


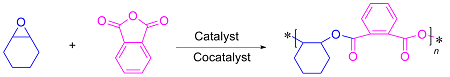
表1 环氧化物和酸酐的开环共聚Table 1 Ring opening copolymerization of epoxides and anhydrides |
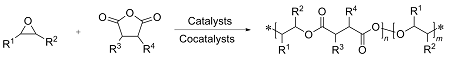
| Entrya | Catalyst/mol% | Cocatalyst/mol% | Epoxide | Anhydride | T/℃ | Time/h | Yieldb/% | Mnc/(kg•mol-1) | PDIc |
|---|---|---|---|---|---|---|---|---|---|
| 1[22] | 卟啉-Al [4.0] | EtPh3PBr [4.0] | EO | PA | r.t. | 168 | 100 | 3.0 | 1.10 |
| 2[23] | β-二亚胺-Zn [0.5] | — | CHO | DGA | 50 | 60 | 91 | 31.0 | 1.20 |
| 3[24] | Salen-CrCl [0.5] | PPNCl [0.5] | PO | CA | 30 | 24 | 100 | 16.9 | 1.20 |
| 4[25] | Salen-Fe [0.4] | PPNCl [0.4] | CHO | PA | 100 | 1 | 100 | 12.3 | 1.26 |
| 5[26] | 氨基三酚-Fe [0.4] | DMAP [0.5] | LO | PA | 65 | 48 | 92 | 10.7 | 1.24 |
| 6[27] | Fe/K异双核 [0.1] | — | CHO | PA | 100 | 1 | 95 | 19.1 | 1.09 |
a Abbreviations: EO, ethylene oxide; CHO, cyclohexene oxide; PO, propylene oxide; LO, limonene oxide; PA, phthalic anhydride; DGA, diglycolic anhydride; CA, camphoric anhydrides. b Mole percentage (mol%) of catalyst and co-catalyst was calculated on the basis of the loading amount of anhydrides. |
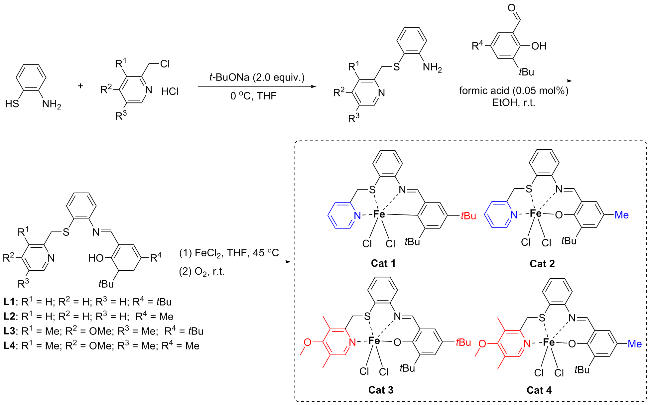
1 结果与讨论
1.1 配合物Cat 1和Cat 3的晶体结构
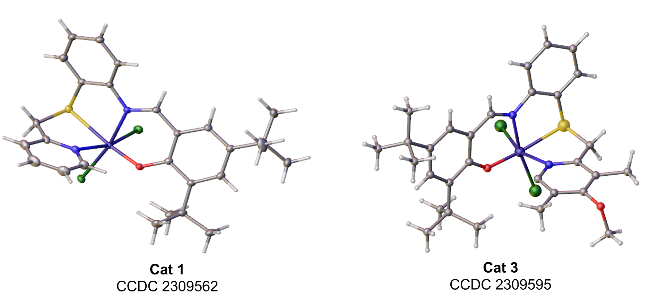
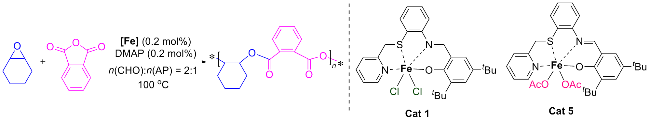
1.2 聚合反应条件分析与讨论
表2 不同催化剂对ROCOP反应的影响aTable 2 Effects of different catalysts on ROCOP reaction |
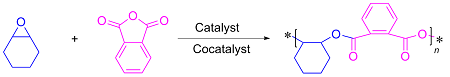
| Entry | Catalyst | Cocatalyst | Yieldb/% | Mnc/(kg•mol-1) | PDIc |
|---|---|---|---|---|---|
| 1 | Cat 1 | PPNCl | 90 | 3.0 | 1.22 |
| 2 | Cat 2 | PPNCl | 88 | 2.9 | 1.19 |
| 3 | Cat 3 | PPNCl | 88 | 3.3 | 1.14 |
| 4 | Cat 4 | PPNCl | 87 | 2.7 | 1.20 |
| 5 | Cat 1 | TBAB | 88 | 2.3 | 1.22 |
| 6 | Cat 1 | TBAC | 92 | 2.6 | 1.23 |
| 7 | Cat 1 | TBAI | 88 | 2.5 | 1.26 |
| 8 | Cat 1 | DMAP | 92 | 3.7 | 1.20 |
| 9 | Cat 1 | Py | 72 | 2.3 | 1.23 |
| 10 | Cat 1 | 4-PPy | 86 | 3.6 | 1.16 |
| 11 | Cat 1 | 4-AP | 89 | 2.6 | 1.26 |
| 12 | Cat 1 | DBU | 90 | 3.0 | 1.33 |
a Reaction conditions: n(CHO)/n(PA)/n(catalyst)/n(cocatalyst)=200/200/1/1, temperature: 100 ℃, time: 3 h. b Isolated yields are given. c Determined by gel permeation chromatography (GPC) in THF. Abbreviations: PPNCl, bis(triphenylphosphoranylidene)ammonium chloride; TBAB, tetrabutylammonium bromide; TBAC, tetrabutylammonium chloride; TBAI, tetrabutylammonium iodide; DMAP, 4-dimethylaminopyridine; Py, pyridine; 4-PPy, 4-pyrrolidinopyridine; 4-AP, 4-aminopyridine; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene. |
表3 催化条件的优化aTable 3 Optimization of catalyst conditions |
| Entry | n(CHO)∶n(PA)∶n(Cat 1)∶n(DMAP) | Yieldb/% | Ester linkage/%c | Mnd/(kg•mol-1) | PDI d |
|---|---|---|---|---|---|
| 1 | 200∶200∶1∶1 | 99 | 82 | 3.6 | 1.23 |
| 2 | 300∶200∶1∶1 | 99 | 79 | 4.5 | 1.27 |
| 3 | 500∶500∶1∶1 | 96 | 83 | 5.1 | 1.28 |
| 4 | 1000∶500∶1∶1 | 99 | 85 | 7.0 | 1.16 |
| 5 | 1000∶500∶2∶1 | 99 | 76 | 9.8 | 1.55 |
| 6 | 1000∶500∶1∶2 | 99 | 82 | 9.5 | 1.37 |
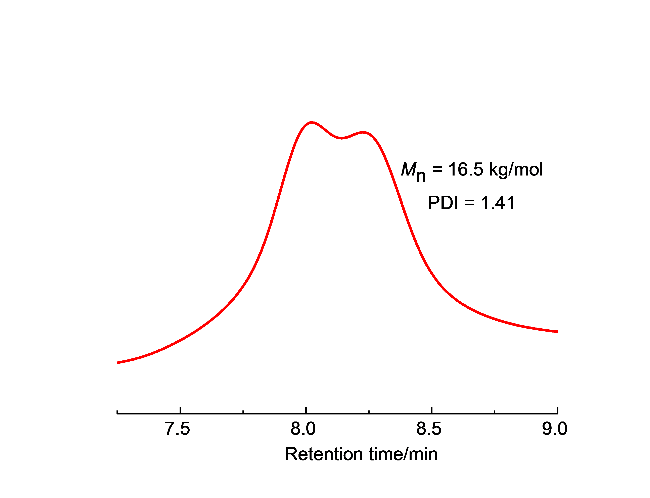
| 7 | 2000∶1000∶1∶1 | 80 | 90 | 12.3 | 1.41 |
| 8e | 2000∶1000∶1∶1 | 99 | 92 | 13.1 | 1.19 |
| 9f | 2000∶1000∶1∶1 | 99 | 90 | 9.5 | 1.29 |
| 10g | 2000∶1000∶1∶1 | 99 | 78 | 7.7 | 1.46 |
a Temperature: 100 ℃, time: 6 h. b Isolated yields are given. c Determined by 1H NMR analysis; d Determined by gel permeation chromatography (GPC) in THF. e Temperature: 100 ℃, time: 10 h. f Catalyst left at room temperature for 3 months. g Using unpurified PA and CHO. |
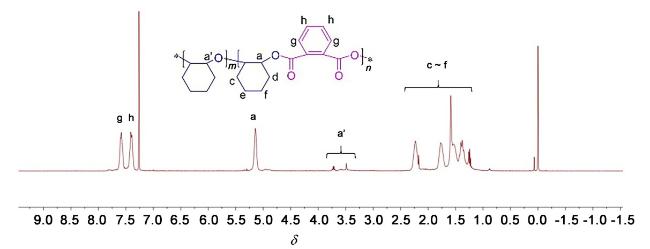
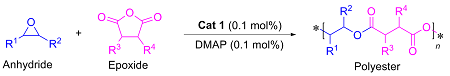
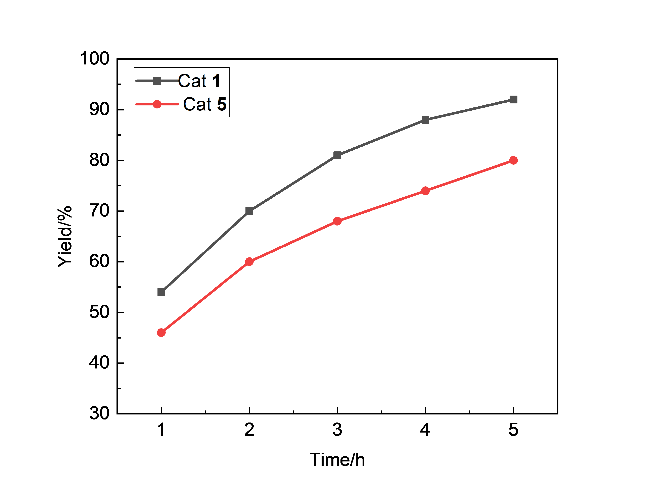
1.3 底物普适性的研究
表4 酸酐和环氧化物的底物拓展aTable 4 Expansion of substrates for acid anhydrides and epoxides |
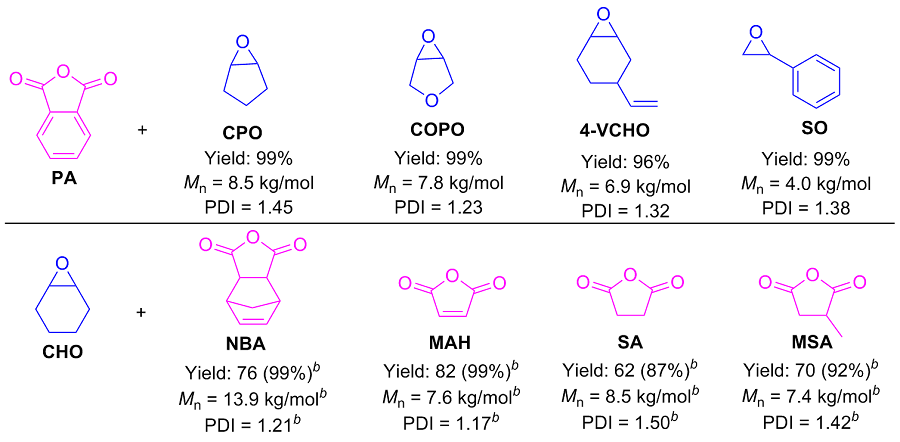 |
a Temperature: 100 ℃, time: 10 h, Cat 1 (0.1 mol%), DMAP (0.2 mol%), n(epoxide)∶n(anhydride)=2∶1, isolated yields are given, determined by gel permeation chromatography (GPC) in THF. b Temperature: 100 ℃, time: 24 h. |
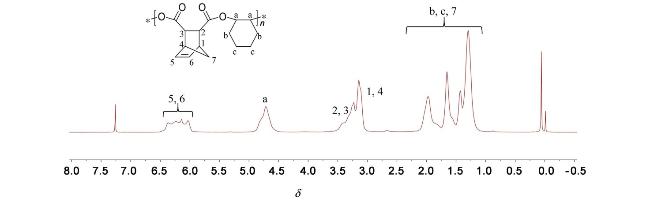
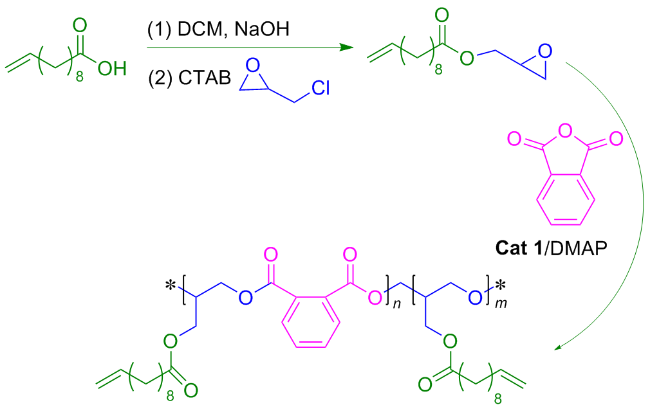
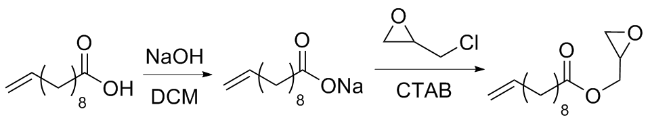
1.4 植物油基聚合物的制备
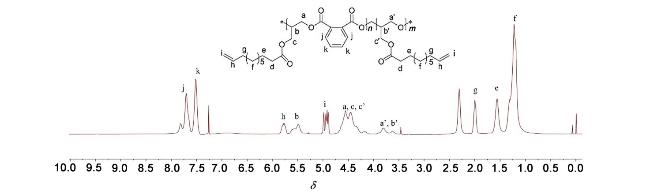
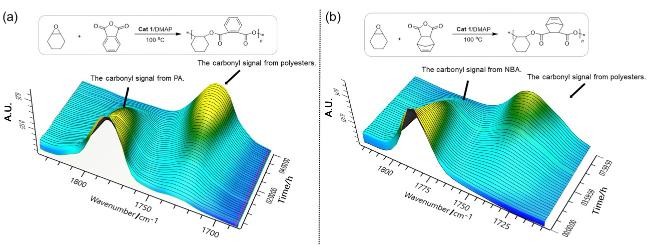
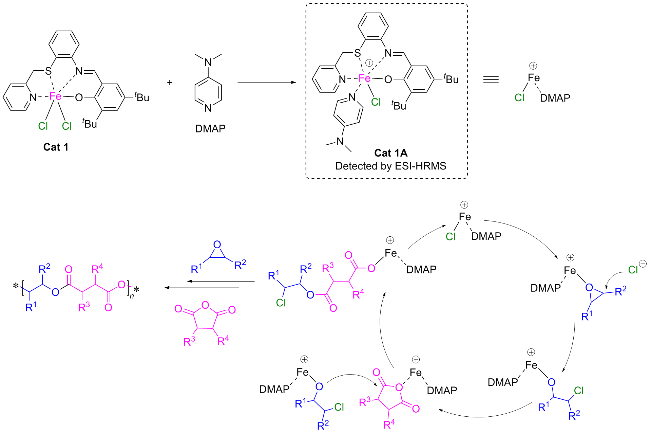
1.5 环氧化物与酸酐共聚的机理探究
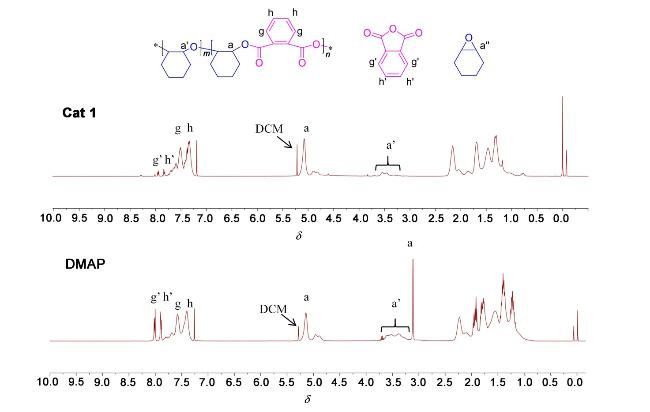
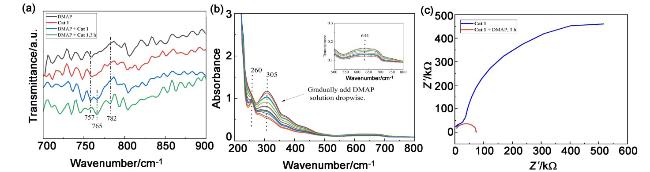
图8 环氧化物和酸酐共聚的机理探索: (a)向配合物Cat 1的二氯甲烷溶液中加入DMAP的FT-IR光谱; (b)向配合物Cat 1的二氯甲烷溶液中加入DMAP的UV-vis光谱; (c) Cat 1的二氯甲烷溶液中加入DMAP的电化学阻抗谱(EIS)Figure 8 Mechanism exploration of copolymerization of epoxides and anhydrides: (a) FT-IR spectra with addition of DMAP to a solution of complex Cat 1 in dichloromethane; (b) UV-vis spectra with addition of DMAP to a solution of complex Cat 1 in dichloromethane; (c) electrochemical impedance spectroscopy (EIS) of DMAP added to dichloromethane solution of Cat 1 |
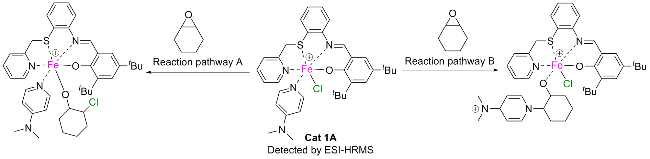
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}