from dicetolic acid (6), cyclized precursor 17 was synthesized over five steps in 62% yield. The core intermediate 20 of dydrogesterone was obtained in 40% yield with an excellent diastereomeric ratio of 48:1. The key advantages of our route include green and efficient carbonyl protection, significantly reducing waste generation, and efficient A- and CD-ring assembly via decarboxylation and Heck coupling, minimizing hazardous reagents and harsh conditions. In the current method, although key intermediate 20 was obtained with a high diastereomeric ratio, the final cyclization yield remains suboptimal, and further investigations are ongoing in our laboratory.
4 Experimental section
4.1 General experimental information
Unless otherwise specified, all reagents and solvents were obtained from commercial suppliers and used without further purification. Solvent was purified according to the procedure from “Purification of Laboratory Chemicals”. Reactions that require heating were performed using an oil bath. The NMR spectra were recorded on Bruker Avance 400, 500 or 600 spectrometers at 400, 500 or 600 MHz in CDCl3 using tetramethylsilane as the internal standard. 1H NMR spectra were calibrated using residual undeuterated solvent as an internal reference (CDCl3 δ 7.26) and 13C NMR spectra were calibrated against the deuterated solvent peak (CDCl3 δ 77.2). Melting points were determined using an OptiMelt capillary melting point apparatus. High resolution mass spectra (HRMS) were acquired in the ESI mode using a Waters TOF mass analyzer. HPLC spectra were acquired in a SHIMADZU LC-20AT with a CHIRALPAK OD-3 column or a Shim-park GIST 3 μm C18 column. Column chromatography was performed on silica gel (300~400 mesh).
4.2 Synthesis of olefins 7 and 8
To a dried 100 mL of round-bottomed flask equipped with a stirrer and a condensation reflux tube were added compound 6 (1.2 g, 5.0 mmol), PdCl2 (18 mg, 2.0 mol%), bis(2-diphenylphosphinophenyl)ether (DPE-Phos) (162.0 mg, 6.0 mol%), Piv2O (2.1 mL, 10.5 mmol, 2.1 equiv.) and trifluoroacetic acid (TEA) (0.1 mL, 0.6 mmol, 12 mol%). The flask was then evacuated and backfilled with N2 for 3 times. Anhydrous DMF (20.0 mL) was added and the resulting solution was stirred in a preheated 130 ℃ oil bath for 2 h, as determined by thin-layer chromatography (TLC). After the reaction mixture was cooled to room temperature, the solid particle was removed by diatomite filtration and the filter cake was washed with ethyl acetate (20.0 mL×3). The organic phase was combined, washed with brine (20 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/ EtOAc, V:V=10:1) to provide 7 (336 mg, 35%) and 8 (346 mg 36%) as colorless oil.
(3aR,7aS,E)-4-Ethylidene-7a-methylhexahydro-1H-indene-1,5(4H)-dione) (7):[13] 1H NMR (500 MHz, CDCl3) δ: 5.76 (qd, J=7.3, 2.4 Hz, 1H), 2.75~2.71 (m, 1H), 2.63~2.45 (m, 3H), 2.36~2.29 (m, 1H), 2.09~1.96 (m, 5H), 1.87~1.76 (m, 2H), 0.93 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 217.8, 201.1, 136.0, 132.3, 48.9, 46.8, 36.6, 35.3, 28.0, 19.7, 14.3, 12.8.
(3aR,7aS,Z)-4-Ethylidene-7a-methylhexahydro-1H-inde- ne-1,5(4H)-dione (8):[13] 1H NMR (500 MHz, CDCl3) δ: 6.85 (qd, J=7.6, 2.7 Hz, 1H), 2.97~2.92 (m, 1H), 2.63~2.46 (m, 4H), 2.33~2.16 (m, 2H), 2.01~1.92 (m, 4H), 1.85~1.78 (m, 1H), 0.96 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 217.5, 199.8, 135.8, 134.9, 47.1, 46.9, 46.6, 35.4, 33.7, 27.2, 22.6, 13.7, 13.0.
4.3 Synthesis of 2-hydroxyethyl 3-((3aS,4S,7aS)-7a-methyl-1-oxooctahydrospiro[indene-5,2'-[1,3]dioxo-lan]-4-yl)propanoate (11)
To a dried 50 mL of round-bottomed flask equipped with a stirrer and a condensation reflux tube were added compound 6 (238 mg, 1.0 mmol), PTSA (5 mg, 1.0 mmol, 0.03 equiv.), and ethylene glycol (186 mg, 3.0 mmol, 3.0 equiv.). The flask was then evacuated and backfilled with N2 for 3 times. Acetonitrile (3 mL) was added and the resulting solution was stirred at room temperature for 5 h (as determined by TLC). After that, the solvent was evaporated and the resulting mixture was suspended in water (20 mL) and extracted with ethyl acetate (10 mL×3). The combined organic layer was washed with brine (20 mL) and dried over anhydrous Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/EtOAc, V: V=5:1) to provide 11 as colorless oil (98 mg, 29%). 1H NMR (500 MHz, CDCl3) δ: 5.31 (s, 1H), 4.31~4.27 (m, 1H), 4.18~4.14 (m, 1H), 4.02~3.96 (m, 4H), 3.83~3.81 (m, 2H), 2.57~2.41 (m, 3H), 2.17~2.10 (m, 1H), 2.03~1.98 (m, 1H), 1.95~1.84 (m, 3H), 1.80~1.76 (m, 1H), 1.70~1.56 (m, 1H), 1.47~1.41 (m, 1H), 0.95 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 219.6, 173.9, 111.5, 66.0, 64.7, 64.5, 61.0, 47.7, 47.6, 42.8, 36.0, 33.5, 30.4, 28.2, 22.4, 21.4, 13.0; HRMS (ESI) calcd for C17H27O6 [M+H]+327.1802, found 327.1805.
4.4 Synthesis of olefins 12 and 13
To a dried 250 mL of round-bottomed flask equipped with a stirrer and a condensation reflux tube were added 6 (8.0 g, 33.5 mmol), PVP-I (2.6 g, 1.0 mmol, 0.03 equiv., based on the content of iodine.), and EG (6.2 g, 100.5 mmol, 3 equiv.). The flask was then evacuated and backfilled with N2 for 3 times. Acetonitrile (150 mL) was added and the resulting solution was stirred in a preheated 60 ℃ oil bath for 5 h (as determined by TLC). After that, the solvent was evaporated, and the resulting mixture was suspended in water (150 mL) and extracted with ethyl acetate (100 mL×3). The combined organic layer was washed with brine (100 mL) and dried over anhydrous Na2SO4. Removal of the solvent under reduced pressure afforded the crude product for next step.
To another dried 250 mL of round-bottomed flask equipped with a stirrer and a condensation reflux tube were added the crude product, PdCl2 (178.7 mg, 3.0 mol%), bis(2-diphenylphosphinophenyl) ether (1.6 g, 9.0 mol%), tertiaryl anhydride (13.1 g, 2.1 equiv.) and triethylamine (408.0 mg, 12.0 mol%). The flask was then evacuated and backfilled with N2 for 3 times. Anhydrous DMF (100.0 mL) was added and the resulting solution was stirred in a preheated 130 ℃ oil bath for 2 h (as determined by TLC). After the reaction mixture was cooled to room temperature, the PdCl2 was removed by diatomite filtration and the filter cake was washed with ethyl acetate (30.0 mL×3). The organic phases were combined and washed with brine (100 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/ EtOAc, V:V=10:1) to provide 12 (4.7 g, 60 %) and 13 (1.7 g, 18%).
(3aS,4S,7aS)-7a-Methyl-4-vinylhexahydrospiro[indene-5,2'-[1,3]dioxolan]-1(4H)-one (12): White solid, m.p. 73.2~74.8 ℃; 1H NMR (500 MHz, CDCl3) δ: 5.72~5.65 (m, 1H), 5.16~5.11 (m, 2H), 3.94~3.87 (m, 4H), 2.48~2.40 (m, 2H), 2.13~2.05 (m, 1H), 1.99~1.93 (m, 1H), 1.81~1.75 (m, 1H), 1.73~1.66 (m, 3H), 1.53~1.42 (m, 2H), 0.94 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 219.6, 134.9, 118.4, 110.7, 65.5, 65.3, 50.8, 47.3, 46.4, 36.0, 31.6, 28.5, 22.3, 13.1; HRMS (ESI) calcd for C14H21O3 [M+H]+ 238.1485, found 238.1488.
(3a'S,4'S,7a'S)-7a'-Methyl-4'-vinylhexahydro-4'H-dispiro-[[1,3]dioxolane-2,1'-indene-5',2''-[1,3]dioxolane] (13): Co- lorless oil. 1H NMR (500 MHz, CDCl3) δ: 5.70~5.63 (m, 1H), 5.10~5.02 (m, 2H), 3.93~3.82 (m, 8H), 2.33~2.29 (m, 1H), 2.13~2.06 (m, 1H), 2.01~1.95 (m, 1H), 1.84~1.77 (m, 2H), 1.69~1.66 (m, 2H), 1.55~1.48 (m, 1H), 1.22~1.15 (m, 1H), 0.94 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 136.0, 118.7, 117.3, 110.9, 65.3, 65.2, 64.6, 51.5, 45.5, 34.2, 31.6, 27.6, 23.0, 13.6; HRMS (ESI) calcd for C16H25O4 [M+H]+ 281.1747, found 281.1755.
4.5 Synthesis of (3aS,4S,7aS)-4-((E)-5-hydroxy-2- methylstyryl)-7a-methylhexahydro-1H-indene-1,5(4H)- dione (16)
To a dried 50 mL of two-necked flask equipped with a stirrer and a condensation reflux tube were added the mixture of 12 and 13 (246 mg, 1.0 mmol), 3-bromo-4-methyl- phenol (14, 187 mg, 1.0 mmol, 1.0 equiv.), anhydrous Na2CO3 (318 mg, 3.0 mmol, 3.0 equiv.), tris(o-toluene)- phosphorus(18 mg, 6.0 mol%) and Pd(OAc)2 (5 mg, 2.0 mol%). The flask was then evacuated and backfilled with N2 for 3 times. DMF (5 mL) was added and the resulting solution was stirred in a preheated 140 ℃ oil bath for 10 h (as determined by TLC). After the reaction mixture was cooled to room temperature, the insoluble materials was removed by diatomite filtration and the filter cake was washed with ethyl acetate (10 mL×3). The organic phases were combined and washed with brine (10 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product for next step.
To another dried 50 mL of round-bottomed flask equipped with a stirrer was added the crude product and tetrahydrofuran (15 mL), and then 1 mol/L hydrochloric acid (5 mL) was added and the resulting solution was stirred in a preheated 50 ℃ oil bath for overnight (as determined by TLC). After the reaction mixture was cooled to room temperature, ethyl acetate (15 mL×3) was added for extraction. The organic phases were combined and washed with brine (15 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/EtOAc, V:V=15:1) to provide 16 (245 mg, 82%). White solid, m.p. 78.2~79.5 ℃; 1H NMR (500 MHz, CDCl3) δ: 6.98~6.96 (m, 2H), 6.65 (dd, J=8.5, 3.0 Hz 1H), 6.55 (d, J=15.5 Hz, 1H), 6.00 (q, J=9.0 Hz, 1H), 5.60 (s, 1H), 3.26 (dd, J=13.0, 9.0 Hz 1H), 2.60~2.53 (m, 3H), 2.28~2.18 (m, 1H), 2.23 (s, 3H), 2.07~1.90 (m, 4H), 1.77~1.69 (m, 2H), 1.23 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 218.1, 210.0, 154.1, 136.9, 131.6, 131.3, 127.2, 125.8, 114.7, 112.5, 55.3, 49.7, 47.3, 37.2, 36.0, 30.2, 22.8, 18.9, 13.5; HRMS (ESI) calcd for C19H23O3 [M+H]+ 299.1642, found 299.1622.
4.6 Synthesis of (3aS,4S,7aS)-4-((E)-5-((tert-butyl- dimethylsilyl)oxy)-2-methylstyryl)-7a-methylhexa-hydro-1H-indene-1,5(4H)-dione (17)
To a dried 50 mL of two-necked flask equipped with a stirrer were added compound 16 (298 mg, 1.0 mmol), and imidazole (140 mg, 2.0 mmol, 2.0 equiv.). The flask was then evacuated and backfilled with N2 for 3 times. Anhydrous DCM (20 mL) was added and the resulting solution was stirred in a 0 ℃ ice bath for 5 min. Then TBSCl (230 mg, 1.5 mmol. 1.5 equiv.) in anhydrous dichloromethane (DCM) (10 mL) was added dropwise into the above reaction solution via a syringe. The reaction solution was stirred at 0 ℃ for 3 min and then brought to room temperature and stirred for 2 h (as determined by TLC). The reaction was quenched with saturated NaHCO3 solution (10 mL), the aqueous phase was extracted with ethyl acetate (10 mL×3) and the combined organic phase was washed with brine (10 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/ EtOAc, V:V=15:1) to provide 17 (400 mg, 97%). Colorless oil. 1H NMR (500 MHz, CDCl3) δ: 6.97 (d, J=8.0 Hz, 1H), 6.95 (d, J=2.5 Hz, 1H), 6.64 (dd, J=8.5, 3.0 Hz, 1H), 6.55 (d, J=16.0 Hz, 1H), 3.25 (dd, J=13.0, Hz, 8.5 Hz, 1H), 2.60~2.53 (m, 3H), 2.27~2.20 (m, 1H), 2.25 (s, 3H), 2.08~2.03 (m, 2H), 1.99~1.93 (m, 1H), 1.78~1.68 (m, 2H), 1.24 (s, 3H), 0.98 (s, 9H), 0.19 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 218.0, 209.4, 154.0, 136.9, 131.9, 131.1, 128.1, 125.8, 119.3, 117.4, 55.4, 49.9, 47.4, 37.3, 36.1, 30.3, 25.9, 23.0, 19.2, 18.3, 13.6, -4.2; HRMS (ESI) calcd for C25H37O3Si [M+H]+ 413.2506, found 413.2512.
4.7 Synthesis of (3aS,7aS)-4-((E)-5-hydroxy-2- methylstyryl)-7a-methyl-1-oxo-2,3,3a,6,7,7a-hexahy- dro-1H-inden-5-yl trifluoromethanesulfonate (18)
To a dried 50 mL of two-necked flask equipped with a stirrer was added compound 17 (677 mg, 1.6 mmol). The flask was then evacuated and backfilled with N2 for 3 times. Anhydrous tetrahydrofuran (THF) (10 mL) was added via a syringe and the resulting solution was stirred in a 0 ℃ ice-water bath. NaHMDS (1.8 mL, 1.0 mol/L, 1.08 equiv.) was slowly added to the above solution via a syringe, and the solution was stirred at this temperature for 15 min, followed by transferring the solution to room temperature and continued to stir for 30 min. After which the solution was returned to 0 ℃ and Tf2NPh (633 mg, 1.8 mmol, 1.08 equiv.) in anhydrous THF (5 mL) was slowly dripped in. The solution was then brought to room temperature again for overnight stirring (as determined by TLC). The reaction was quenched with saturated NH4Cl solution (5 mL), the aqueous phase was extracted with ethyl acetate (20 mL×3), the combined organic phase was washed with brine (20 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product.
To another 50 mL round-bottomed flask containing a stirrer were added KF (48 mg, 0.8 mmol, 1.5 equiv.) and tetraethylene glycol (2 mL). The flask was then evacuated and backfilled with N2 for 3 times. The crude product in anhydrous ethyl acetate (10 mL) was slowly added dropwise in and the mixture was stirred at room temperature for 1 h (as determined by TLC). The reaction was quenched with water (5 mL), the aqueous phase was extracted with ethyl acetate (5 mL×3), the combined organic phase was washed with brine (20 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/EtOAc, V:V=15:1) to provide 18 (569 mg, 83%). White solid, m.p. 136.6~137.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.04~6.97 (m, 3H), 6.81 (d, J=16.4 Hz, 1H), 6.70 (dd, J=8.4, 2.8 Hz, 1H), 2.97~2.92 (m, 1H), 2.72~2.63 (m, 3H), 2.54~2.47 (m, 1H), 2.38~2.31 (m, 1H), 2.29 (s, 3H), 2.06~1.98 (m, 2H), 1.73~1.65 (m, 1H), 1.05 (s, 3H), 0.99 (s, 9H), 0.20 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 217.2, 152.9, 145.1, 137.0, 133.5, 131.0, 128.6, 128.2, 121.8, 118.67 (q, 1JC-F=313.1 Hz), 115.0, 47.7, 45.2, 36.3, 27.5, 25.8, 20.7, 18.8, 13.7; 19F NMR (471 MHz, CDCl3) δ: -74.69; HRMS (ESI) calcd for C20H21F3O5SNa [M+Na]+ 453.0954, found 453.0951.
4.8 Synthesis of (10R,13S,14S)-10,13-dimethyl- 7,10,11,12,13,14,15,16-octahydro-3H-cyclopenta[a]-phenanthrene-3,17(6H)-dione (20)
To a dried 50 mL of round-bottomed flask containing a stirrer were added compound 18 (215 mg, 1 mmol) and 5% Pd/C (22 mg). The flask was then evacuated and backfilled with N2 from a balloon for 3 times. Anhydrous ethyl acetate (10 mL) was added via a syringe, and then the N2 in flask was replace with H2 by a balloon. After which the reaction solution was reacted for 5 h at room temperature (as determined by TLC). The Pd/C was filtered out with diatomite filtration and the filter cake was washed with ethyl acetate (10 mL×3). The combined organic phases were washed with brine (15 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product for next step.
To a 50 mL round-bottomed flask equipped with a stirrer and a condensation reflux tube were added the crude product, Pd(OAc)2 (11.2 mg, 0.05 equiv.), ligand (37.0 mg, 0.1 equiv.) and anhydrous K2CO3 (165.6 mg, 1.2 mmol, 1.2 equiv.) under the protection of N2. The flask was then evacuated and backfilled with N2 from a balloon for 3 times. Anhydrous toluene (20 mL) was added and the resulting solution was stirred in a preheated 140 ℃ oil bath for 12 h (as determined by TLC). After the reaction mixture was cooled to room temperature, the insoluble material was removed by diatomite filtration and the filter cake was washed with ethyl acetate (10 mL×3). The organic phases were combined and washed with brine (10 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product, which was purified by flash chromatography on silica gel (heptane/EtOAc, V:V=10:1) to provide 20 (112.8 mg, 40%). White solid, m.p. 70.2~71.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.17 (d, J=10.1 Hz, 1H), 6.25 (dd, J=10.1, 1.7 Hz, 1H), 6.13 (s, 1H), 2.76~2.68 (m, 1H), 2.57~2.42 (m, 5H), 2.33~2.25 (m, 2H), 2.22~2.13 (m, 1H), 2.07~2.01 (m, 1H), 1.94 (dd, J=13.0, 6.7 Hz, 1H), 1.71~1.63 (m, 1H), 1.59~1.52 (m, 1H), 1.45 (s, 3H), 0.77 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 218.7, 185.9, 166.6, 153.1, 131.2, 129.3, 127.9, 123.6, 47.3, 47.0, 44.2, 36.4, 30.5, 29.7, 28.9, 28.6, 24.5, 20.7, 12.9; HRMS (ESI) calcd for C19H23O2 [M+H]+ 283.1693, found 283.1710.
Supporting Information 1H NMR, 13C NMR and chiral HPLC spectra of compounds 7~20. Crystal structure and data refinement of 20. The Supporting Information is available free of charge via the Internet at http://sioc- journal.cn.
(Zhao, C.)