


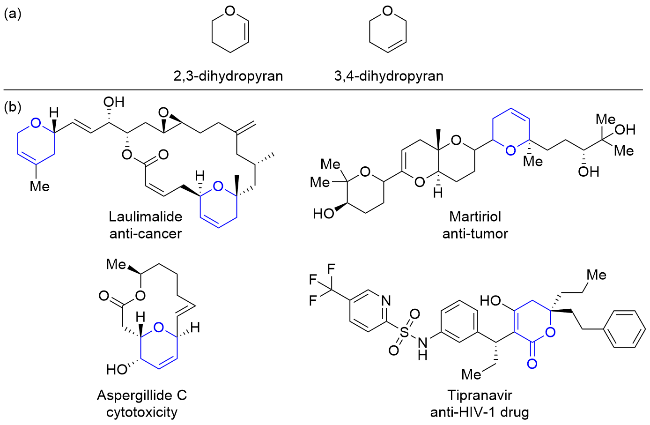
吡喃是含有一个氧原子和两个双键的不饱和六元杂环化合物, 而二氢吡喃是吡喃的重要饱和衍生物之一, 在吡喃环上存在一个双键. 按照双键位置不同, 主要分为2,3-二氢吡喃和3,4-二氢吡喃两大类(图1a). 二氢吡喃结构是大量存在于天然产物和生物活性分子中的一类重要的结构单元, 在有机合成领域也是一类应用广泛的合成前体, 主要用于糖类化合物及天然产物的合成领域[1]. 例如, 从海洋海绵体中分离得到的天然产物Laulimalide[2], 含有两个二氢吡喃结构, 能促进微管蛋白聚合, 具有抗癌活性; 从红藻中分离出具有二氢吡喃结构的天然产物Martiriol[3], 具有抗肿瘤的作用; 从海洋真菌中分离出来的含有二氢吡喃的大环内酯类化合物 Aspergillide C[4], 具有显著的细胞毒性; 由二氢吡喃环氧化得到的二氢吡喃内酯Tipranavir[5], 是一种人类免疫缺陷病毒1型(HIV-1)蛋白酶抑制剂(图1b). 因此, 对于二氢吡喃类化合物的合成尤其是对其环上碳原子的立体选择性控制一直以来吸引了许多化学家的密切关注.
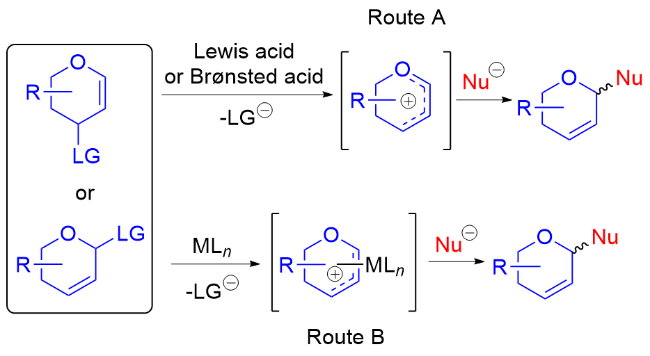
传统上合成二氢吡喃类化合物的方法主要包括: (1)通过Ferrier重排反应或Achmatowicz重排反应构建二氢吡喃骨架, (2)通过Hetero-Diels-Alder (HAD)反应构建二氢吡喃骨架, (3)通过烯烃复分解(RCM)构建二氢吡喃骨架等. 上述方法已经被包括作者在内的国内外诸多学者进行过系统梳理和全面总结[6], 但是目前较少有专门关于二氢吡喃骨架修饰构建其官能化衍生物的系统性综述总结. 二氢吡喃类化合物可在一定条件下与各种含氧、碳、氮基亲核试剂反应, 得到相应的氧基、碳基、氮基二氢吡喃类衍生物, 这些衍生物可进一步转化为相应的氧苷糖、碳苷糖和氮苷糖等. 此外, 对于硫基二氢吡喃类衍生物的合成与转化制备硫苷糖也有少量文献报道[7]. 一般来说, 使用二氢吡喃类衍生物作为糖基供体, 大致通过两种反应机理制备相应衍生物[8](如Scheme 1): 路径A使用路易斯酸或者布朗斯特酸促进二氢吡喃类衍生物离去基团离去, 形成碳正离子或氧鎓离子中间体, 之后被亲核试剂进攻得到相应的产物; 路径B在金属催化剂的作用下, 通过离去基团的离去得到π-烯丙基-金属中间体, 之后再被亲核试剂进攻得到相应的产物.
在本文中, 我们将主要综述氧基二氢吡喃类衍生物、碳基二氢吡喃类衍生物和氮基二氢吡喃类衍生物的合成研究进展, 探讨相关反应机理及应用, 并对该领域的发展前景进行总结与展望.
1 氧基二氢吡喃类衍生物的研究进展
1.1 氧基2,3-二氢吡喃类衍生物的研究进展
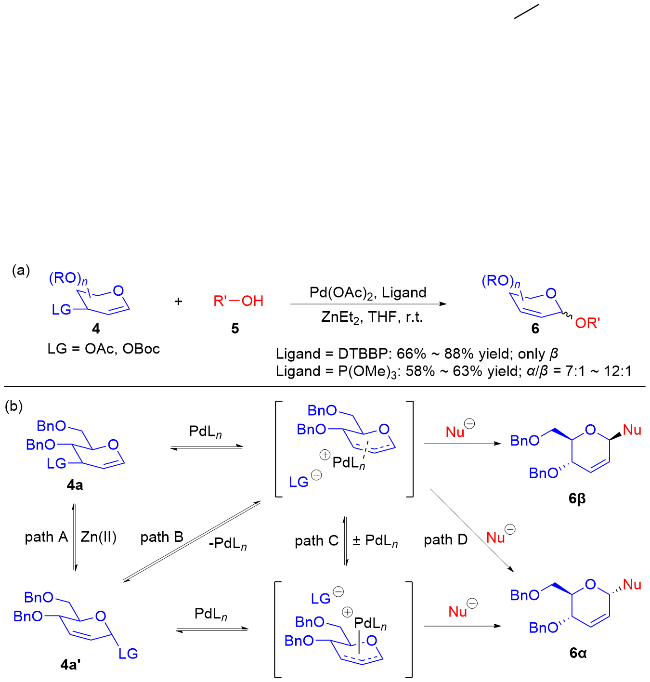
2004年, Lee等[10]使用2,3-二氢吡喃衍生物作为糖基供体, 在钯催化下实现了O-糖基化反应. 其中, 使用Pd(OAc)2作为催化剂, 引入ZnEt2添加剂既能促进亲核加成又有助于离去基团的离去. 另外, 该反应的立体选择性完全由配体结构控制, 当使用2-(叔丁基)-2-联苯膦(DTBBP)作为配体时, 得到了完全β构型的O-苷糖; 而使用P(OMe)3作为配体时, 以α-构型的O-苷糖为主要产物(Scheme 3a). 从反应机理可知, 获得产物的构型主要由钯-配体与底物的配位方式, 以及亲核试剂进攻方式所决定. 此外, 作者推测了四条路线A~D获得α-构型产物可能的反应路径: 使用P(OMe)3作为配体, 这些反应路径可能会相互竞争; 而当DTBBP为配体时, 只保留了直接从4a→6β的反应机制(Scheme 3b).
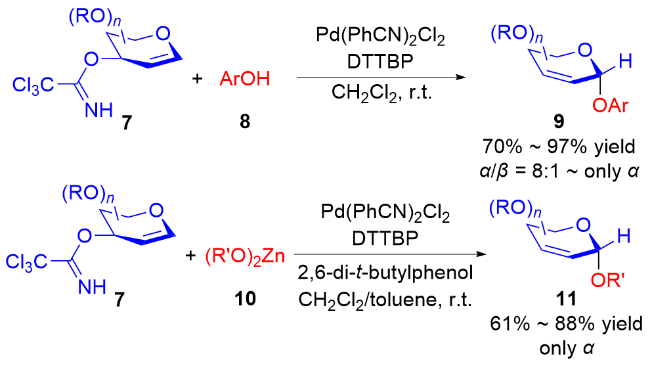
2007, Nguyen小组[11]使用钯-配体-糖基供体络合物的策略来控制二氢吡喃底物上的立体化学, 以较高的立体选择性合成了α-O-苷糖. 以Pd(PhCN)2Cl2作为催化剂, 2-二-叔丁膦基-2',4',6'-三异丙基联苯(DTTBP)作为配体, C(3)-三氯乙酰亚胺酸酯基作为离去基团, 多种芳基醇8作为糖基受体直接参与反应, 以较好的产率和良好的α-选择性获得O-苷糖9. 对于活性较低的脂肪醇, 则需要使用更为活泼的锌(II)烷氧基化合物10作为反应底物, 并且以2,6-二叔丁基苯酚作为质子供体促进反应进行, 最终伯醇、仲醇等均可顺利参与反应, 得到单一立体选择性的α-构型产物11 (Scheme 4).
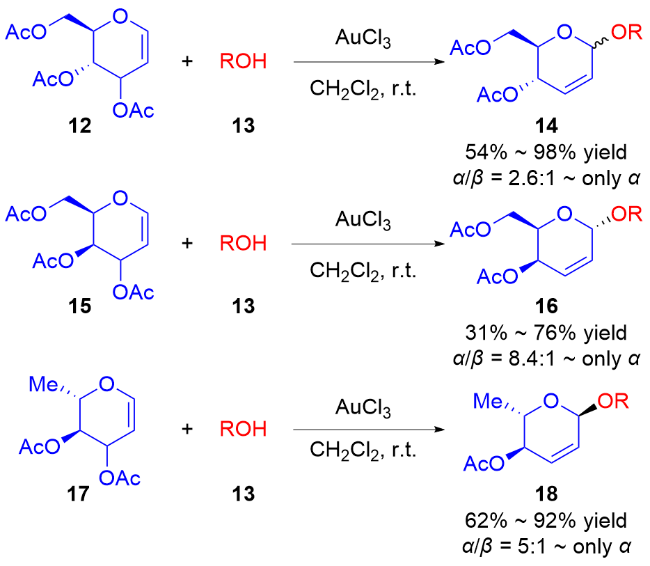
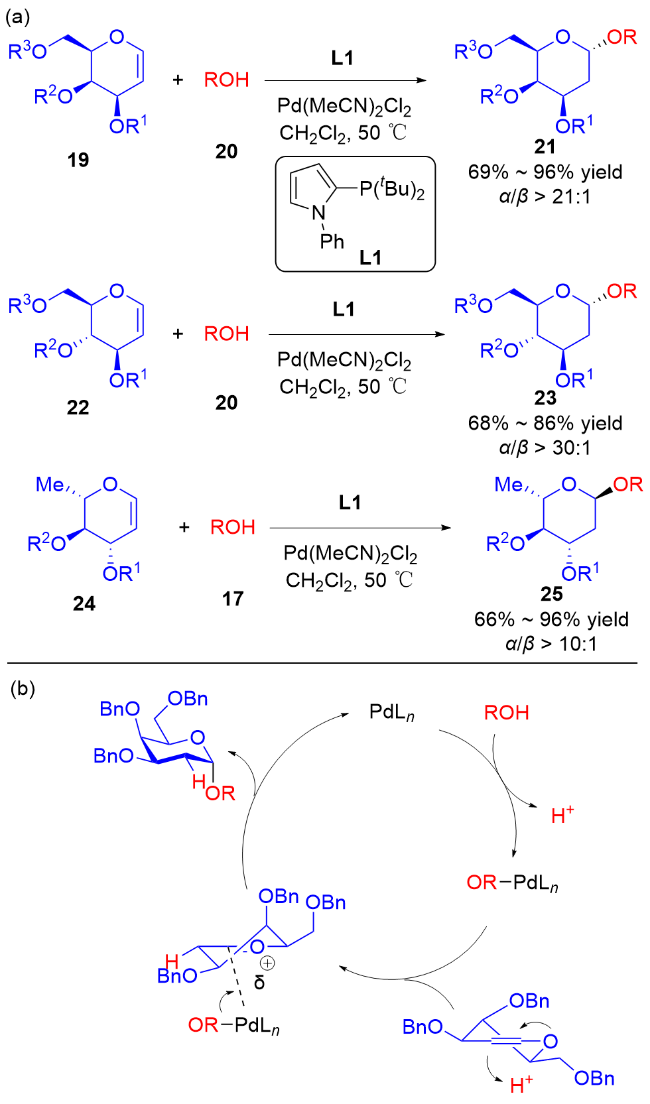
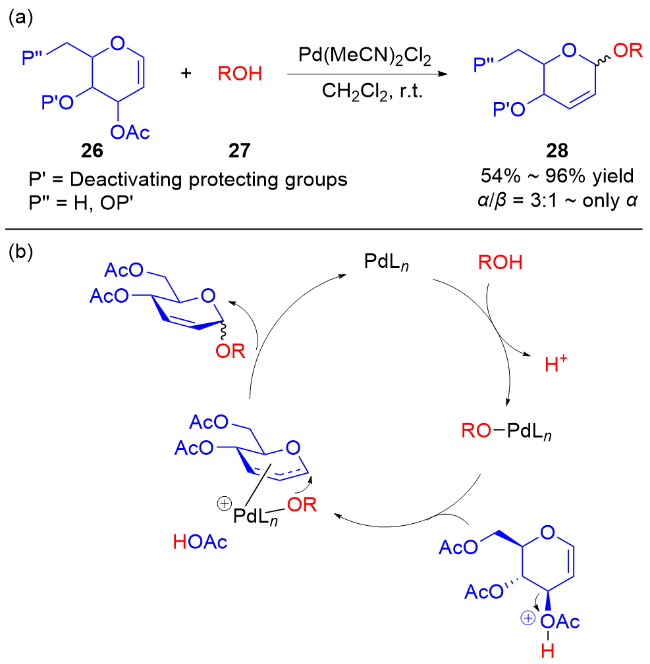
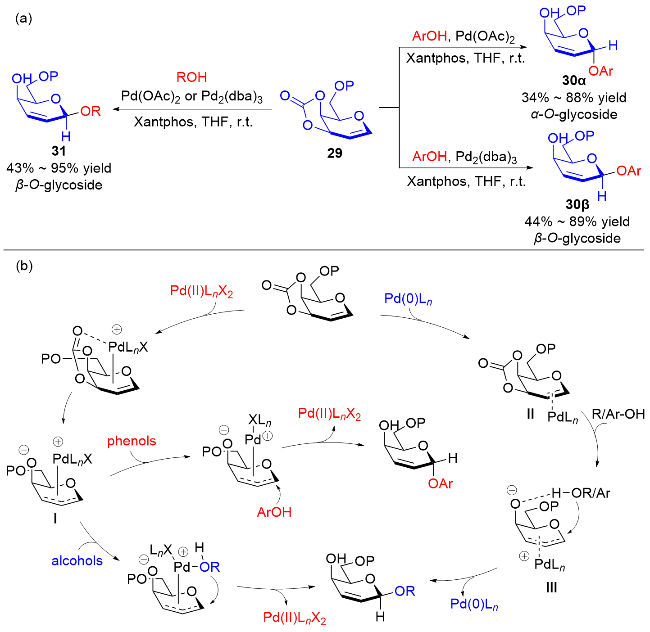
2017年, Liu课题组[15]使用3,4-O-碳酸酯半乳苷糖烯29为糖基供体, 通过使用不同的价态的钯催化剂[Pd(0)和Pd(Ⅱ)]实现了各种糖苷键的立体选择性构建. 当使用Pd(OAc)2为催化剂时, 脂肪醇会与Pd(Ⅱ)在β面上配位, 从而得到β-O-苷糖产物31, 使用芳香醇则会获得α-O-苷糖产物30α; 以Pd2(dba)3作为催化剂时, 由于空间位阻效应只能在β面进行配位, 无论是脂肪醇还是芳香醇都只能得到β-O-苷糖产物30β和31 (Scheme 8a). 根据实验结果, 他们提出了可能的反应机理: Pd(Ⅱ)更倾向于与二氢吡喃的双键直接配位, 通过碳酸酯的导向作用脱羧生成中间体Ⅰ, 钯配合物会阻碍氢键的生成, 并通过两个路径获得不同构型的产物. 与之相反, Pd(0)与膦配体配位后体积太大, 无法在β面与二氢吡喃的双键配位, 因此Pd(0)配合物与二氢吡喃的双键在反面配位形成中间体Ⅱ, 脱羧后由于氢键的作用使脂肪醇或酚类受体在β面进攻π-烯丙基钯中间体Ⅲ, 得到β-O-苷糖 (Scheme 8b).
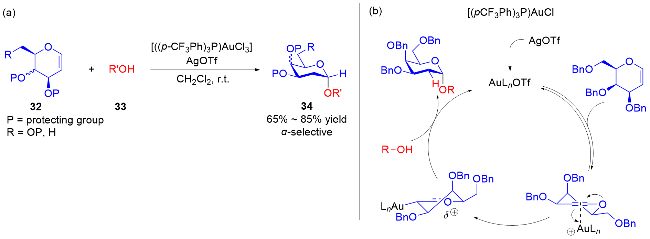
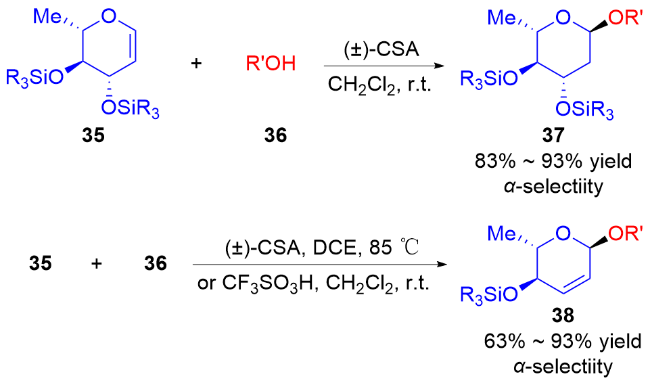
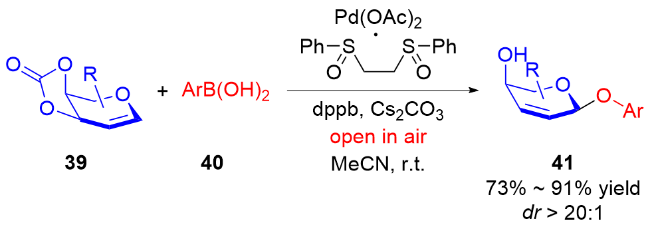
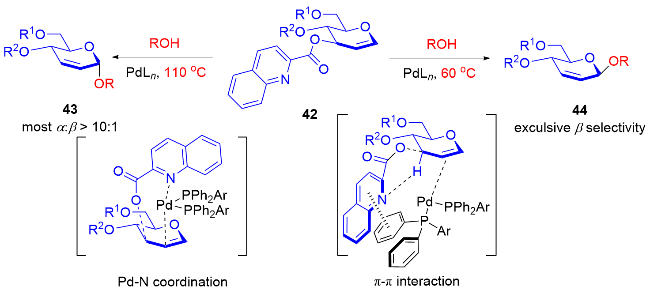
随着自由基化学在有机合成领域的复兴, 最近, 叶新山、熊德彩等[20]使用光诱导或电促进等方式, 通过产生活性自由基中间体制备了一系列氧基2,3-二氢吡喃类衍生物.
1.2 氧基3,4-二氢吡喃类衍生物的研究进展
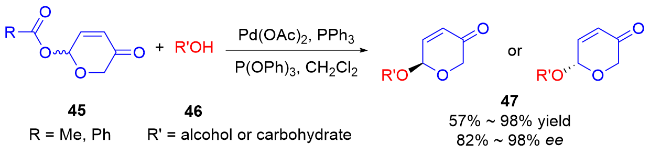
2003年, O'Doherty等[22]采用与Feringa课题组类似的方法, 在三(二亚苄基丙酮)二钯/氯仿加合物[Pd2-(dba)3•CHCl3]的催化作用下, 选择性地将α-2-取代6-酯基-2H-吡喃-3(6H)-酮48α转化为立体构型完全保持的α-2-取代6-烷氧基-2H-吡喃-3(6H)-酮50α, 同样地将含有β-酯基的吡喃酮48β转化为含有β-烷氧基的吡喃酮50β (Scheme 14a). 该类二氢吡喃酮碳酸酯是通过Achmatowicz重排产物进行酰化得到的. 首先, 糠醇51在N-溴代琥珀酰亚胺(NBS)的作用下发生Achmatowicz重排, 得到羟基无保护的二氢吡喃酮52, 之后进行酰化反应获得相应的二氢吡喃酮碳酸酯类化合物53 (Scheme 14b).
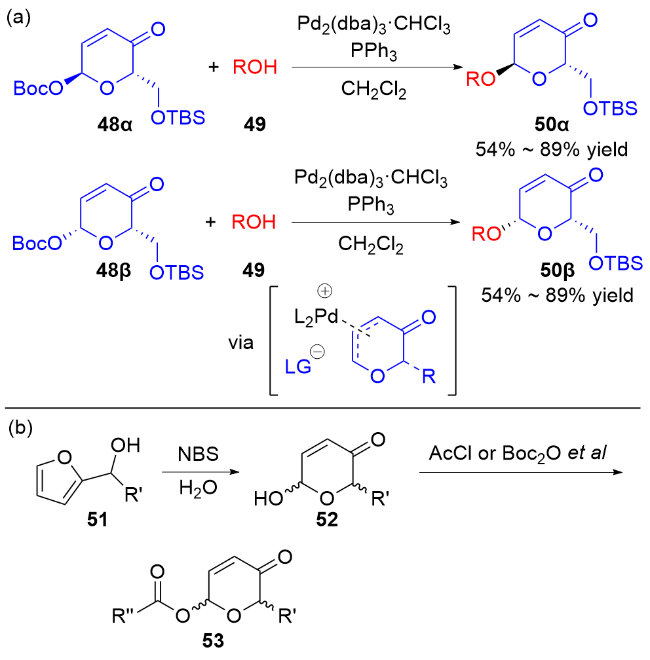
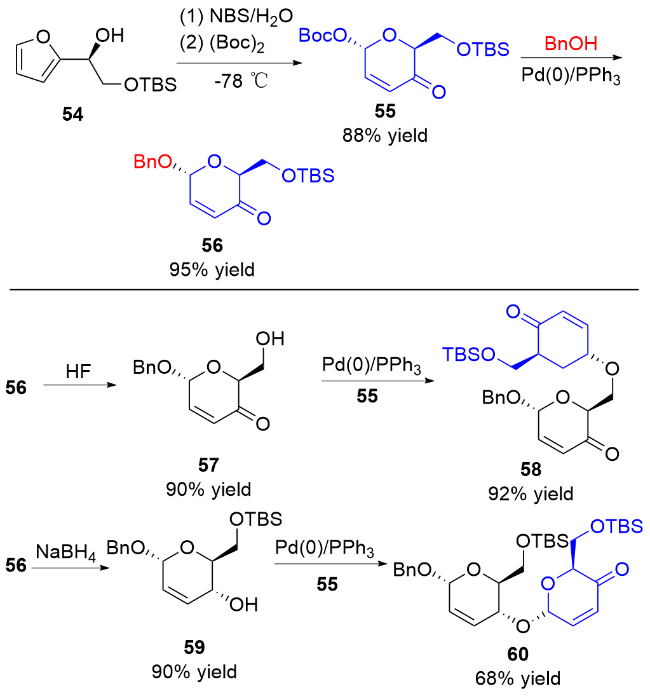
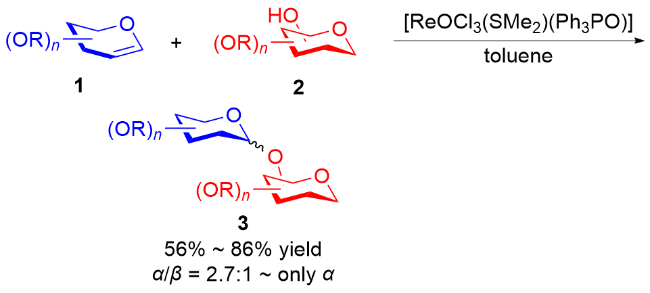
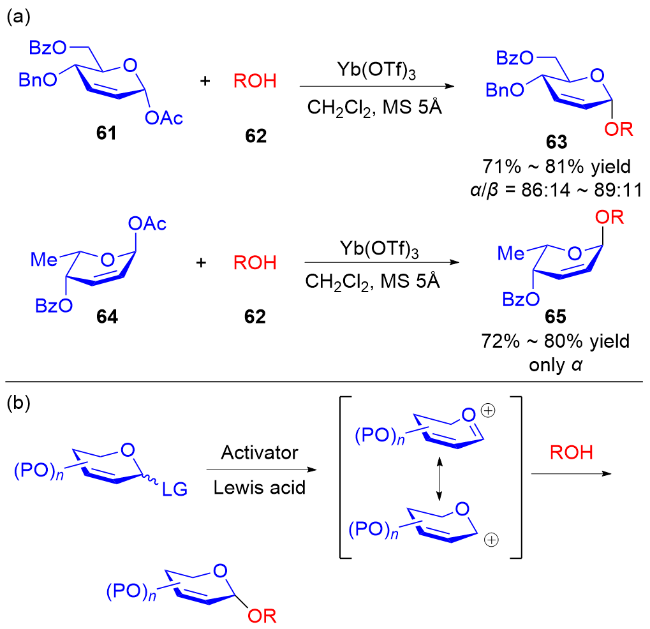
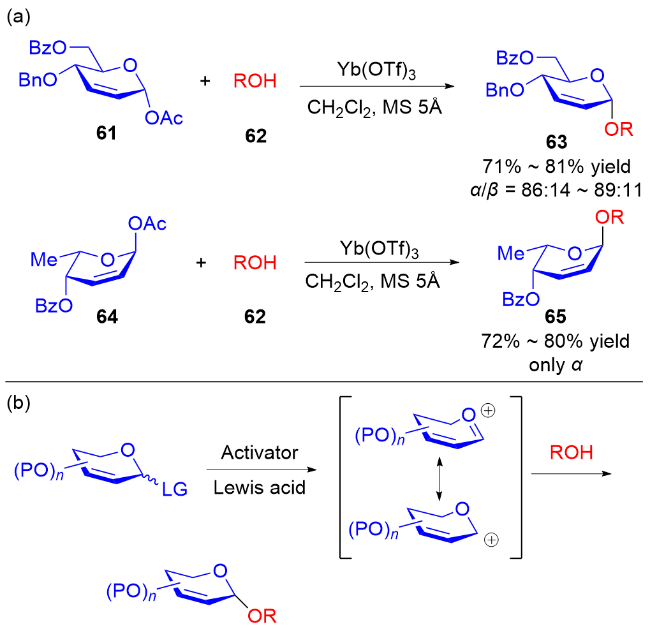
2006年, Toshima等[24]通过模拟溶菌酶引发的水解反应, 设计了以3,4-二氢吡喃类衍生物为糖基供体的策略. 在Yb(OTf)3的活化下, 醇62作为亲核试剂与3,4-二氢吡喃化合物61和64反应, 以良好的立体选择性获得了相应的O-苷糖63和65 (Scheme 16a). 他们认为二氢吡喃乙酸酯经过路易斯酸的活化, 生成稳定的氧鎓离子或碳正离子, 之后被亲核试剂捕获得到最终产物 (Scheme 16b).
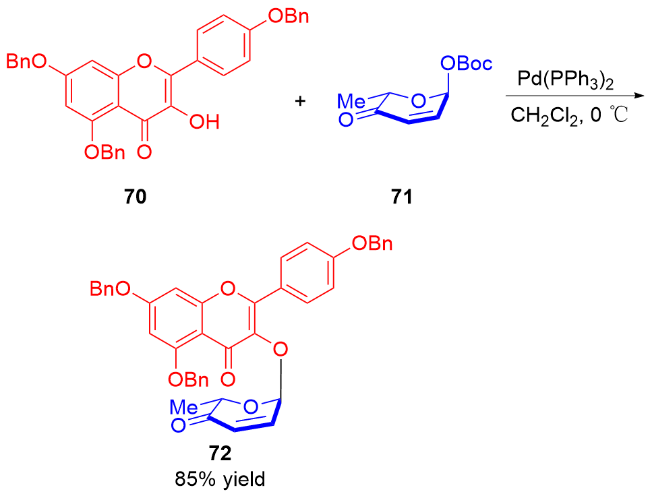
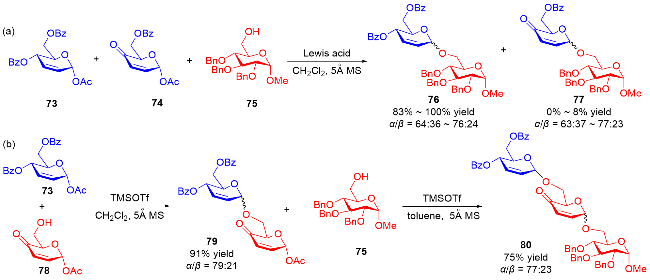
2010年, Toshima课题组[27]发现使用路易斯酸作为催化剂, 75作为糖基受体选择性地与3,4-二氢吡喃73(非3,4-二氢吡喃酮74)发生糖基化反应(Scheme 19a). 于是, 他们利用该特性, 在TMSOTf的催化作用下, 通过两步串联反应, 成功实现了三糖80的合成. 通过改变溶剂防止了第一个糖苷键的断裂, 并提高了第二步α-糖苷化的立体选择性(Scheme 19b).
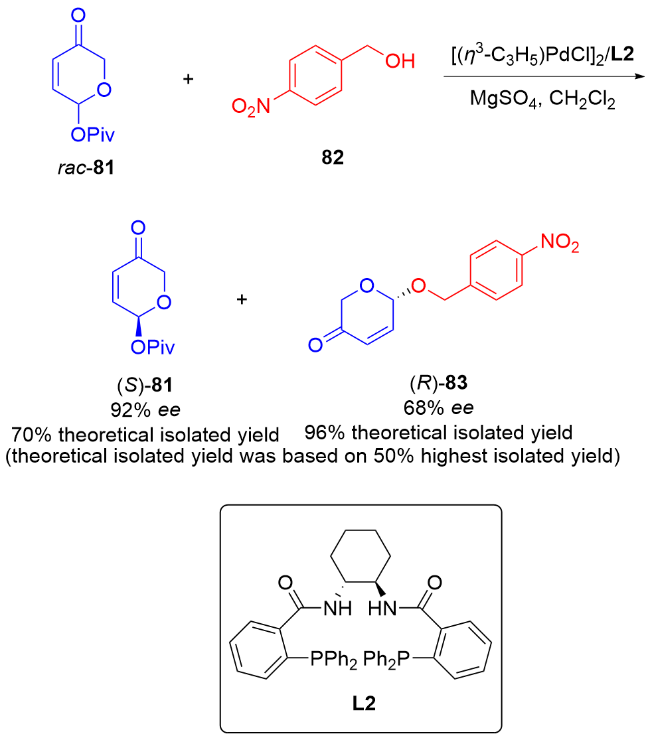
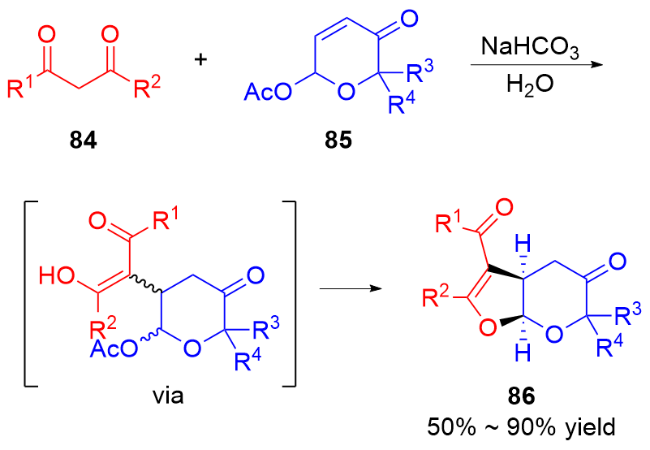
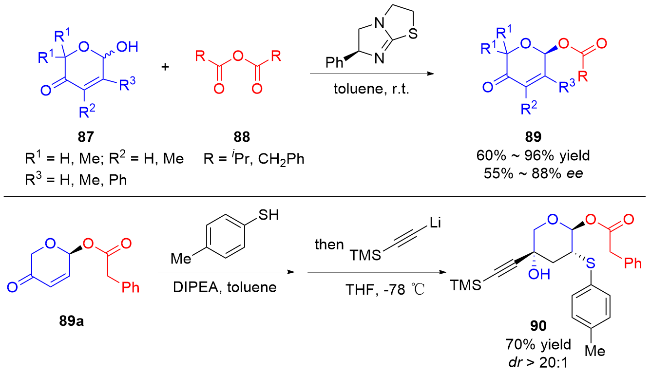
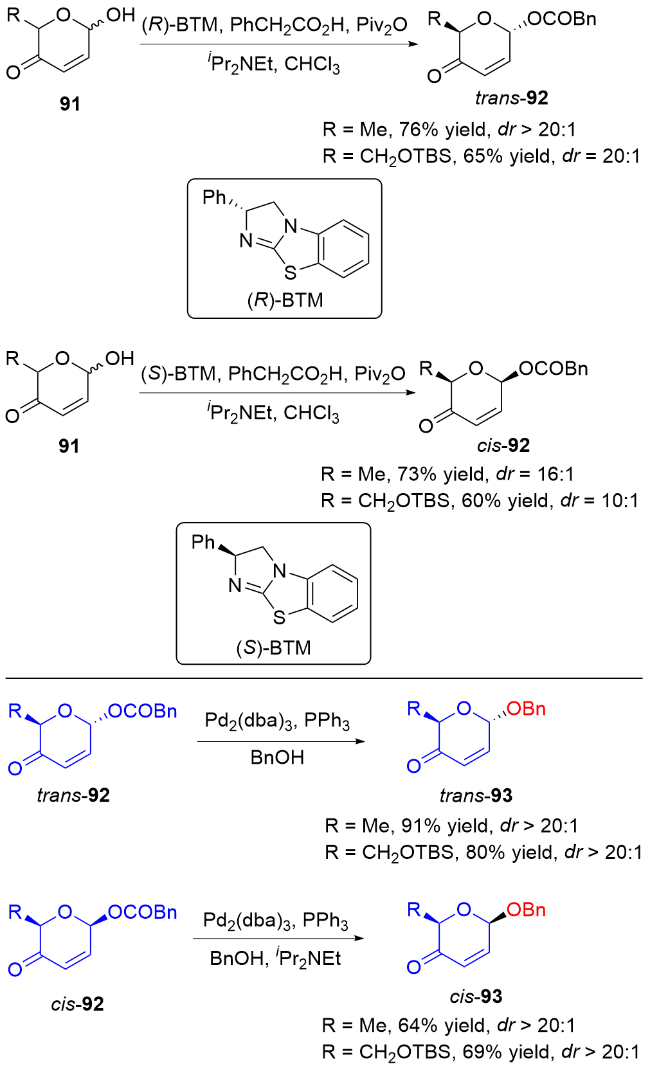
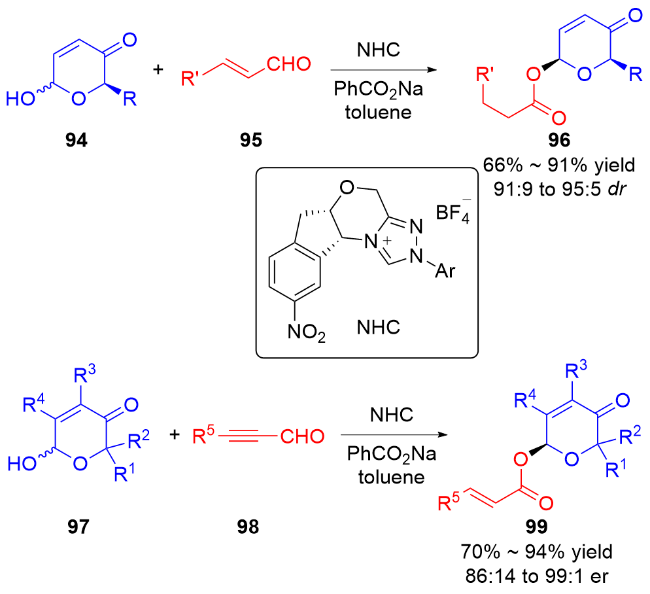
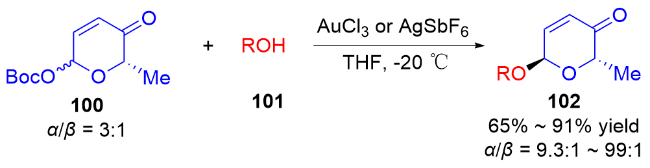
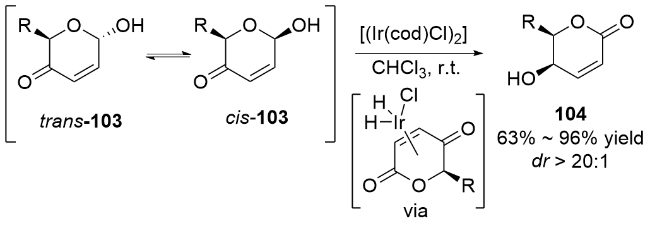
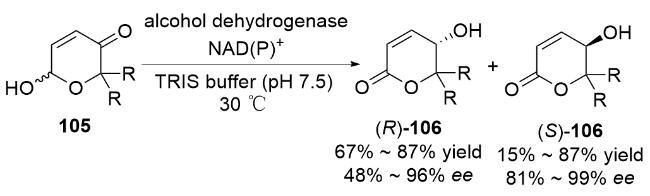
2 碳基二氢吡喃类衍生物的研究进展
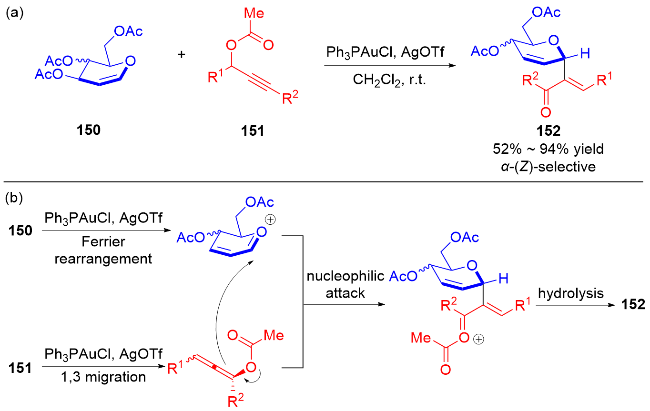
目前, 已有综述详细总结了基于糖烯的碳苷合成方法研究进展[36], 本文仅总结烯烃结构处无额外取代基或官能团的碳基二氢吡喃类衍生物的研究进展.
2.1 碳基2,3-二氢吡喃类衍生物的研究进展
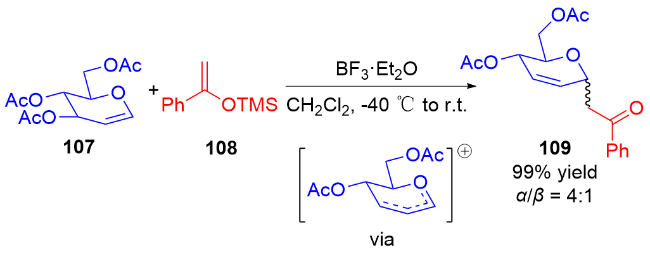
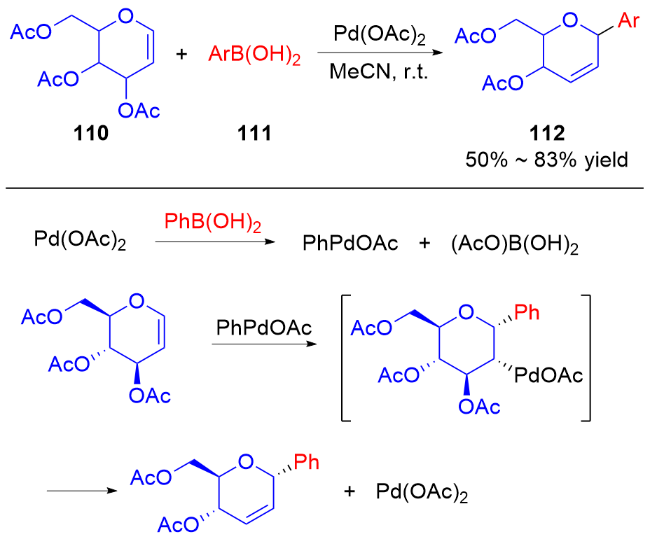
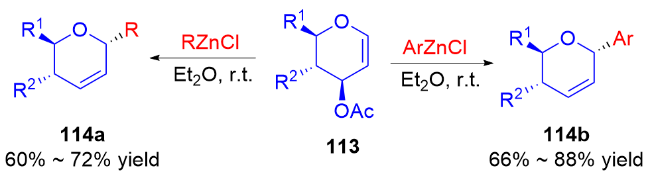
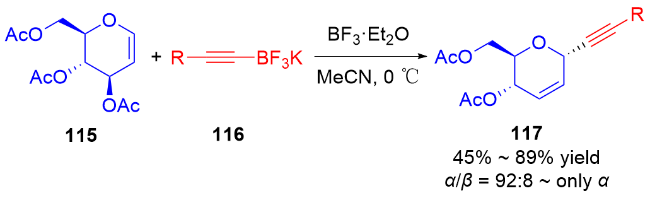
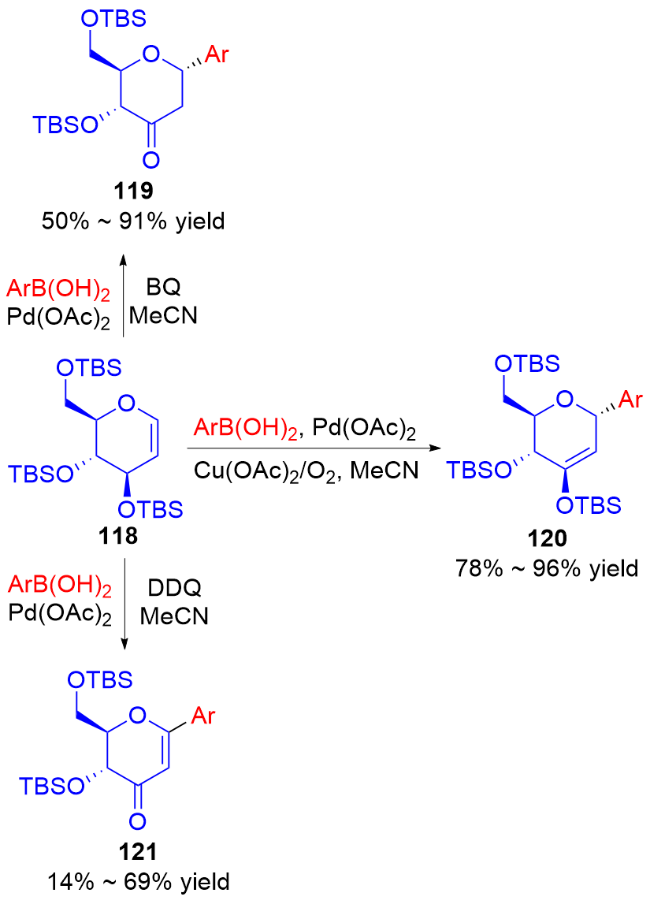

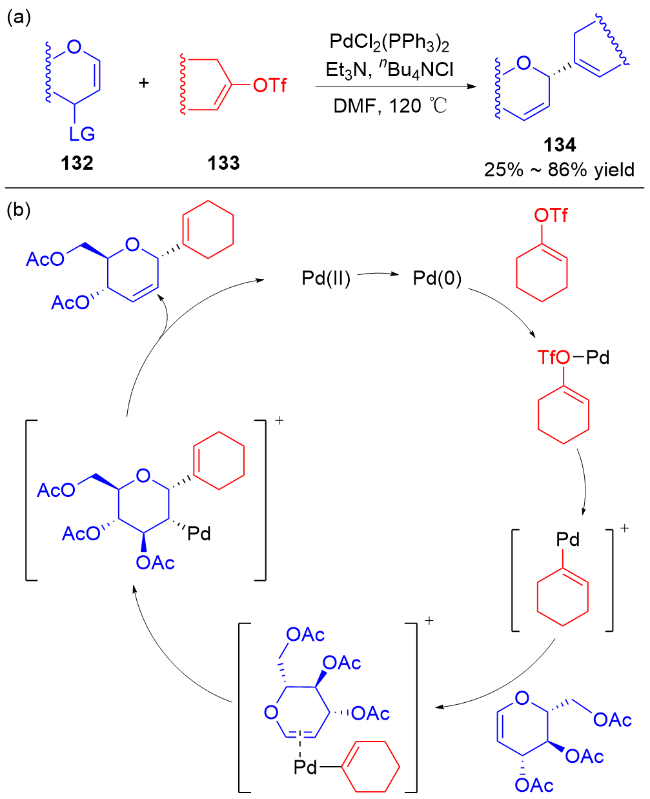
2011年, Liu等[45]开发了一种高效的钯催化三氟甲磺酸烯醇酯糖基化反应的方法. 使用PdCl2(PPh3)2为催化剂, 在Et3N和nBu4NCl作用下, 多种具有离去基团的2,3-二氢吡喃132与各种三氟甲磺酸烯醇酯133发生偶联反应, 以较高区域和立体选择性得到一系列α-碳基二氢吡喃类衍生物134 (Scheme 36a), 烯基官能团的引入也为后续转化提供了可能. 作者认为在该反应中, 首先Pd(II)还原Pd(0), 之后通过氧化加成形成烯基-Pd阳离子络合物, 然后该络合物与二氢吡喃上的双键配位从而形成Pd-糖烯络合物, 最后发生反式-β-消除得到α-C-苷糖并释放Pd(II) (Scheme 36b).


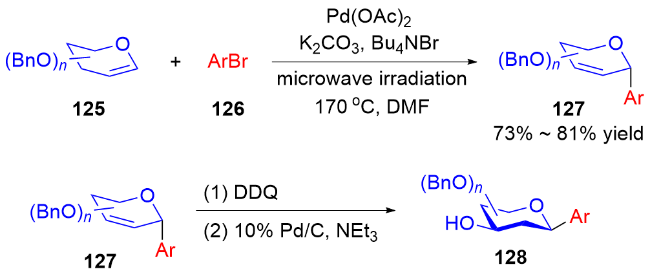
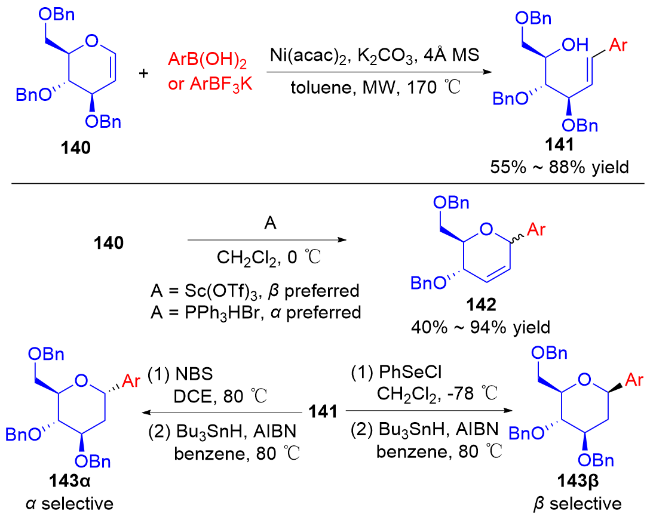
2014年, 叶新山课题组[48]采用“开环-闭环”的策略, 实现了多种C-芳基糖苷的合成. 首先, 在微波辅助下, 使用Ni(acac)2作为催化剂, 二氢吡喃化合物140与不同的芳基硼酸或芳基三氟硼酸钾反应, 区域选择性地消除糖醛的β-O, 得到开环产物141. 从开环产物出发, 可以在Sc(OTf)3或Ph3PHBr作用下发生闭环反应, 立体选择性地合成α或β-C-芳基-Δ2,3-苷糖142; 或者通过PhSeCl介导的环化反应, 用Bu3SnH/AIBN成功合成β-D-芳基-C-苷糖143β. 而α构型的苷糖143α可由NBS介导的环化反应, 经Bu3SnH/AIBN作用后被立体选择性合成(Scheme 39).

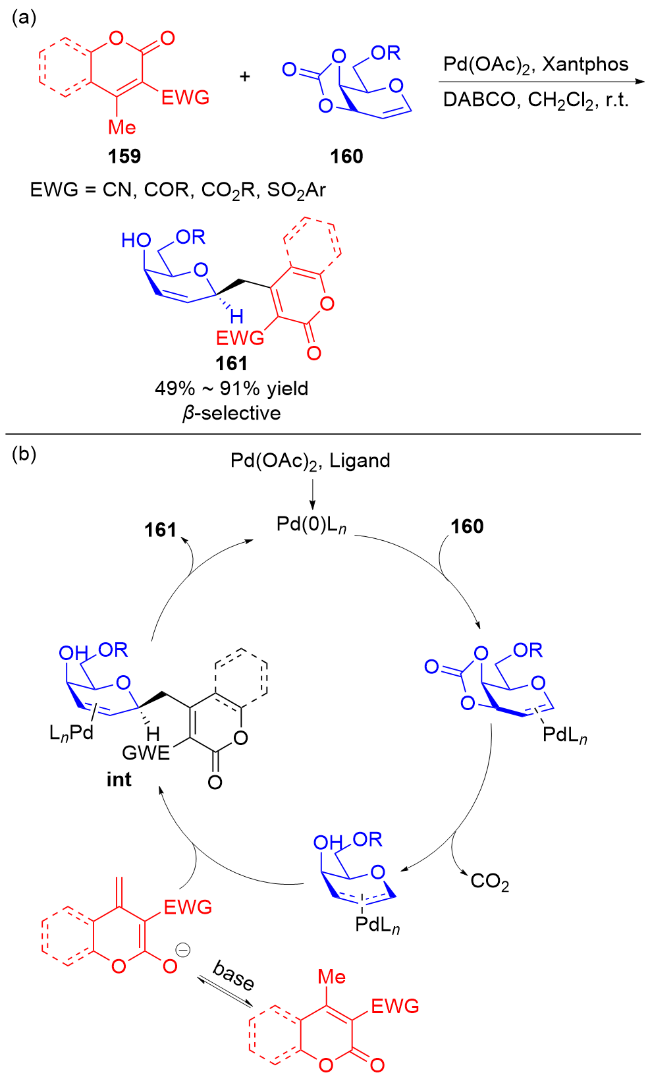
2019年, Zhang课题组[55]报道了钯催化香豆素159与二氢吡喃化物160的区域和立体选择性反应. 室温下在二氯甲烷中, 以及Pd(OAc)2、Xantphos和三乙烯二胺(DABCO)存在下, 多种香豆素159与二氢吡喃化物160均可发生反应, 得到β-C-烯基苷糖161 (Scheme 45a), 并实现了克级制备. 机理研究表明: Pd(II)首先被膦配体还原为Pd(0), Pd(0)在二氢吡喃的α-面与双键进行配位, 之后经脱羧形成π-烯丙基-Pd(II)中间体, 香豆素与π-烯丙基-Pd(II)中间体发生亲核加成反应生成络合物int. 最后释放产物161并实现Pd(0)的再生(Scheme 45b).
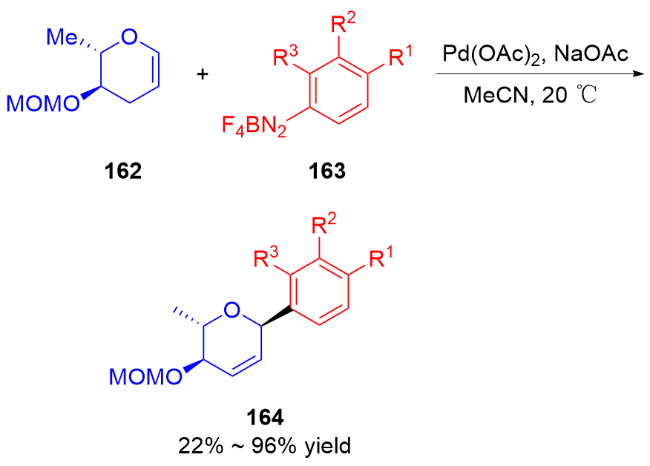
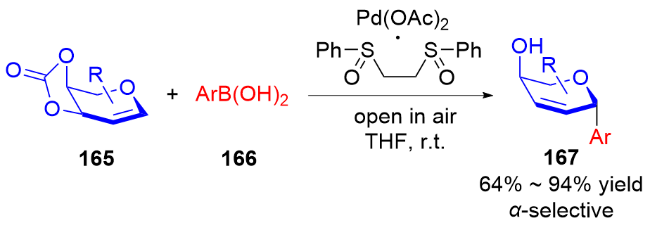

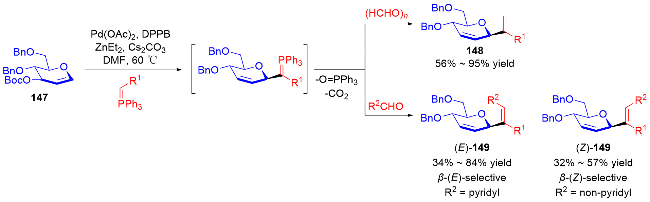
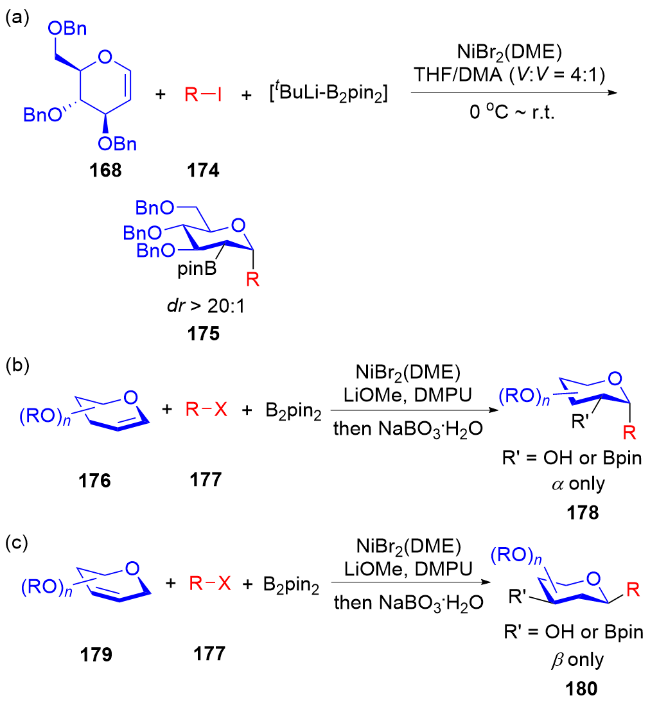
2024年, Brown课题组[59]报道了镍催化二氢吡喃168、碘代烃174和叔丁基锂-联硼酸频那醇酯(tBuLi-B2pin2)的多组分偶联反应, 实现了高立体选择性地在糖烯类化合物的C2、C3位同时形成C—C键和 C—B键的反应, 制备含硼α-C-苷糖衍生物175. 该反应具有底物范围广, 产物可以转化为多种单糖化合物等优点(Scheme 49a). 随后, 钱德云等[60]和李阳阳、阴国印 等[61]分别独立报道了底物范围更广泛的镍催化2,3-二氢吡喃176、卤代烃177和联硼酸频那醇酯(B2pin2)的多组分偶联反应, 高选择性地在糖烯类化合物的C2、C3位同时形成C—C键和C—B键的反应, 制备含硼α-C-苷糖衍生物178 (Scheme 49b). 值得注意的是, 当以3,4-二氢吡喃179为反应底物时, 能够实现高选择性地在糖烯类化合物的C2、C4位同时形成C—C键和C—B键的反应, 制备含硼β-C-苷糖衍生物180 (Scheme 49c).
2.2 碳基3,4-二氢吡喃类衍生物的研究进展
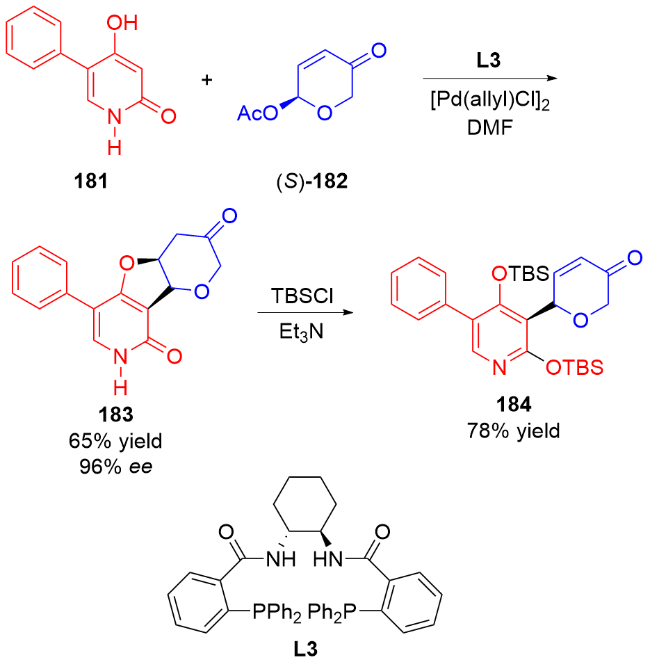
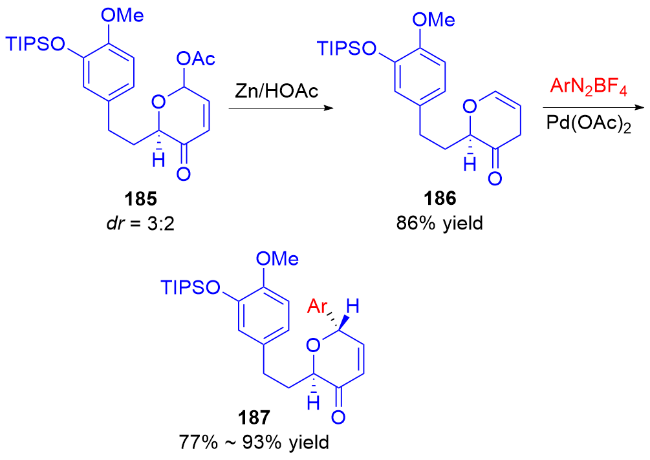
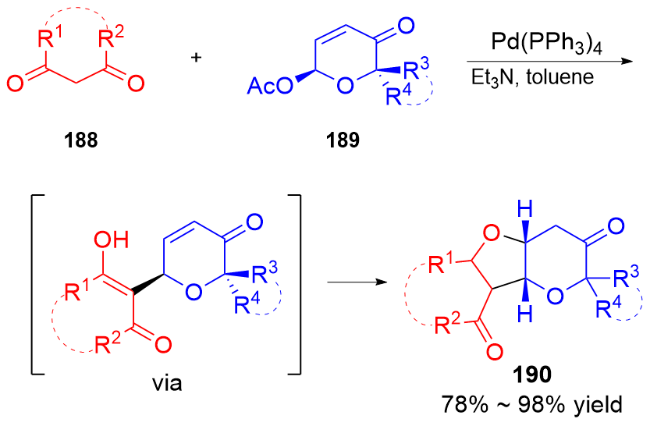
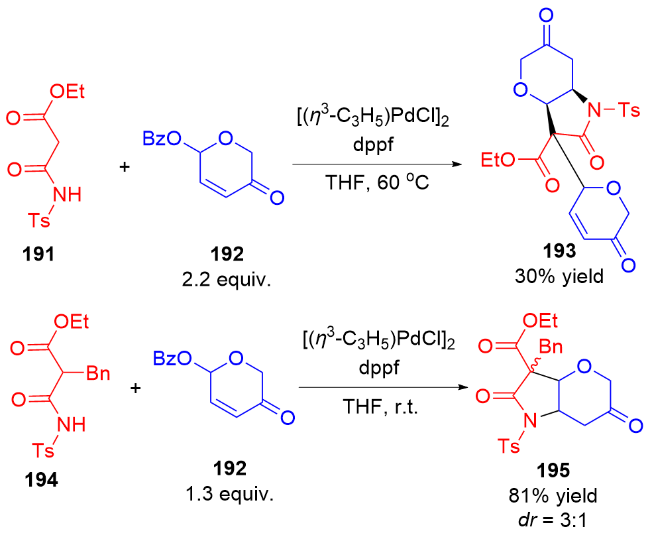
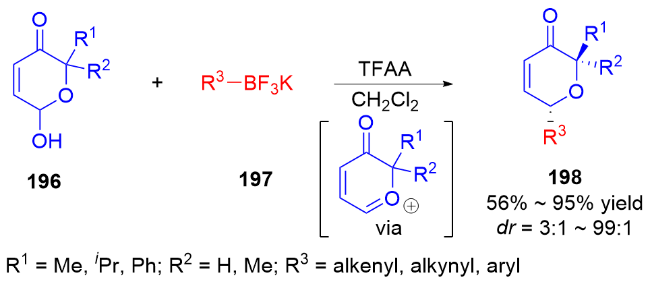
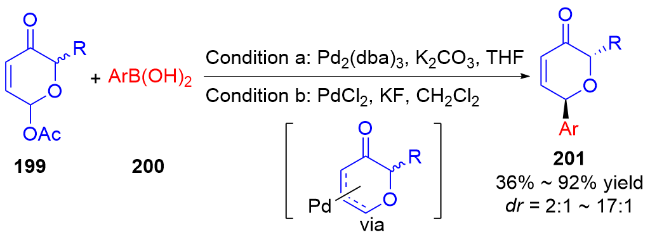
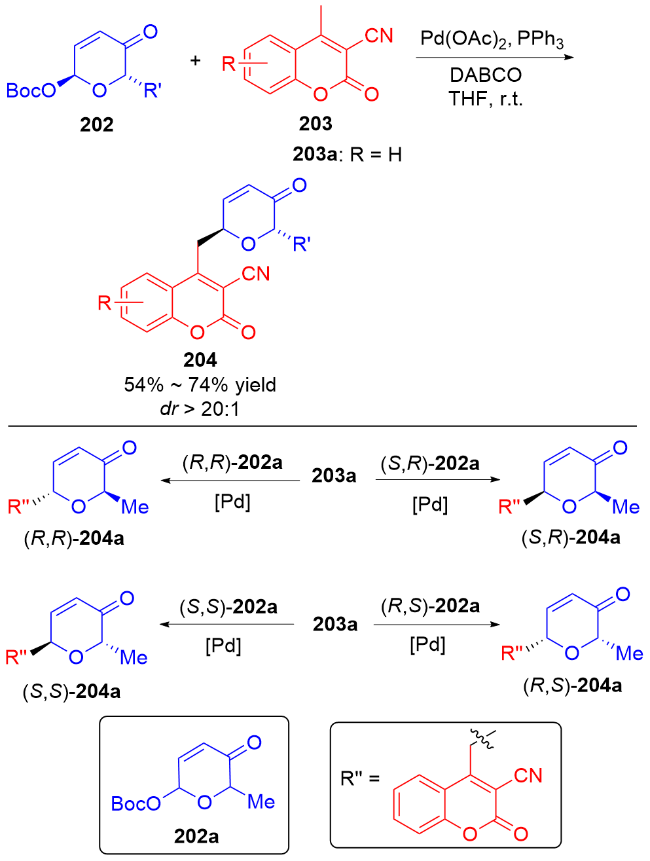
3 氮基二氢吡喃类衍生物的研究进展
3.1 氮基2,3-二氢吡喃类衍生物的研究进展
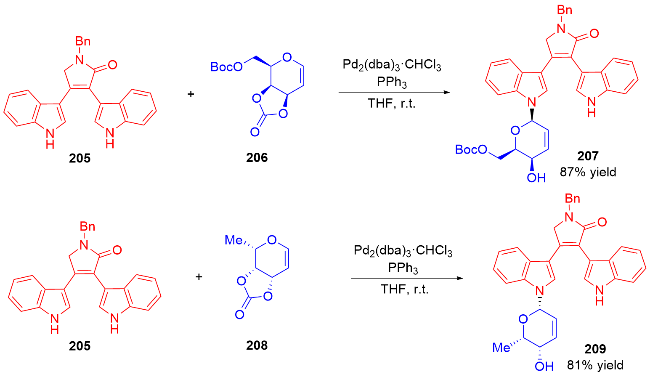
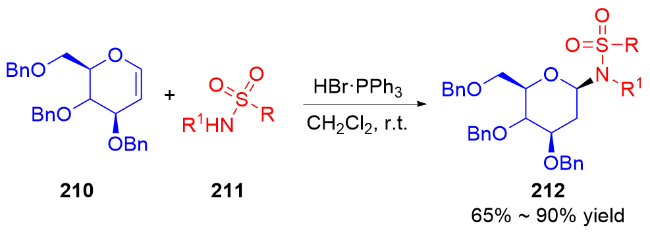
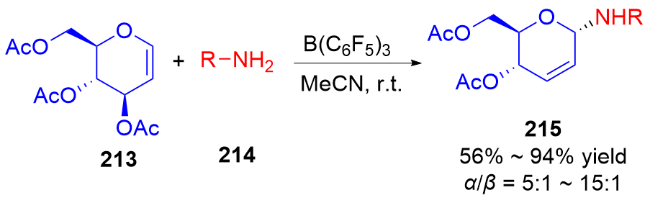
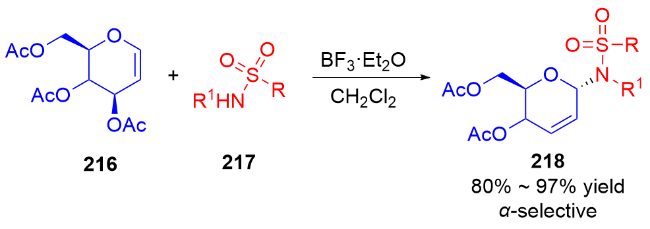
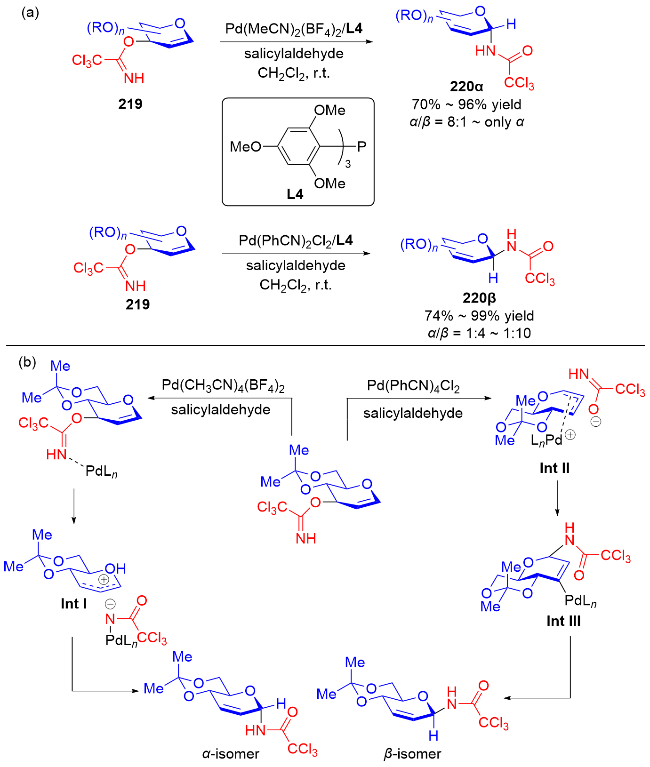
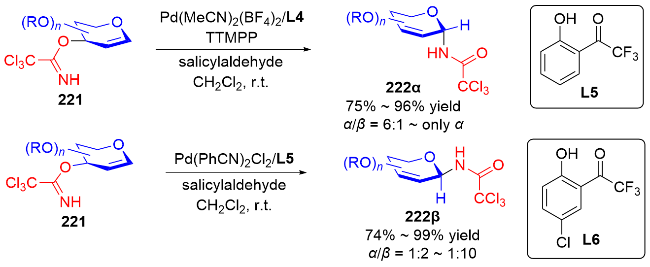
2007年, Nguyen课题组[73]发展了一种Pd(II)催化立体选择性合成α-和β-N-二氢吡喃基三氯乙酰胺的方法. 反应在二氯甲烷中进行, 使用水杨醛作为添加剂, 以三氯亚胺酯219为底物, 通过钯-配体作用控制异头碳的立体选择性, 使用阳离子型钯催化剂Pd(MeCN)4(BF4)2提高了产物的α-选择性, 而中性钯催化剂Pd(PhCN)2Cl2更有利于β构型产物的生成(Scheme 61a). 机理研究表明: 对于阳离子型催化剂, Pd(MeCN)4(BF4)2-水杨醛配合物与三氯亚胺酯的氮原子配位, 随后离去生成烯丙基阳离子Int I, 三氯酰胺区域选择性地加成到Int I的α面上, 最终得到α-构型产物220α. 相反, 使用Pd(Ph-CN)2Cl2-水杨醛配合物会促进分子内环化诱导的重排反应发生, 钯催化剂与底物的双键配位形成π-烯丙基-钯络合物Int II, 该络合物被亚胺亲核进攻, 随后Int II发生环化得到σ-复合物Int III, 最后发生Grob断裂反应解离释放β-构型产物220β (Scheme 61b).
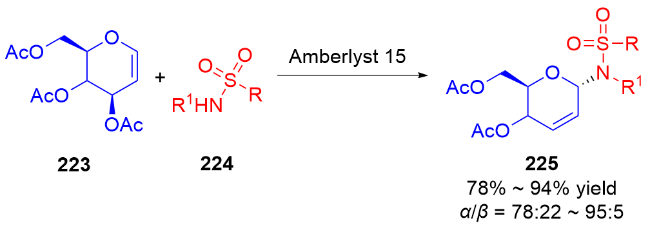
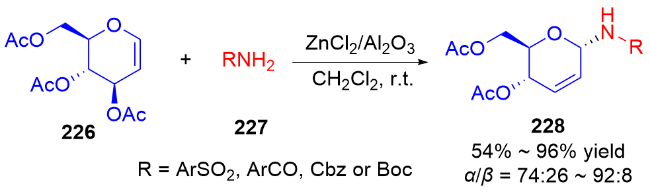
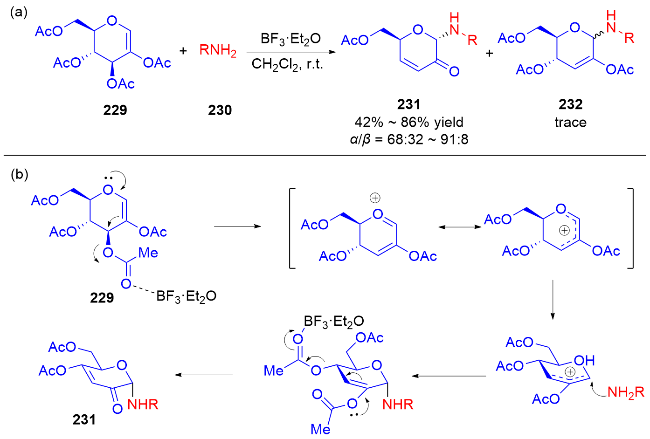
2013年, Liu课题组[77]报道了一种路易斯酸催化合成N-氨基烯酮类化合物的方法. 在CH2Cl2中, BF3•Et2O作为催化剂, 实现了二氢吡喃229与各种含氮亲核试剂230的反应, 结果没有得到预期的2-氨基-3-OAc取代的不饱和吡喃232, 而是观察到生成重排后的N-氨基烯酮类化合物231, 并具有优异的α-立体选择性(Scheme 65a). 他们推测可能的反应机理: 首先, 化合物229与路易斯酸配位, 可以容易地消除乙酰氧基, 同时双键发生迁移(费里尔重排), 在C-1处产生阳离子, 得到氧鎓离子或碳正离子中间体. 含氮亲核试剂从α-面进攻, 得到具有α-构型的二氢吡喃. 最后该中间体经过酯解得到二氢吡喃酮231 (Scheme 65b).
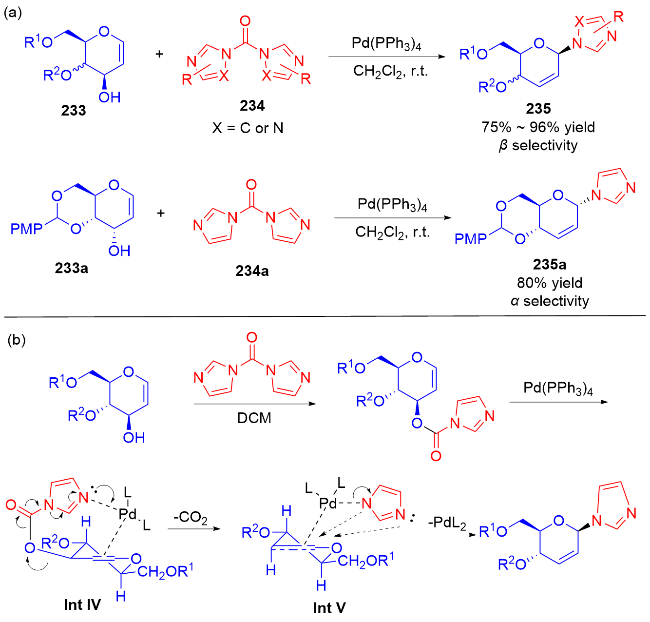
2014年, Liu等[78]通过一锅法合成了β-N-咪唑基二氢吡喃衍生物. 该反应以Pd(PPh3)4催化的脱酸烯丙基取代反应为基础, 经过酯化、脱羧和烯丙取代三步反应, 对含不同的保护基团的糖烯223和不同的羰基二咪唑类似物234都能以良好的产率和立体选择性, 得到了相应的产物235. 另外, 通过改变二氢吡喃C3位的手性可实现对底物α-和β-选择性的控制(Scheme 66a). 根据实验结果, 他们提出了该反应的机理: 首先, 二氢吡喃233与羰基二咪唑234a反应生成酯基中间体. 之后, 在Pd(PPh3)4催化下, 由于二氢吡喃双键和氮孤对电子与钯催化剂的双重配位效应, 导致关键中间体Int IV与钯催化剂在β-面配位. 随后, 在钯催化剂的促进下通过脱羧反应生成中间体Int V. 最后, 通过亲核加成得到具有β-选择性的产物(Scheme 66b).
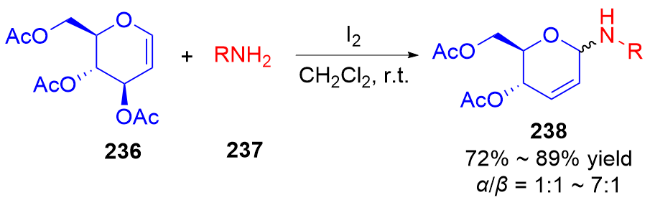
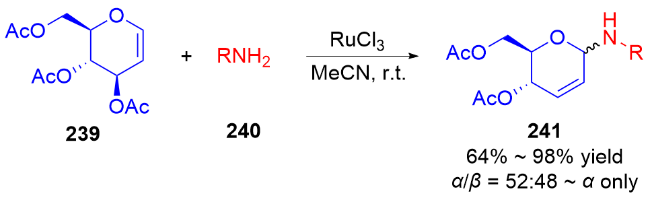
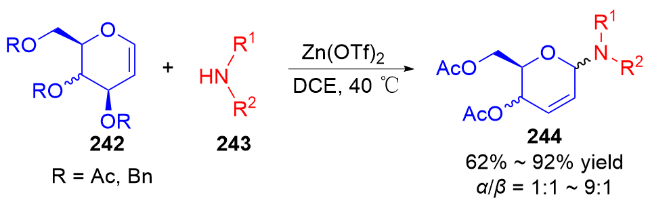
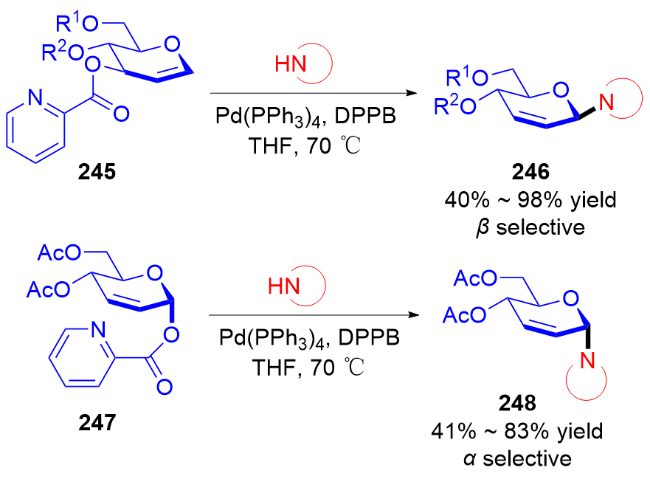
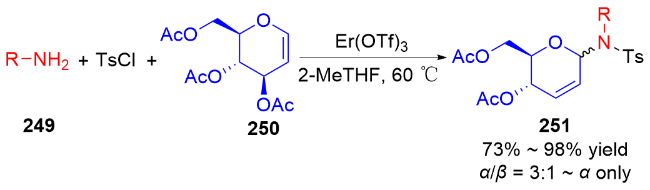
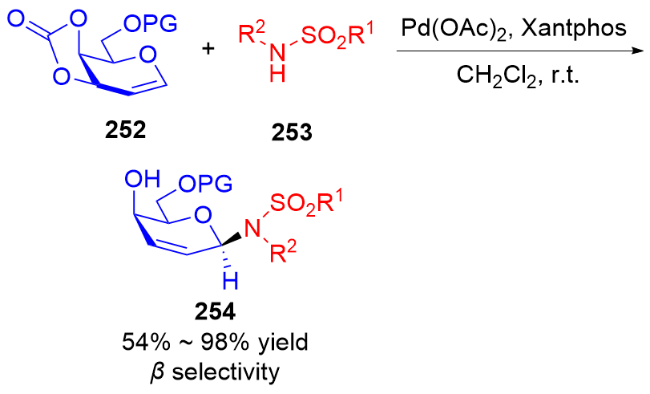
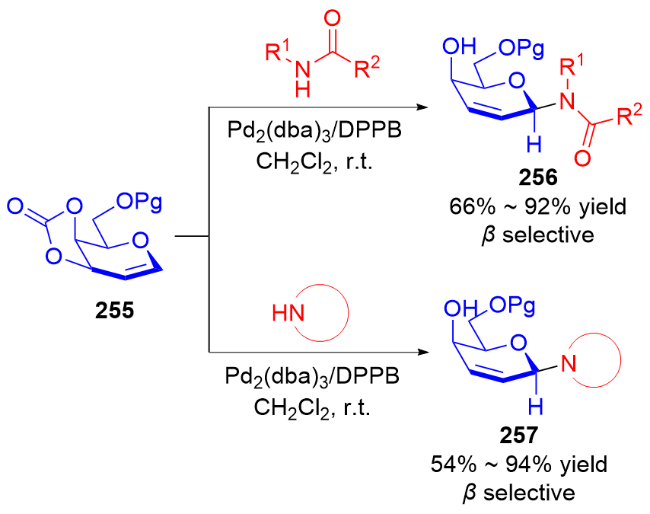
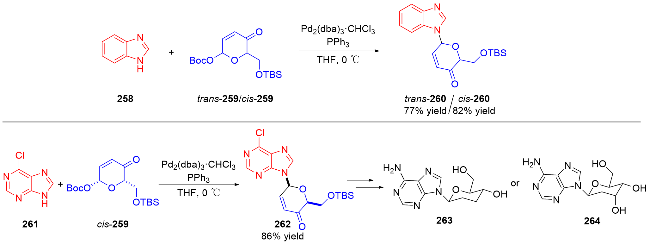
3.2 氮基3,4-二氢吡喃类衍生物的研究进展
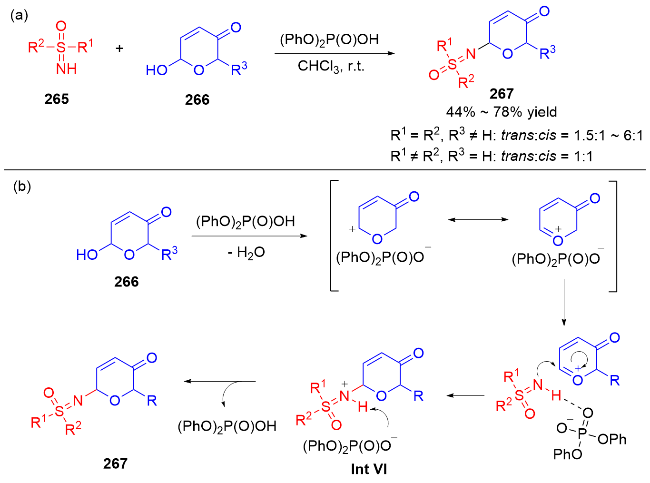
2021年, 我们课题组[88]使用布朗斯特酸磷酸二苯酯为催化剂, 以Achmatowicz重排产物二氢吡喃酮266为原料, 通过与亚砜亚胺265的亲核取代反应, 合成了一系列亚砜亚胺基取代的二氢吡喃酮类衍生物267. 当亚砜亚胺上的取代基相同时, 二氢吡喃酮底物C6上有取代基时, 得到trans-结构为主的产物; 而当亚砜亚胺上取代基不同时, 二氢吡喃酮C6上无取代基时, trans-产物和cis-产物的比例1∶1 (Scheme 75a). 该方法条件温和、操作简单, 避免了传统上使用钯、铱等贵金属催化剂, 进一步丰富了“de novo”从头合成糖苷类化合物的策略. 机理研究表明: 磷酸二苯酯作为布朗斯特酸类催化剂, 能够促进二氢吡喃酮类化合物266的羟基脱水形成碳正离子中间体或氧鎓离子中间体. 亚砜亚胺类化合物265通过磷酸酯活化促进亚砜亚胺的氮原子亲核进攻氧鎓离子中间体, 生成的中间体Int VI随后脱除质子, 得到亚砜亚胺基取代的二氢吡喃酮类衍生物267 (Scheme 75b).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论与展望
二氢吡喃类化合物的合成研究在有机合成和药物化学领域具有重要的地位和广泛的应用前景. 由于篇幅和能力所限, 本文仅详细总结了氧基二氢吡喃类衍生物、碳基二氢吡喃类衍生物和氮基二氢吡喃类衍生物的合成与转化研究进展, 这些反应为合成含二氢吡喃结构的农药、医药、功能材料和精细化学品等提供了新方法. 其中, 基于钯、铱等过渡金属催化以及路易斯酸催化合成二氢吡喃类衍生物反应研究较多, 发展最为成熟. 但是该领域仍有一些未来需要解决的问题: (1)需进一步发展高效、高选择性的合成方法, 提高二氢吡喃类化合物的合成效率和产率; (2)探索新的反应途径和催化剂体系, 实现对二氢吡喃环上各个碳原子的立体选择性精确控制; (3)应用计算化学和理论模拟等手段, 深入理解反应机理和反应动力学; (4)进一步研究二氢吡喃类化合物的生物活性或药理学特性, 探索其在药物研发中的潜在应用. 随着新的合成方法和策略不断涌现, 我们相信二氢吡喃类化合物的合成方法将得到进一步发展, 为化学合成和药物研发等相关领域带来更多的机遇.
(Cheng, F.)