


1,3-烯炔是一类同时具有烯基和炔基的有机分子, 是一种多用途的不饱和化合物, 不仅存在于许多天然产物中[1], 还在药物化学、材料科学和有机合成等领域有着广泛的应用[2]. 自1965年Arens课题组[3]实现了1,3-烯炔制备乙炔醚化合物的开创性工作以来, 1,3-烯炔由于其结构独特及具有多种化学反应位点等特性, 已被用作多种反应的基本单元, 以获得各种特殊的分子结构.
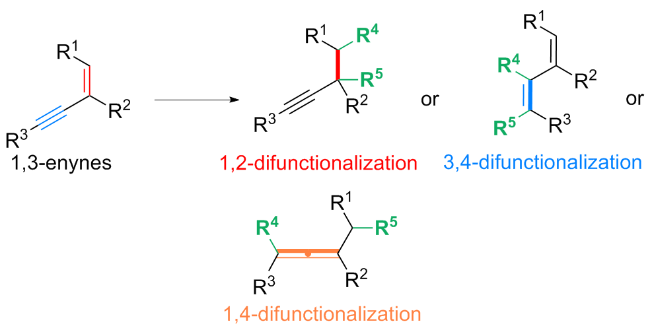
烯烃和炔烃的双官能化反应是使用简单而丰富的化学原料简洁高效地合成各种高附加值分子的基本策略[4]. 1,3-烯炔具有特殊的共轭结构, 具有多个反应位点, 控制其选择性双官能化一直是重要的研究热点. 1,3-烯炔的双官能化反应主要包括1,2-、3,4-和1,4-双官能化(Scheme 1): 1,3-烯炔的1,2-双官能化是对烯烃部分的官能化, 被广泛应用于构建炔基化合物; 1,3-烯炔的3,4-官能化是对炔烃部分的官能化, 是构建1,3-二烯型化合物、噻吩和吡咯等芳香杂环的重要合成策略; 1,3-烯炔的1,4-官能化是同时对烯基和炔基官能化, 是合成多取代丙二烯衍生物的便捷途径. 近几年, 1,3-烯炔的双官能化策略被广泛报道, 相继实现了1,3-烯炔的1,2-双烷基化、1,2-羟基化磷酸化、3,4-硼氰化、3,4-/1,4-芳基化磺酰化及1,4-卤化三氟甲基化等各种双官能化反应.
由于1,3-烯炔的特殊结构, 控制1,3-烯炔化学和区域选择性双官能化的挑战一直存在. 实现1,3-烯炔中烯烃、炔烃部分双官能团化或烯烃与炔烃部分的同时双官能化备受挑战. 2020年, Procter课题组[5]对铜催化的烯炔官能化做了详细的综述. 近几年, 光/电催化策略在1,3-烯炔的双官能化反应中迅猛发展, 为1,3-烯炔的双官能团化反应提供了新策略. 本文综述了近几年1,3-烯炔的1,2-、3,4-和1,4-双官能化反应的研究进展, 并对部分机理做了详细解析.
1 1,3-烯炔的1,2-双官能化反应
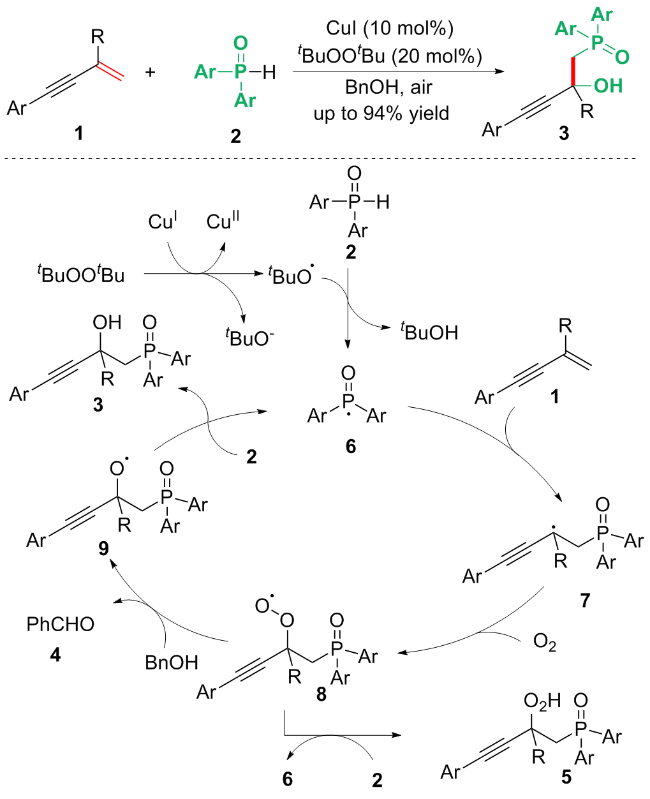
为了探究反应机理, 作者在反应体系中加入1 equiv.的自由基捕获剂2,2,6,6-四甲基-1-哌啶基氧基(TEMPO), 无产物3生成, 表明该反应是通过自由基途径进行的. 随后作者在标准条件下进行反应体系的高分辨质谱分析(HRMS), 发现该反应体系中存在产物3、副产物苯甲醛4和过氧化合物5. 基于上述机理研究, 作者提出反应可能的机理(Scheme 2): 首先, CuI与自由基引发剂DTBP (tBuOOtBu)发生氧化还原反应, 得到叔丁氧基自由基与CuII物种. 随后, 叔丁氧基自由基直接从二芳基氧化膦中掘取一个氢原子生成二芳基磷化物自由基中间体6. 中间体6进攻底物1的双键端侧, 得到自由基中间体7, 中间体7进一步捕捉空气中的氧气得到过氧化合物自由基中间体8. 中间体8从磷试剂2中攫取氢原子, 生成过氧化产物5; 中间体8也可以从BnOH中攫氢, 得到羟基自由基中间体9. 最后中间体9与二芳基氧化膦反应得到最终产物3.
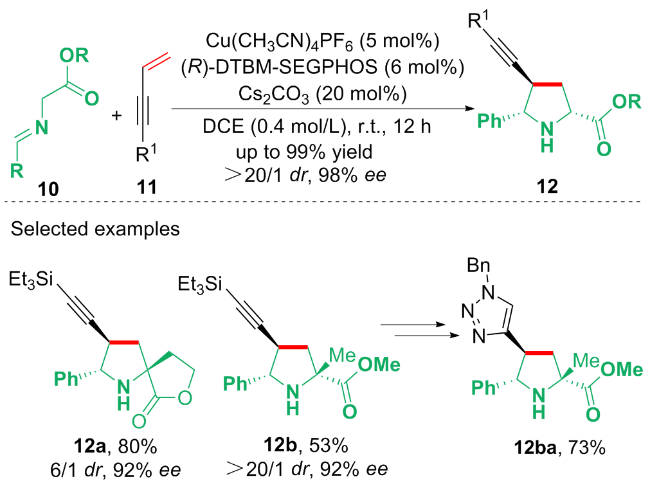
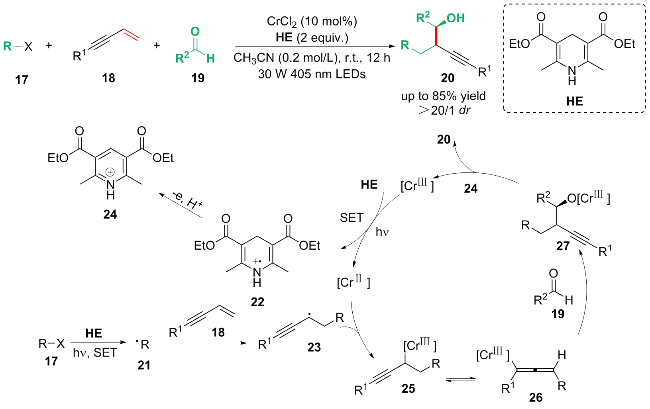
基于开关灯实验、荧光量子产率以及自由基捕获等机理探究, 作者提出了可能的光催化循环反应路径: 首先, 在蓝光照射下, HE同时通过单电子转移过程还原卤代烃17与CrIII, 得到烷基自由基中间体21和22. 烷基自由基中间体21进攻1,3-烯炔18的双键的端位, 形成丙炔自由基中间体23. 中间体22进一步失去一个电子和一个质子形成吡啶24. 随后, 自由基中间体23被CrII捕获, 生成亲核的丙炔基CrIII配合物25, 配合物25与丙烯基CrIII配合物26以共振形式达到平衡. 配合物26与醛19反应生成具有高非对映选择性的产物27. 最后, 通过吡啶24水解27中的O—Cr键, 生成最终的产物20.
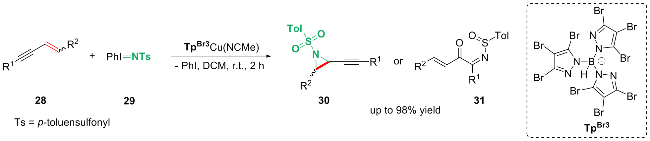
2 1,3-烯炔的3,4-双官能化反应
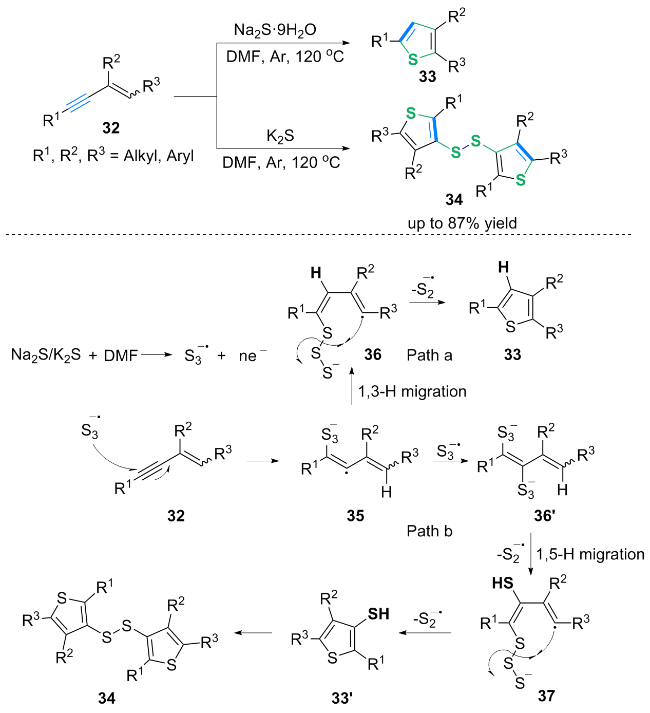
基于自由基抑制实验及前期文献报道[12], 作者提出了两种可能的反应路径: 首先由K2S/Na2S生成的三硫基自由基阴离子($\text{SO}{{_{3}^{}}^{-\ \bullet }}$)进攻底物32的炔基端侧, 生成自由基阴离子中间体35. 在合成2,3,5-三取代噻吩(Path a)时, 中间体35通过1,3-氢迁移转化为中间体36; 中间体36通过S—S键均裂和分子内自由基偶联得到环化产物33. 在合成3-噻吩基二硫醚(Path b)时, 三硫基自由基阴离子($\text{SO}{{_{3}^{}}^{-\ \bullet }}$)与中间体35反应生成阴离子中间体36', 中间体36'通过S—S键均裂和1,5-氢迁移得到中间体37, 中间体37通过S—S键均裂和分子内自由基偶联得到中间体33'. 最后, 中间体33'被氧化生成二硫产物34.
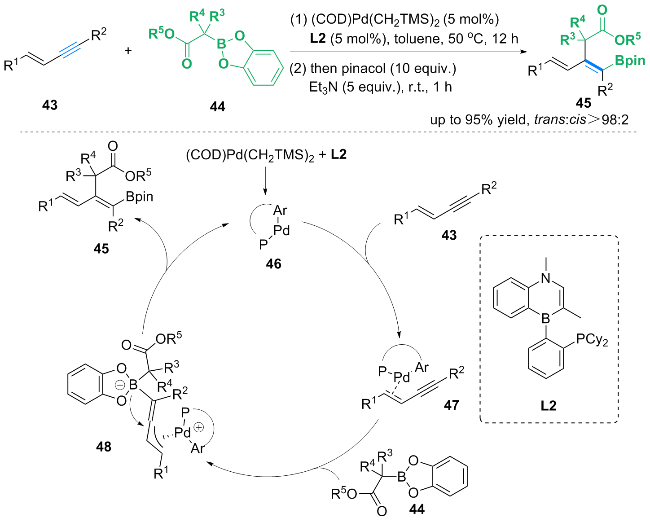
经过DFT计算等机理研究, 作者提出了可能的反应机理: 在Senphos配体L2的存在下, (COD)Pd- (CH2TMS)2还原消除1,2-双(三甲基硅基)乙烷, 得到Pd0配合物46. 随后配合物46与1,3-烯炔43反应, 得到配合物47. 然后, 配合物47被C-硼烯醇化合物经历outer- sphere的氧化加成得到化合物48. 随后, 化合物48中的烯丙基钯被烯醇化合物进攻得到最后产物45, 同时重新得到零价钯配合物46.

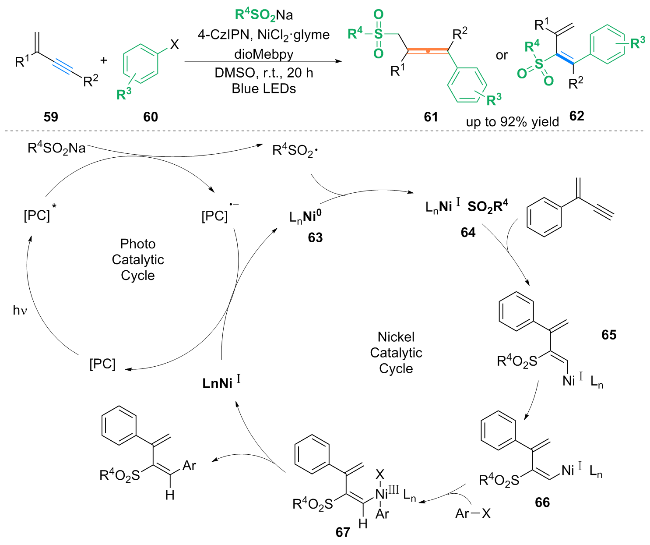
作者结合自由基实验等机理研究, 提出了可能的反应机理. 在光催化循环中, 光催化剂PC (4-CzIPN)被光照激发得到激发态PC*, 然后PC*被亚磺酸钠还原, 生成磺酰基自由基以及PC•. 在镍催化循环中, Ni0物种63捕获磺酰自由基并进行自由基加成, 得到NiI磺酰基64. 随后NiI磺酰基64插入1,3-烯炔的叁键上, 生成中间体65, 65通过Ni辅助的异构化转化为中间体66. 随后芳基卤化物氧化加成生成NiIII中间体67, 接着中间体67被还原消除生成3,4-芳基化磺酰化的1,3-二烯产物.
3 1,3-烯炔的1,4-双官能化反应
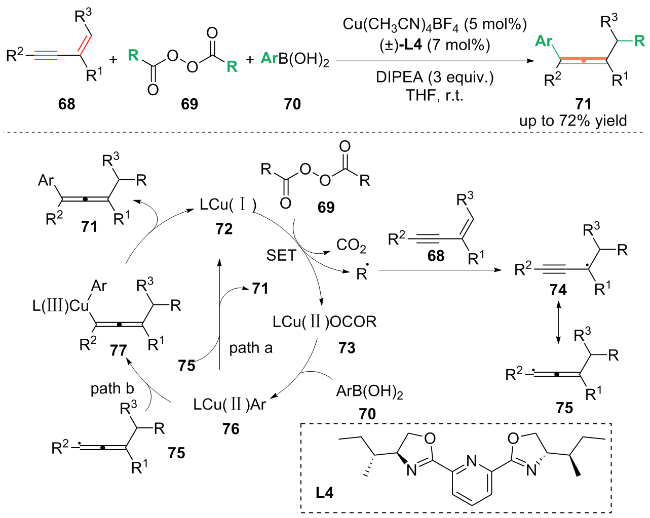
作者通过自由基抑制实验及自由基钟摆实验等机理研究, 提出了可能的反应机理: Cu(I)配合物72通过单电子转移(SET)过程与烷基双酰过氧化物69反应, 生成烷基自由基和Cu(II)配合物73. 烷基自由基与1,3-烯炔68发生自由基加成, 得到丙炔基自由基中间体74. 随后, 中间体74通过共振得到烯丙基自由基中间体75. 配合物73可以与ArB(OH)2 (70)进行配体交换生成Ar-Cu(II)配合物76. 结合机理实验, 该策略可能存在两种反应路径: Path a是Ar-Cu(II)配合物76与丙烯基自由基中间体75发生自由基取代反应, 生成最终产物71; Path b中Ar-Cu(II)配合物76与丙二烯基自由基中间体75结合形成Cu(III)中间体77, 通过还原消除, 生成最终产物71.
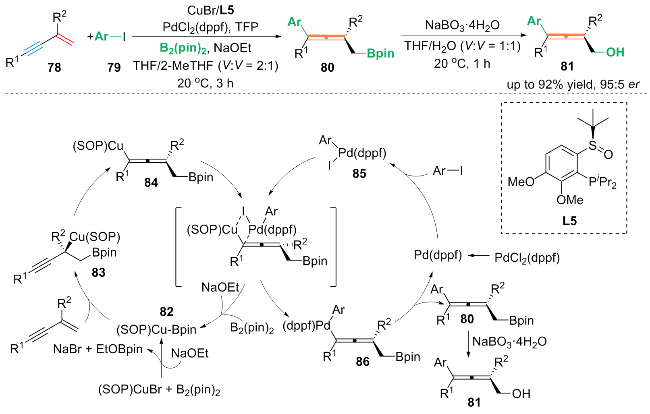
基于DFT计算以及其它机理验证, 作者提出了可能的反应机理: 在铜催化循环中, (SOP)CuBr与B2(pin)2在NaOEt存在下生成(SOP)Cu-Bpin配合物82, 82加成到1,3-烯炔上得到炔丙基铜中间体83. 83发生立体专一异构化生成轴手性的连烯基铜中间体84, 84与芳基钯配合物85发生立体定向的金属转移生成了联烯基钯配合物86. 然后, 86通过还原消除得到轴手性的丙二烯产物80. 最后, 产物80被氧化得到最终产物81.
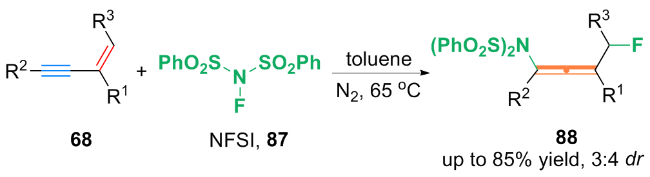
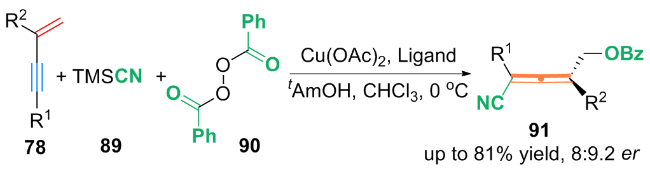
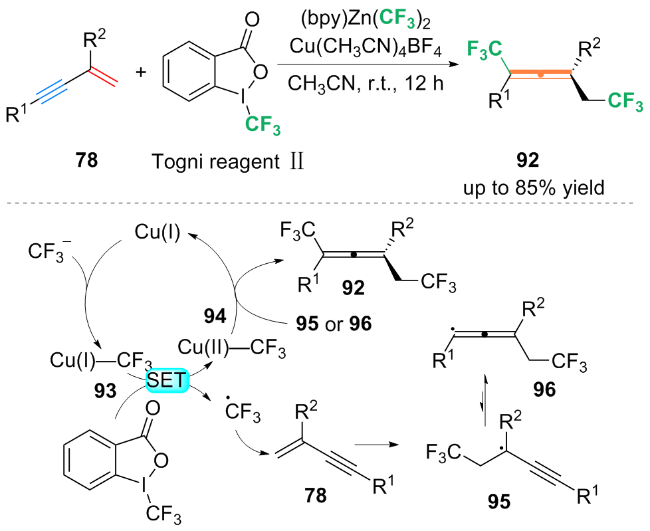
基于自由基捕获实验等机理探究, 作者提出了一种可能的机理: 首先, (bpy)Zn(CF3)2中的$\text{CF}_{3}^{}$与铜催化剂络合, 生成了Cu(I)-CF3配合物93. 配合物93与Togni试剂Ⅱ发生了单电子转移(SET)过程, 生成三氟甲基自由基(CF3•)和Cu(II)-CF3中间体94. 随后, CF3•进攻1,3-烯炔78上烯基的端侧, 得到丙炔自由基95. 自由基95通过互变异构得到丙烯基自由基96. 最后, 丙炔自由基95或丙烯基自由基96被Cu(II)-CF3进攻, 得到双(三氟甲基化)产物92. 对于丙烯基自由基96和丙炔基自由基95与Cu(II)-CF3中间体94的反应性强弱问题, 还需进一步的机理研究.
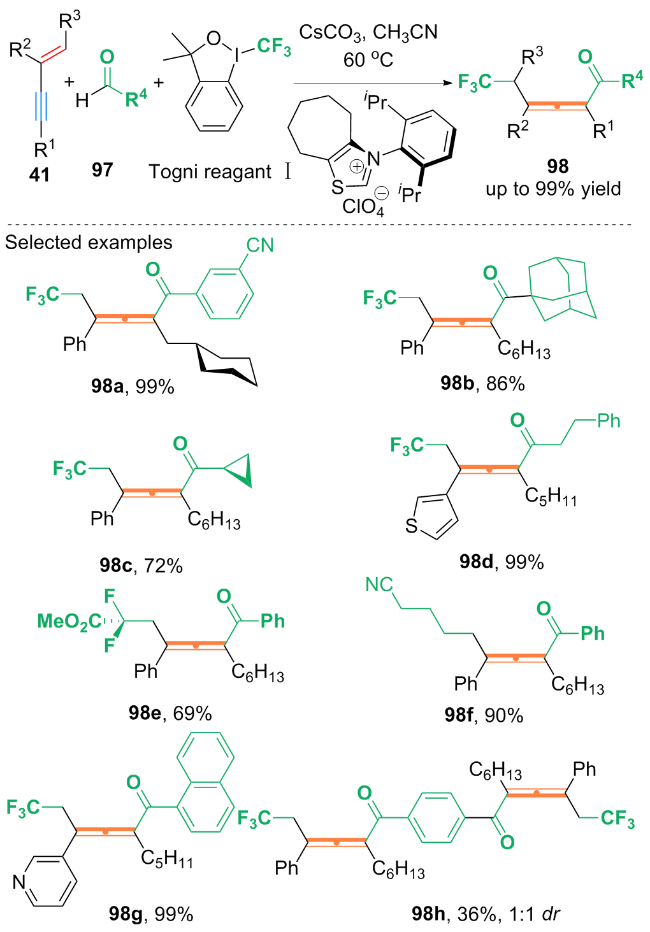
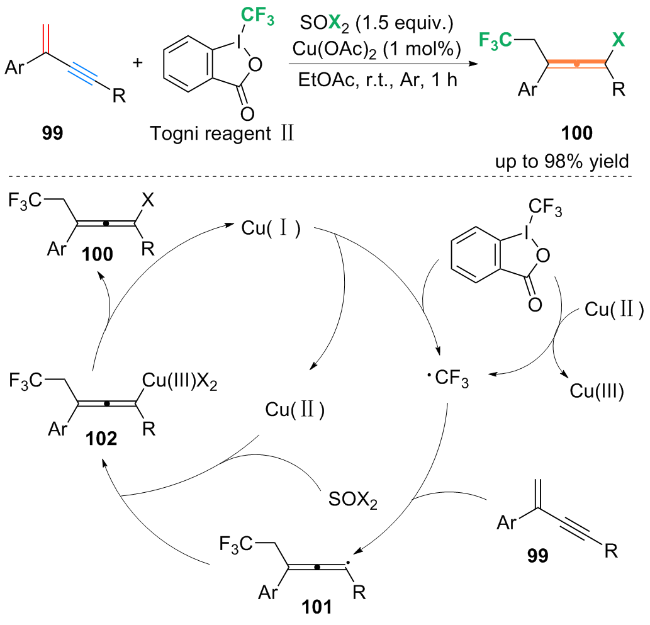
作者通过自由基捕获实验及自由基钟摆实验, 得到自由基捕获产物与开环产物, 提出了可能的反应机理: 首先, Togni试剂Ⅱ与铜(II)配合物发生单电子转移(SET)过程, 生成亲电的三氟甲基自由基(CF3•). CF3•随后被1,3-烯炔99捕获, 生成三氟甲基化的丙烯基自由基中间体101. 中间体101与Cu(II)配合物和SOX2结合, 生成CF3-丙烯基-Cu(III)X2配合物102, 然后102还原消除得到最终产物100.
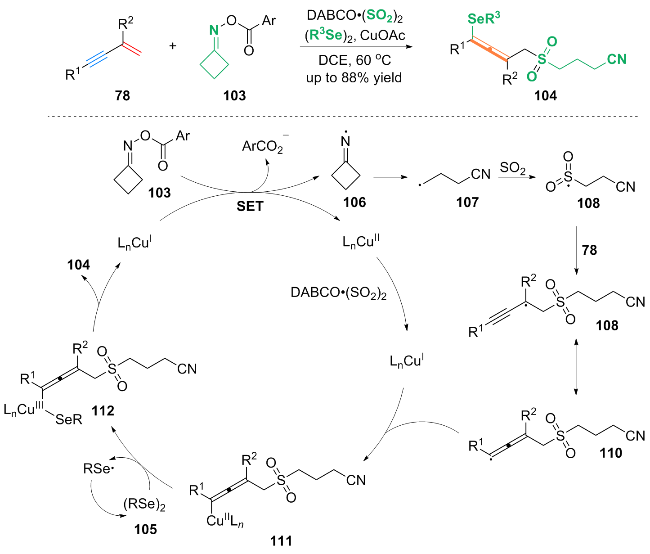
基于自由基实验及高分辨质谱分析(HRMS)等机理研究, 作者提出了可能的反应机理: 首先, 环酮肟酯103与Cu(I)配合物发生单电子转移(SET)过程, 得到亚胺基自由基中间体106和Cu(II)配合物. 紧接着, 中间体106通过β-C—C键的裂解开环生成氰烷基自由基中间体107, 中间体107被DABCO•(SO2)2中的SO2迅速捕获, 生成氰基烷基磺酰自由基中间体108. 中间体108与1,3-烯炔78发生自由基加成得到丙炔基自由基中间体109, 中间体109通过共振得到其共振形式的丙烯基自由基中间体110. 中间体110与Cu(I)配合物配位生成Cu(II)配合物111, 随后Cu(Ⅱ)配合物111与二硒醚105反应生成Cu(III)配合物112. 最后, Cu(III)配合物112通过还原消除得到最终产物104.
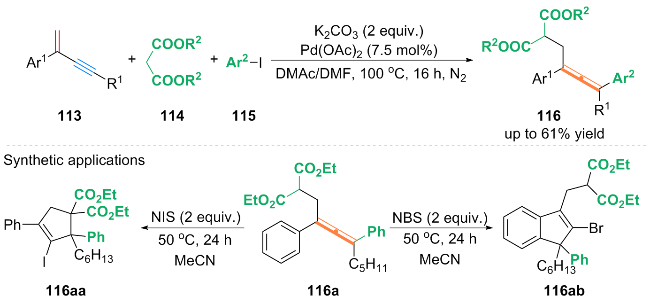

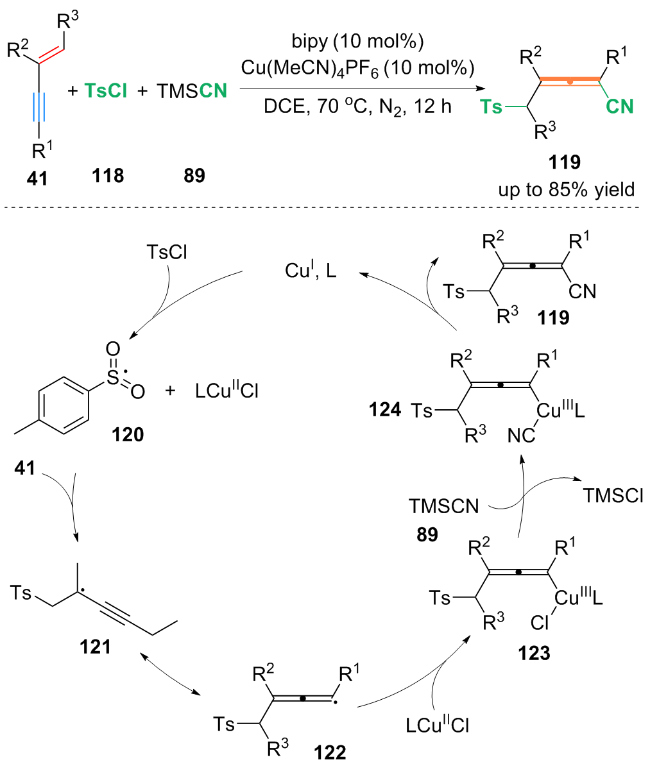
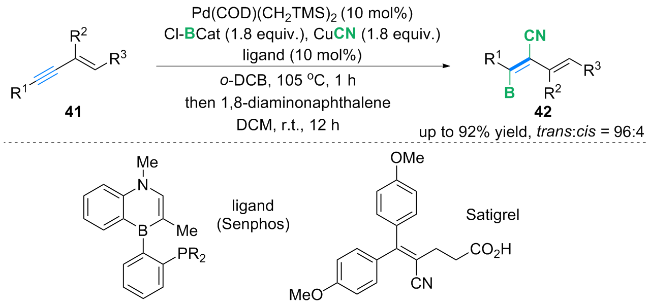
自由基捕获实验表明, 在反应过程中产生了磺酰基和丙烯基自由基. 作者提出了含磺酰基和丙烯基自由基的铜催化循环机理: 首先, Cu(MeCN)4PF6催化剂在bipy配体存在下, 通过单电子转移(SET)过程与TsCl反应, 生成磺酰基自由基中间体120和LCuⅡCl配合物. 中间体120进攻1,3-烯炔41的烯基端侧, 得到自由基中间体121, 中间体121发生共振得到丙烯基自由基122. 随后, 丙烯基自由基122与LCuⅡCl配合物反应生成配合物123, 123与TMSCN 89发生配体交换, 得到氰基中间体124. 最后氰基中间体124通过还原消除生成最终产物119, 并生成铜催化剂完成铜催化循环.
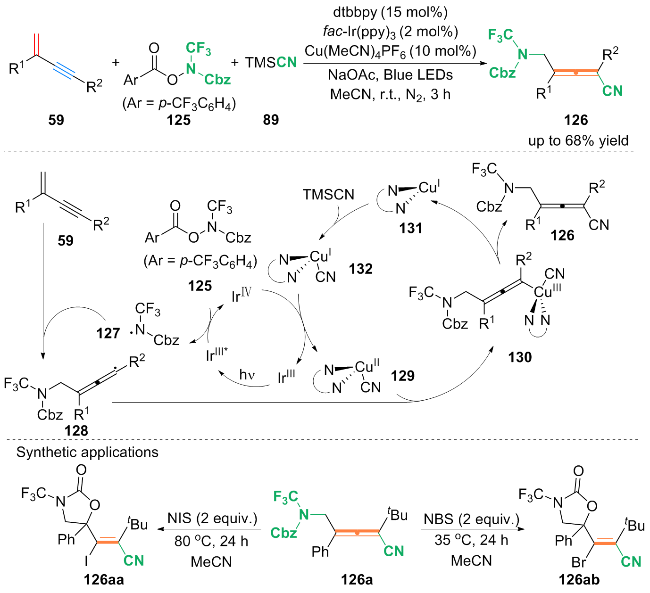
基于自由基捕获等实验, 提出反应可能的机理: 首先, 光催化剂fac-IrⅢ(ppy)3被蓝光激发得到激发态的fac-IrⅢ(ppy)3*, fac-IrⅢ(ppy)3*与N-CF3羟胺试剂125发生单电子转移(SET)过程, 得到自由基中间体127和fac- IrⅣ(ppy)3. 中间体127进攻1,3-烯炔59中的烯烃外侧, 得到烯丙基自由基中间体128. 中间体128与CuⅡ(CN)配合物129发生偶联生成CuⅢ配合物130. 最后, 配合物130还原消除得到最终产物126和CuⅠ配合物131. CuⅠ配合物131与TMSCN (89)发生配体交换得到CuⅠ(CN)配合物132. 随后, 配合物132被fac-IrⅣ(ppy)3氧化为CuⅡ(CN)配合物129和fac-IrⅢ(ppy)3, 完成催化循环.

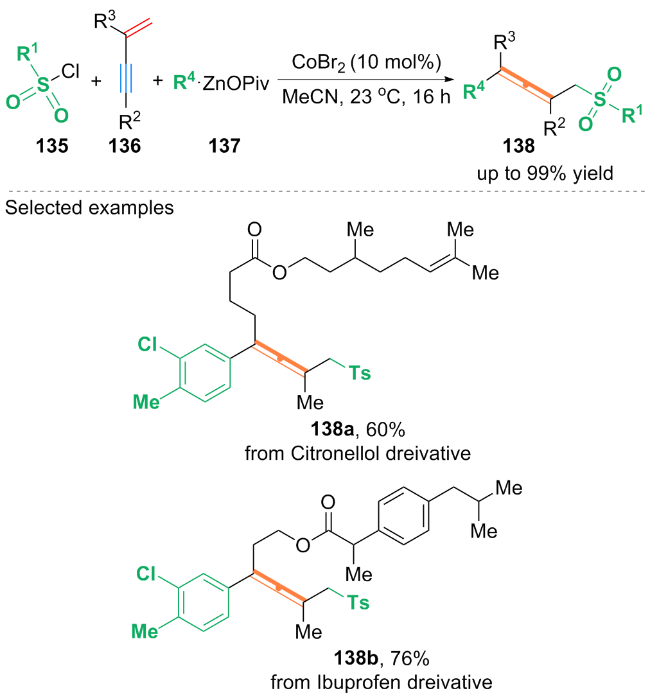
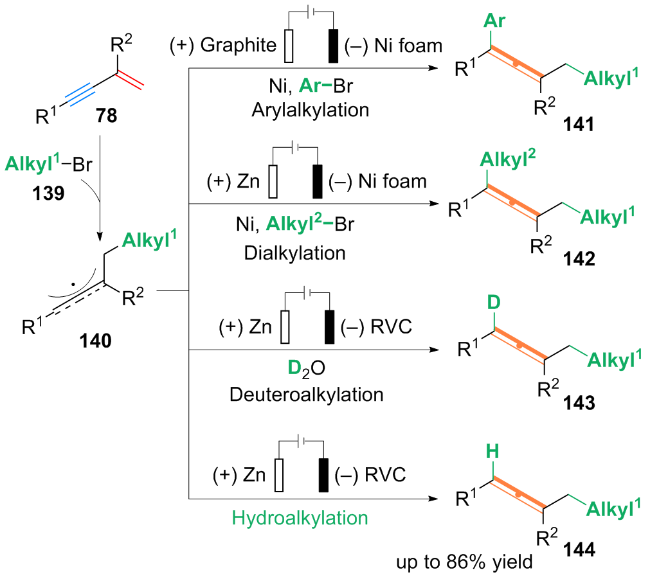
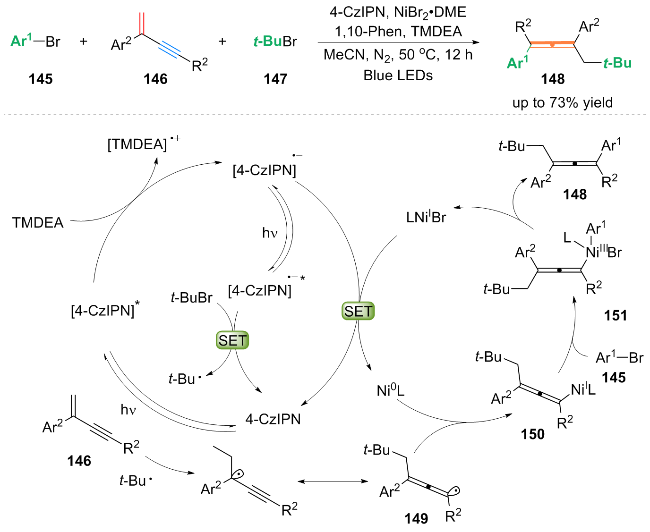
作者基于自由基捕获等实验, 提出了可能的双重催化反应路径: t-BuBr 147与[4-CzIPN]•*通过单电子转移过程得到叔丁基自由基t-Bu•. 随后, t-Bu•加成到1,3-烯炔146的双键上发生异构化, 得到丙二烯基自由基中间体149, 其中间体149被Ni0L配合物捕获, 形成丙二烯基-NiIL配合物150. 随后, 芳基溴化物氧化加成到配合物150上得到NiIII配合物151, 配合物151还原消除产生芳基烷基化的丙二烯产物148.
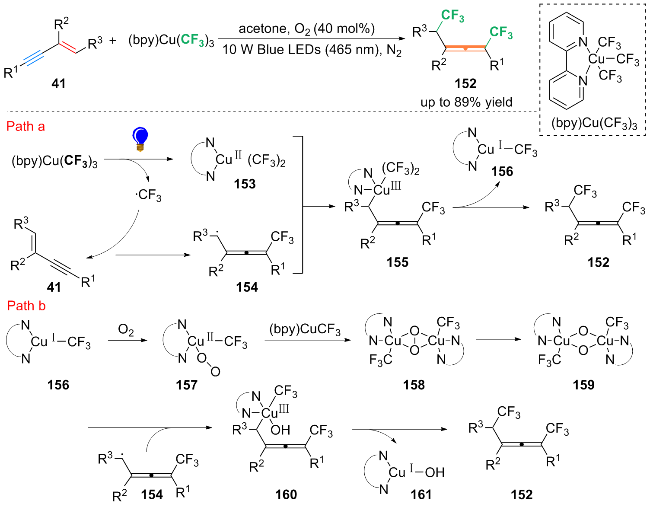
基于自由基捕获实验, 作者提出了光和铜协同催化的反应机理: 在蓝光照射下, (bpy)CuⅢ(CF3)3释放三氟甲基自由基(•CF3), 生成中间体(bpy)CuⅡ(CF3)2 (153). 随后, •CF3进攻1,3-烯炔41的炔基外侧, 得到自由基加成产物丙二烯基自由基154. 然后, 154与中间体153发生偶联得到CuIII配合物155. 155具有高度反应性, 经过快速的还原消除得到产物152和CuI配合物156 (Path a). 配合物156可被O2氧化为CuⅡ配合物157, 配合物157发生自身的聚合得到配合物158. 随后, 158快速异构化为CuIII配合物159, 159捕获丙二烯基自由基154得到配合物160. 最后, 160通过还原消除生成最终产物152, 并释放CuI配合物161 (Path b).
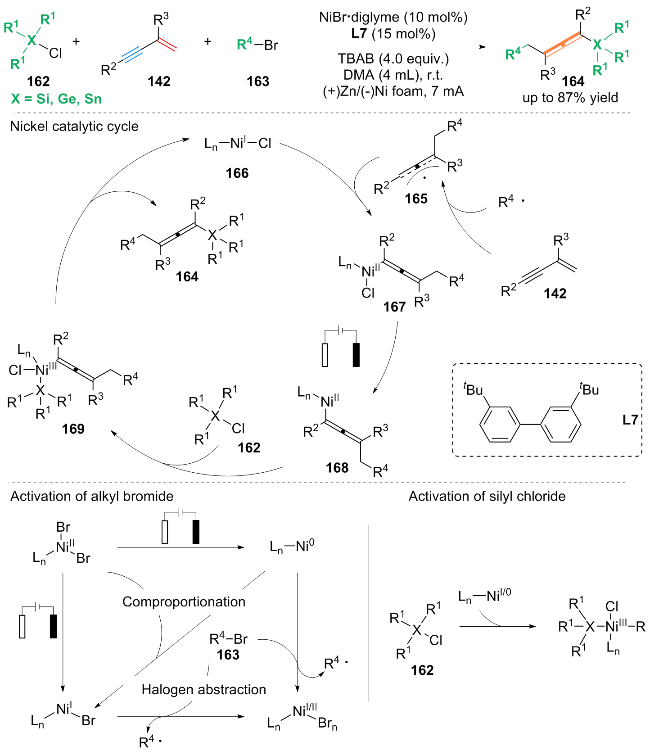
基于自由基验证实验及循环伏安实验等一系列控制实验, 该方案机理分为烷基溴活化的自由基部分、硅氯活化的非自由基部分与镍催化循环部分. (1)烷基溴活化的自由基途径: 配合物Ln-NiII-Br2被还原为NiI或Ni0, NiI或Ni0可以从烷基溴中提取卤素原子生成烷基自由基和NiI/II配合物, 也可以直接在阴极还原生成烷基自由基. (2)电化学条件下, 硅氯活化的非自由基途径: 氯硅烷与阴极还原的镍配合物协同氧化加成. (3)镍催化循环机理: 首先, 生成的烷基自由基加入到1,3-烯炔142的末端sp2碳上, 形成烯基自由基中间体165, 随后165被NiI配合物166捕获, 生成NiII中间体167. 中间体167由阴极还原形成NiI中间体168. 紧接着, 氯化硅配合物氧化加成到中间体168上得到NiIII中间体169. 最后, 中间体169发生还原消除, 产生三组分交叉偶联产物164, 并再生NiI配合物166.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
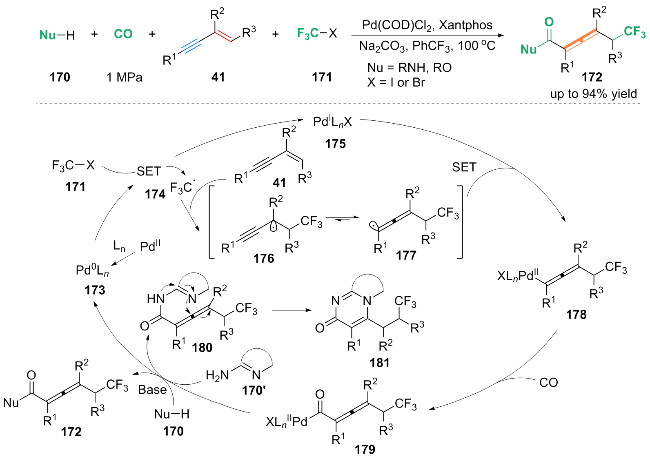
基于机理研究与前期文献报道[39], 作者提出了一种钯介导的自由基循环机理: 首先, Pd(COD)Cl2与配体Ln配位, 生成Pd0Ln配合物173. 然后, 配合物173诱导CF3X发生单电子转移(SET)过程, 生成三氟甲基自由基中间体174和PdILnX配合物175. 随后, 中间体174被1,3-烯炔41捕获, 生成叔丙炔自由基中间体176, 中间体176可以异构化得到相对稳定的丙烯基自由基中间体177. PdILnX物配合物175与丙烯基自由基中间体177配位生成PdII络合物178, 一氧化碳(CO)插入到络合物178中生成中间体179. 中间体179与亲核试剂170反应生成三氟甲基羰基化产物172. 值得注意的是, 双亲核试剂170'与中间体179反应生成中间体180, 180发生丙烯亲核加成得到环化产物181.
4 总结与展望
综上所述, 1,3-烯炔的双官能化反应已取得很大的进展. 本文以1,3-烯炔的1,2-, 3,4-, 1,4-双官能化为主线, 对1,3-烯炔的1,2-双烷基化、1,2-羟基化磷酸化、1,2-烷基化磺酰化、3,4-硼氰化、3,4-碳硼化、3,4-烷基化胺化、3,4-/1,4-芳基化磺酰化、1,4-卤化三氟甲基化、1,4-三氟甲基氨基氰化、1,4-芳基烷基化及1,4-羧化磺酰基化等各种双官能化反应进行总结. 这些反应为构建炔基化合物、丙二烯衍生物及1,3-二烯等重要有机化合物提供了通用而有效的方法. 就目前而言, 1,3-烯炔的双官能团化依旧需要在体系中加入氧化还原剂、过渡金属催化剂或光催化剂等. 在今后的研究中, 开发更加经济、绿色且高效的催化策略, 高化学和区域选择性地实现1,3-烯炔的双官能团化值得期待, 也将更高效绿色地合成炔基化合物、丙二烯衍生物和1,3-二烯等一系列重要的有机化合物.
(Zhao, C.)