


有机合成广泛应用于药物、化工产品及功能材料等生产中, 在人类社会发展中占有极为重要的地位. 通常来说, 有机反应具有反应速度缓慢和副产物多等特点, 而催化剂的使用则能显著提高有机反应的效率, 并减少副反应的发生, 催化合成已成为有机化学中最为重要的研究策略之一. 催化合成分为均相催化反应与异相催化反应. 均相催化反应效率高, 但催化剂难以回收. 异相催化反应因催化剂与反应物及产物处于不同的相态, 反应结束后, 可通过简单的过滤、离心或沉降等物理方法将催化剂从反应体系中分离出来, 易于回收. 大多数异相催化剂具有较高的热稳定性和机械稳定性, 即使在较苛刻的反应条件下仍然能保持其催化活性和选择性, 且经受多次反应循环而不发生明显的性能下降. 异相催化剂的回收再利用减少了对环境的污染和对资源的消耗, 符合绿色化学的发展理念, 特别是对于大规模反应, 在节约成本及减少污染方面具有重要意义. 近年来, 随着现代经济的高速发展, 环境污染以及生态恶化等问题日渐严重, 探索温和、绿色且高效的催化策略成为化学工作者们追求的目标和前进的方向, 异相催化因其特有的优势, 已成为“绿色化学”中最活跃、最有前途的研究领域之一.
遵循“碳达峰、碳中和”的发展理念, 利用绿色环保型催化剂实现有机转化是最理想的合成途径. 基于此, 一种受海洋生物贻贝启发人工合成的仿生材料聚多巴胺(Polydopamine, 简称PDA), 因具有廉价无毒、环境友好和稳定可回收的特点, 近年来受到异相催化领域的关注. PDA的合成灵感来自于贻贝分泌的粘附蛋白, 此类蛋白几乎能够粘附在任何表面[1]. 2007年, Messersmith课题组[2]以多巴胺为原料, 在碱性条件下空气中聚合, 首次合成了与粘附蛋白结构相似的高分子聚合物PDA, 自此以后, 其粘附特性被广泛研究, 主要应用于表面修饰领域[3-5]. 而作为“贻贝激发”的仿生材料, PDA也具有良好的生物相容性, 在随后的十几年间, 被广泛开发应用于生物医学、纳米医学和药物传递等领域[6-12]. PDA由多巴胺氧化聚合而成, 目前关于PDA结构的讨论主要围绕着结构单元的共价-非共价组装展开[13-20]. 研究表明, 多巴胺及其氧化衍生物5,6-二羟基吲哚、5,6-吲哚醌和5,6-二羟基吲哚啉等结构单元通过自身的苯环以共价键连接形成四聚体、八聚体等, 除了共价结构, PDA分子中还包含了氢键作用及π-π堆积作用(图1). 由于π-π堆积作用的存在, 其成键π轨道和反键π*轨道可分别构成价带和导带, 在可见光照射下能够吸收光子能量激发电子从最高被占轨道(HOMO)跃迁到最低空轨道(LUMO) (π-π*跃迁), 产生光生电子(e-)-空穴(h+)对, 故PDA具有一定的捕光能力, 在水的光化学氧化和有机染料的光催化降解等领域也有诸多应用[21].
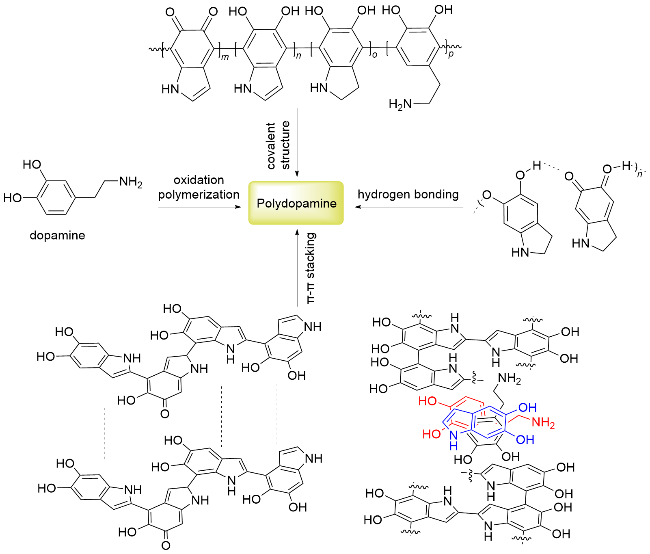
目前, 研究者们报道了大量关于PDA的合成、结构及其在医药、水处理等领域应用的综述, 而关于PDA在催化转化中的应用综述较少. 事实上, PDA分子中含有的大量儿茶酚结构能够将贵金属离子还原(如Au[22-33]、Ag[34-40]、Pd[41-45]或Pt[46]等), 或通过其广泛存在的邻二酚/邻醌结构与廉价金属(Fe3+[47-48]、Ni2+[49]、Co2+[50]或 Cu2+[51-53]等)配位进行负载, 构成M@PDA催化剂(图2), 可应用于多种有机反应中[54]. 另外, PDA结构中含有的酚羟基与氨基基团具有酸性和碱性作用, 其自身也可以作为催化剂使用. 尽管相关文章已有报道, 但总结性的论文仍然缺乏. 本文针对近年来M@PDA及PDA作为催化剂在不同类型有机反应中的应用进行综述, 具体以M@PDA催化的反应, 包括偶联反应、还原反应、三组分反应、分子间环合反应及烯烃的双官能团化反应等, 以及PDA自身催化的反应为分类, 从催化剂的活性及回收利用等内容进行详细的归纳和总结, 以期这种无毒环保的仿生材料能够受到更多关注.
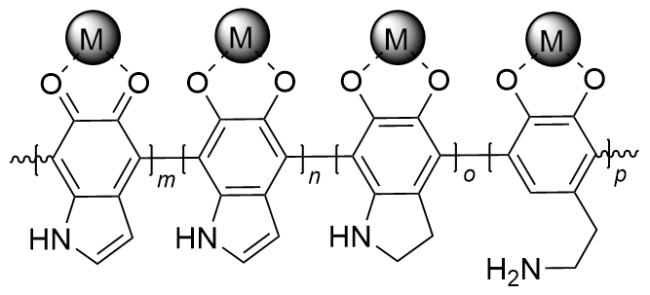
1 M@PDA催化的反应
1.1 M@PDA催化的偶联反应
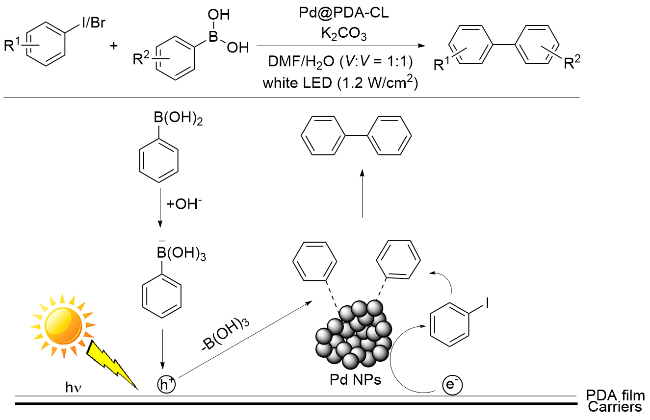
偶联反应是有机合成中最重要的反应类型之一, 常作为考察各类催化方法适用性的首选研究对象. 2016年, 谢阿明课题组[21]利用PDA包覆碳化丝瓜(CL), 并使钯纳米颗粒(Pd NPs)在PDA膜上原位生长, 形成Pd@PDA-CL催化剂, 并探索了其在Suzuki-Miyaura偶联反应中的应用. 研究结果证实, Pd@PDA-CL在光照条件下能够高效地催化Suzuki-Miyaura偶联反应(Scheme 1), 且催化剂重复使用五次, 活性未见下降. 在该反应过程中, PDA良好的粘附性使得反应物聚集在PDA界面附近, 在光照条件下, PDA吸收可见光产生光生电子(e-)- 空穴(h+)对, 光生电子转移到Pd NPs上催化芳基卤化物形成芳基Pd络合物; 同时, 芳基硼酸的C—B键被光生空穴氧化, 形成另一种芳基Pd络合物, 最后通过转金属化和还原消除得到偶联产物.
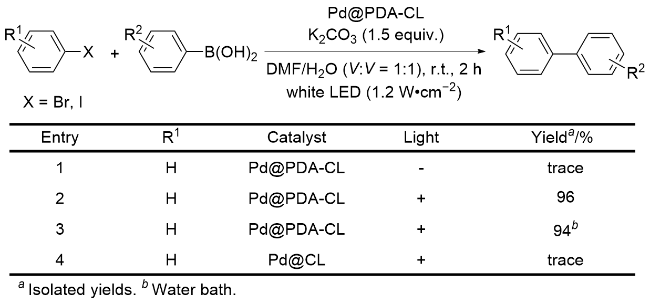
为了验证光照及PDA在反应中的作用(Scheme 2), 将Pd@PDA-CL催化的反应体系放置于黑暗中, 结果显示, 只有痕量产物产生, 由此表明反应中光照是必需条件(Entry 1); 若将反应在光照下置于水浴加热中, 并未观察到产率有明显改变, 进而排除热效应影响(Entry 3). 另外, 将钯纳米颗粒直接负载在碳化丝瓜上, 所得的Pd@CL催化剂在Suzuki-Miyaura偶联反应中几乎无催化作用(Entry 4), 通过电子顺磁共振(EPR)测试可知, Pd@CL比Pd@PDA-CL中的光生电子-空穴对寿命短, 说明在Pd NPs催化的偶联反应中, PDA的使用可以显著增强光的吸收与转化, 起到提高催化活性的作用.
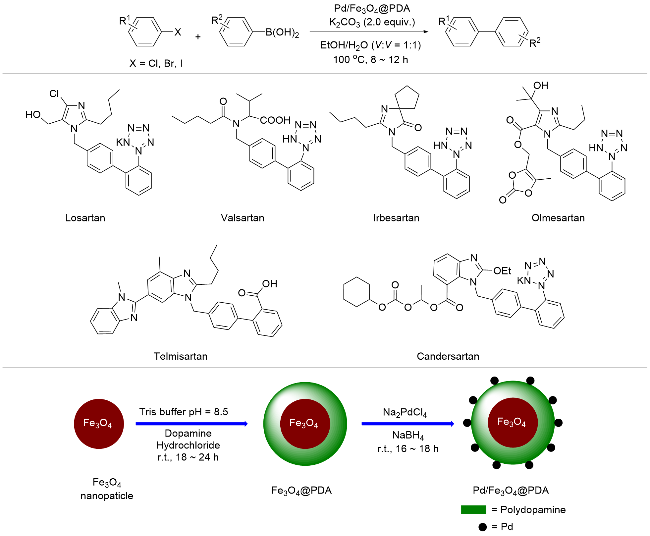
PDA通常以粉末或薄膜碎片的形式存在, 在过滤回收的过程中会不可避免地受到损失. 将PDA与磁性氧化铁基纳米材料相结合, 利用磁铁回收催化剂是实现催化剂循环使用的有效方法. 2016年, Kumar等[55]在纳米氧化铁颗粒上包覆PDA, 再将Pd负载于PDA上, 得到Pd的质量分数为3.88%的Pd/Fe3O4@PDA磁性催化剂, 用其催化芳基碘化物或芳基重氮盐与芳基硼酸的Suzuki-Miyaura偶联反应, 产物收率可以达到99% (Scheme 3). 研究表明, 在催化循环过程中, Pd纳米颗粒会脱离PDA表面, 发生氧化加成得到Pd(II), 而在最后的还原消除步骤后, 又生成Pd(0)重新沉积在PDA表面. 此催化剂在重复使用五次后催化活性没有任何损失, 反应得到的产物收率只存在轻微的偏差(2%左右), 展现了良好的可循环性. Pd/Fe3O4@PDA催化能力较高, 即使以活性较低的芳基溴化物为底物, 反应也可以顺利进行, 以中等偏上的产率获得目标产物, 但是, 对于芳基氯化物还没有展现出催化活性. 值得一提的是, 偶联产物4'-甲基-[1'-联苯]-2-乙腈是合成沙坦类药物分子的重要原料[56], 这种高效可回收的异相催化策略或将在药物大量生产中得以应用.
磁性催化剂的高效回收优势引起化学工作者的极大关注, 2017年, London课题组[57]再次报道了磁性催化剂在有机反应中的应用. London等选择Pd催化的Heck偶联反应为研究对象, 在研究过程中制备了无磁性的Pd-PDA和有磁性的Fe3O4@Pd-PDA两种催化剂, 用于比较二者的活性差别. 测试显示, 两种催化剂的Pd的质量分数分别为3.03%和1.86%, 二者都表现出优异的催化活性(Scheme 4, a), 但磁性催化剂Pd/Fe3O4@PDA在重复使用后没有得到相应产物, 这是由于金属Pd从载体表面发生了不可逆的浸出. 相比较而言, Pd-PDA的催化应用范围更广, 还可以用于硝基的催化转移加氢(CTH)反应中. 基于此, London课题组尝试了Pd-PDA催化的串联Heck偶联-催化转移加氢(Heck-CTH)反应(Scheme 4, b), 但此串联反应体系兼容性较差, 没有产物生成, 可能主要归因于Heck偶联反应过程伴随着Pd浸出, 反应结束后Pd没有二次沉积, 从而阻碍了CTH反应的发生.
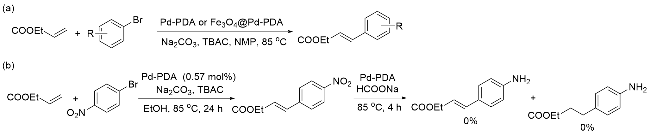
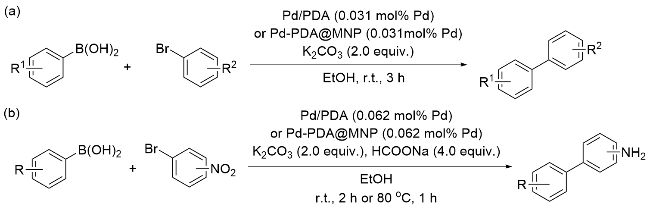
基于前期的研究工作基础, 2018年, London课题 组[58]报道了Pd/PDA催化的Suzuki偶联反应(Scheme 5, a)和串联的Suzuki偶联-催化转移加氢(Suzuki-CTH)反应(Scheme 5, b). 将多巴胺在碱性条件聚合制备PDA, 利用儿茶酚结构将Pd(II)还原为Pd(0), 并将其负载, 构成Pd/PDA催化剂. Pd/PDA在Suzuki偶联反应中展现出极高的催化活性, 无论是苯硼酸还是杂芳环硼酸底物, 都能顺利获得目标产物, 且可用于放大量实验. 将其与磁性纳米材料结合, 合成Pd-PDA@MNP磁性催化剂用于回收再利用, 循环实验显示催化剂在循环使用五次后未见明显失活. 机理研究显示, 催化过程主要依靠Pd(0)浸出在溶液中进行, 这是反应能够顺利实现的关键. 需要指出的是, 在后续的串联Suzuki偶联-催化转移加氢反应中, 为使反应按预期顺序进行, 催化转移加氢过程所需的还原剂需要延迟加入, 以避免脱卤化副产物的产生.
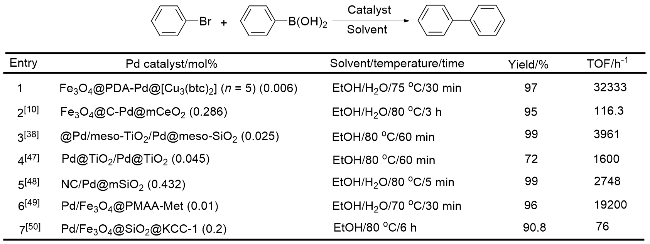
将PDA类催化剂与多孔材料相结合, 是制备多功能催化剂的重要手段, 使用多孔的金属有机框架(MOF)与PDA催化剂结合, 能够显著提高催化剂的活性. 最近, 卞凤玲课题组[59]报道了一种新型的分层组装的Fe3O4@PDA-Pd@[Cu3(btc)2]纳米复合材料. 将Fe3O4@PDA-Pd纳米复合材料加入Cu(CH₃COO)₂•H₂O的乙醇溶液中, 在70 ℃下搅拌15 min, 利用磁场分离, 并使用乙醇洗涤. 随后, 将复合材料分散在均苯三甲酸(H3btc)的乙醇溶液中加热搅拌, 分离洗涤. 通过不同的循环次数, 制备了一系列MOF壳厚度不同的Fe3O4@PDA- Pd@[Cu3(btc)2]纳米复合材料. 该材料在Suzuki偶联及硝基还原和反应中都显示出高催化活性, 这可能是由于含铜MOF与Pd纳米颗粒之间的协同作用. 在Suzuki偶联反应中, 此催化剂的活性高于多种Pd复合材料, 转化频率(TOF)高达32333 h-1 (Scheme 6), 即使使用反应活性较低的芳基氯化物为原料, 反应也可以顺利进行; 同样, 在硝基还原反应中, 其比Fe3O4@PDA-Pd或Cu(OAc)2具有更高催化活性. 同时, 该纳米复合材料分离方便, 可回收性高, 对于以上两种反应, 重复使用8次, 产率仍然很高. 另外, 将多孔硅材料与Fe3O4@PDA结合也可用于催化Suzuki偶联反应, 多孔硅与Fe3O4纳米颗粒结合构成核-壳结构, PDA聚合在Fe3O4/SiO2表面, 通过还原Pd(II)制备Fe3O4/SiO2@PDA/Pd催化剂, 可回收性高, 能够重复使用十二次[60].
对于上述偶联反应, 主要通过循环体系来完成, 即将催化剂回收后再重新利用. 在研究异相催化的偶联反应过程中, 化学家们还发展了“非循环”体系, 即将催化剂固定在反应瓶内壁, 化学反应在瓶壁上发生. 2017年, Fei课题组[61]设计将PDA修饰在多孔硅纳米材料(SNFs)包覆的玻璃反应瓶表面, 并利用儿茶酚的还原能力, 在PDA表面沉积Pd纳米颗粒, 构建SNFs-Pd催化体系, 将反应置于瓶壁上进行. 该体系在Heck反应中展现出极高的催化活性(TOF=8520 h-1)、底物兼容性及可循环性. 在碘苯与丙烯酸叔丁酯的偶联反应中, 反应瓶重复使用二十次, 产率依然较高. 并且, 该体系具有良好的反应条件兼容性, 能够实现连续的Heck偶联反应(Scheme 7). 作者还对SNFs-Pd体系的催化应用范围进行了考察, 研究显示, 该体系具有高度通用性, 适用于烯烃和芳香硝基化合物的催化加氢反应、碘苯与异腈的酰胺化反应以及碘苯、二氨基苯酚与异腈的三组分反应等, 反应效率高, 体系可循环性好其中, 在烯烃的催化加氢中, 反应器重复使用十次, 产率依然高达97%.
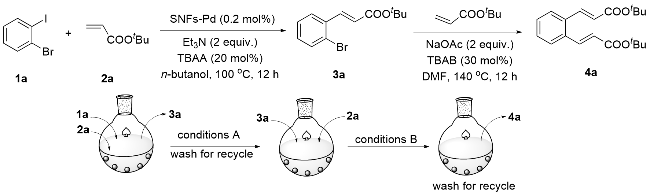
以上报道表明, PDA负载金属催化的Suzuki偶联与Heck偶联反应得到了充分发展, 而PDA负载金属催化的其它偶联反应也需要去探索. 2020年, Kumar等[62]报道了磁性催化剂Pd/Fe3O4@PDA在水相中催化Ullmann偶联反应, 经测定, 催化剂中Pd质量分数为6.92%. 对于水相中的Ullman偶联反应, Kumar团队利用甲基化β-环糊精(RM-β-CD)来改善催化剂的性能, 以芳基硼酸或芳基卤化物为底物, 成功构建了对称或不对称的联芳基化合物. 反应范围扩展研究证明, 各类芳基卤化物(溴、氯、碘)的偶联反应均可实现, 得到的产物产率高达92% (Scheme 8, a, b), 该磁性催化剂可以通过磁铁方便地回收, 5次循环后回收率依然高达98%. 但是, 使用芳基硼酸为底物, 反应活性有所下降, 只能以中等偏上的产率得到目标产物(Scheme 8, c, d); 而当使用芳基重氮盐为底物时, Pd/Fe3O4@PDA表现出了更低的催化活性, 得到的产物产率均较低(Scheme 8, e).
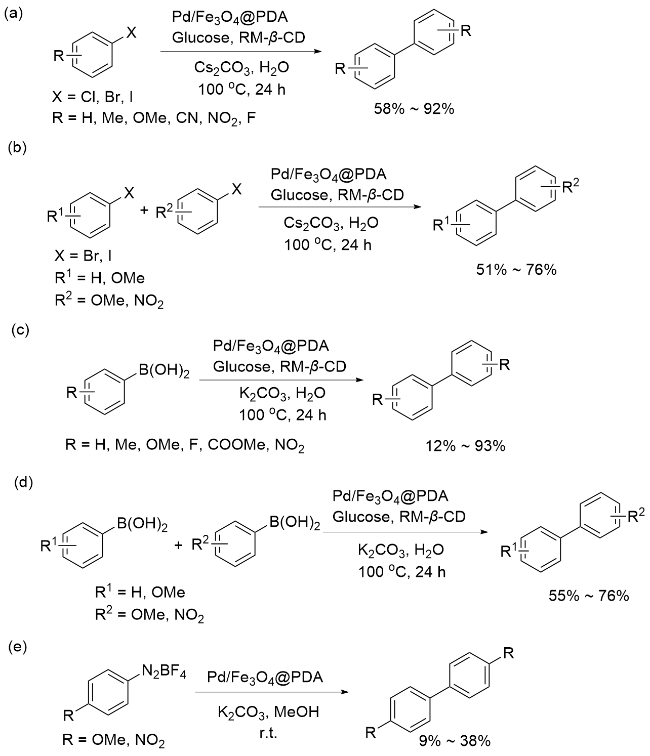
除了上述报道的碳-碳偶联反应, PDA负载金属也可以用于催化碳-杂原子键的偶联. 2018年, Veisi等[63]通过将Fe3O4@PDA浸泡在镀钯溶液中, 使钯离子吸附在Fe3O4@PDA表面, 得到了可以磁性分离的Fe3O4@PDA/Pd(II)催化剂, 测得Pd质量分数为2.09%. 材料表征显示, 在催化剂的制备过程中, PDA中的儿茶酚基团未将Pd(II)还原成Pd(0), X射线光电子能谱技术(XPS)分析中发现的Pd(II)特征峰给予了充分证明. Fe3O4@PDA/Pd(II)在Buchwald-Hartwig的C—N键偶联反应中表现出优异的催化性能(Scheme 9), 能以高产率合成多种芳胺(65%~98%), 且催化剂可重复使用六次, 催化活性不会有明显变化.
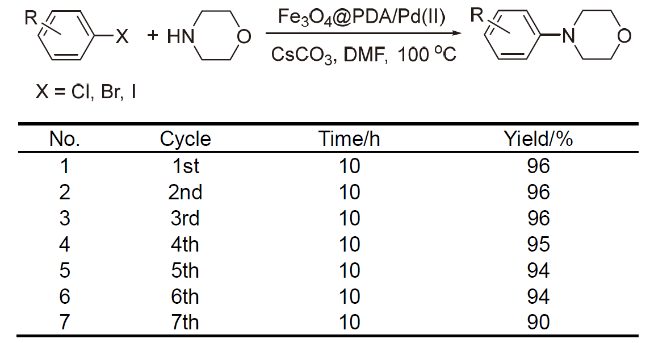
1.2 M@PDA催化的还原反应
PDA负载金属在多种偶联反应中展现了较高的催化活性, 除此之外, M@PDA在催化转移加氢过程中也具有一定的活性. 2017年, London课题组[57]报道了Pd-PDA催化的硝基化合物及羰基化合物的加氢反应(Scheme 10), 经测定, 催化剂的Pd质量分数为3.03%, 在该反应中, 所采用的氢源为HCOONa. 研究显示, Pd-PDA催化的硝基还原反应官能团兼容性较高, 当底物中含有甲基、羟基、氨基、甲氧基、酰胺、酯和乙酰基等官能团时, 均可以以较高产率获得目标产物. 但是, 对于羰基化合物的还原反应来说, 底物的适用范围则较窄, 除了苯乙酮外, 其它取代的苯乙酮类底物反应活性均不高, 通常需要延长反应时间来提高转化率, 而醛类化合物则不能反应, 这可能是由于醛羰基与PDA中的氨基形成的亚胺阻隔了催化位点. 进一步地, 为了方便地回收催化剂, 作者将Pd-PDA与磁性纳米材料Fe3O4结合, 制备了磁性的Fe3O4@Pd-PDA催化剂, 经测定, Pd质量分数为1.86%, 此催化剂在催化加氢反应中连续使用5次后, Pd质量分数仅略有下降(1.55%), 催化活性不受影响.
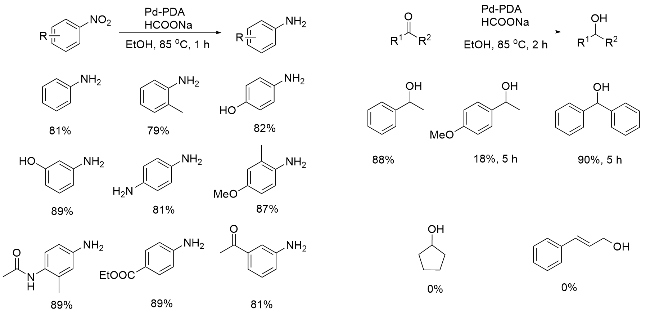
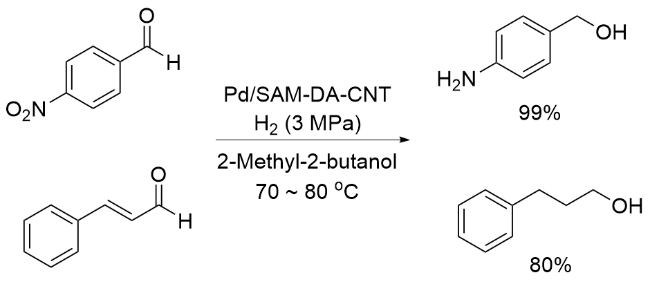
2018年, 李国柱课题组[45]对Pd-PDA催化的还原反应进行了深入研究, 以4-硝基苯甲醛在水中的加氢反应作为模板反应, 通过筛选不同的支撑材料, 如SBA-15、氧化铝、石墨烯和碳纳米管(CNT)等, 获得了用羧酸烷烃硫醇单层修饰的、碳纳米管为支撑材料的最佳催化剂Pd/SAM-DA-CNT. 其催化活性高的原因在于PDA通过引入表面锚定基团, 促进了金属纳米颗粒的吸收和原位分散沉积; 羧酸单分子层通过提高载体与Pd之间的静电作用, 有利于Pd纳米颗粒的有序排列, 改性后的催化剂Pd纳米颗粒的平均尺寸可以达到1.0~2.0 nm. 该体系具有广泛的适用性, 在羰基和双键等的催化加氢反应中都展现了良好的性能, 即使是可能导致催化剂失活的醛类化合物也可以被还原成相对应的醇类产物(Scheme 11).
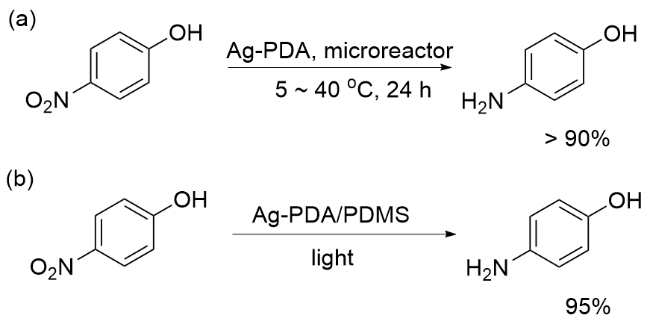
将Ag纳米颗粒负载于PDA表面制备的Ag-PDA催化剂, 在硝基的还原反应中也展现了良好的催化性能. 2017年, 刘壮和褚良银等[64]构建了微通道表面涂层为PDA的玻璃微反应器, 将其浸没在硝酸银溶液中, 使Ag纳米颗粒原位固定在PDA层表面. 以形成的Ag- PDA为催化剂, NaBH4为氢源, 在微反应器中催化还原对硝基苯酚. 反应底物在24 h内的转化率始终保持在90%以上(Scheme 12, a), 而催化剂在使用24 h后, Ag NPs仍牢固地固定在表面微通道表面. 研究表明, 对硝基苯酚在微反应器中的转化率会随着温度的升高而增大, 这是由于升高温度增加了Ag的负载量. 随后, 为了进一步明确温度对Ag-PDA催化活性的影响, 朱恂和杨扬等[65]进行了深入探索. 结果显示, 纳米银-聚多巴胺复合材料(Ag-PDA/PDMS)在光照条件下, 能够通过光热转换升高反应温度, 并与银纳米颗粒产生的热电子共同作用, 进而实现对对硝基苯酚的高效还原(Scheme 12, b).
1.3 M@PDA催化的其它反应
1.3.1 三组分反应
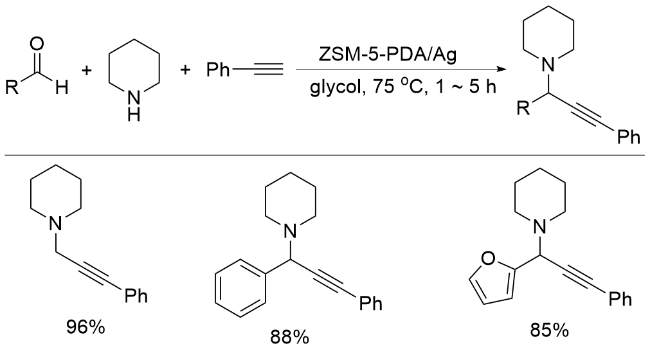
多组分反应体系复杂, 不易控制, Yang和Kim课题组[66]报道了唯一一例PDA负载银催化的醛、炔烃与胺的串联反应(Scheme 13). 将PDA包覆在ZSM-5型沸石表面, 通过儿茶酚结构将Ag(I)还原, 构成ZSM-5-PDA/ Ag催化剂, 经XPS测定, Ag质量分数为4.8%. 利用ZSM-5-PDA/Ag催化醛、炔烃与胺的串联反应, 得到了芳基炔丙胺产物, 产率为96.5%. 较高的反应效率可归因于沸石的大比表面积, 使Ag纳米颗粒得到良好分散, 这为乙炔的C—H键活化反应提供了足够的反应空间. 该催化剂在第二次和第三次循环中活性略有下降, 产物的产率分别为95%和94.2%, 经测定, 催化剂中Ag的质量分数分别下降到4.5%和4.1%, 因此, ZSM-5-PD/Ag的活性损失源于载体表面银纳米颗粒数量的减少.
1.3.2 分子间环合反应
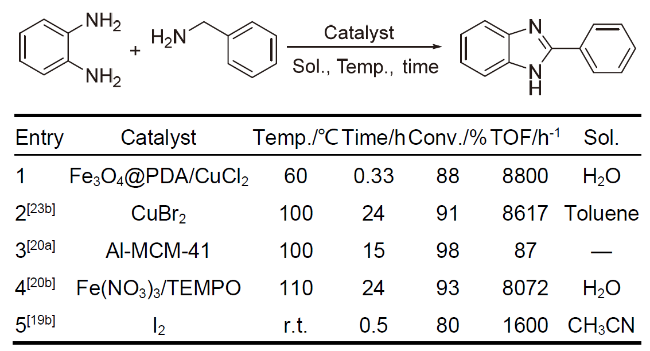
在上述反应中, 多数以PDA负载贵金属如Pd、Ag等进行催化研究, 而利用PDA负载廉价金属催化的反应研究相对较少, 仅在最近两年才有相关报道. 2023年, Badbedast等[67]使用PDA包覆磁性纳米颗粒Fe3O4, 随后负载Cu(II)制备了Fe3O4@PDA/CuCl2催化剂, 该催化剂在邻苯二胺与苄胺的分子间环合反应中展现了良好的催化性能, TOF值高达8800 h-1 (Scheme 14). 同时, 该催化剂也展现了良好的可回收性, 能够通过磁性方便地从反应体系中分离, 其在重复使用五次后, 反应产物的产率依然高达84%. 该方法使用廉价可循环的Fe3O4@PDA/CuCl2为催化剂, 空气为氧化剂, 水为溶剂, 是可持续化学的典型研究, 或将成为该领域未来发展最有潜力的方向之一.
1.3.3 烯烃的双官能团化反应
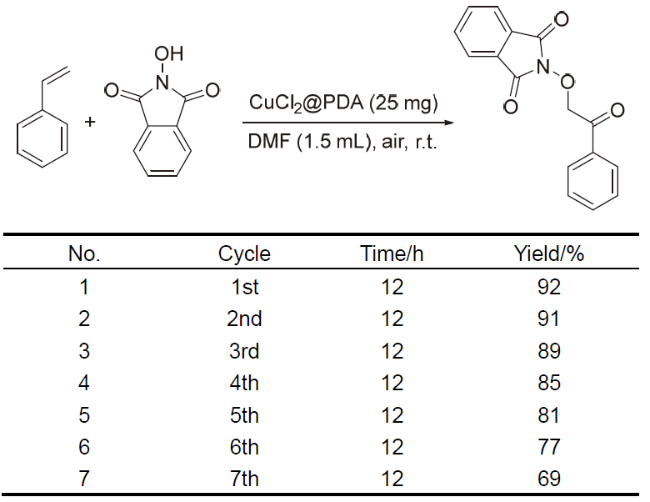
PDA负载廉价金属实现的催化转化因具有经济性优势而受到关注. 2024年, 我们课题组[68]报道了另一例PDA负载廉价金属铜催化的有机转化, 发展了CuCl2@PDA催化的烯烃的双氧化反应(Scheme 15). 以苯乙烯和N-羟基邻苯二甲酰亚胺(NHPI)为原料, 空气中的氧气为氧源, 在CuCl2@PDA催化条件下, 成功制备了α-氧代酮类化合物. 该反应效率高, 官能团兼容性好, 且催化剂制备过程简单, 直接将CuCl2与多巴胺共聚即可获得. 电感耦合等离子体(ICP)测定显示, 该催化剂中的Cu的质量分数为24.4%, 相较于其它均相铜催化体系, CuCl2@PDA在烯烃的双氧化反应中展现的催化性能更加优异, 产物的产率可以达到92%, 而使用相同用量的铜盐, 则不能达到目前的产率. 同时, 该催化剂也展现了良好的可回收性, 在循环使用第七次时, 产物产率依然能达到69%, 催化活性的下降主要归因于催化剂在多次循环使用后活性Cu的流失. 需要指出的是, 这是首次通过异相催化的途径实现烯烃的双氧化反应, 该策略具有经济与绿色的优势, 将为更多类型的烯烃双官能团化反应提供研究思路.
2 PDA自身催化的反应
在PDA结构中, 既存在酸性(OH)基团, 又存在碱性(NHR, NH2)基团, 因此, 在相关转化中PDA可以同时活化亲电试剂与亲核试剂, 起到协同催化的作用.
2.1 PDA催化的Aldol反应
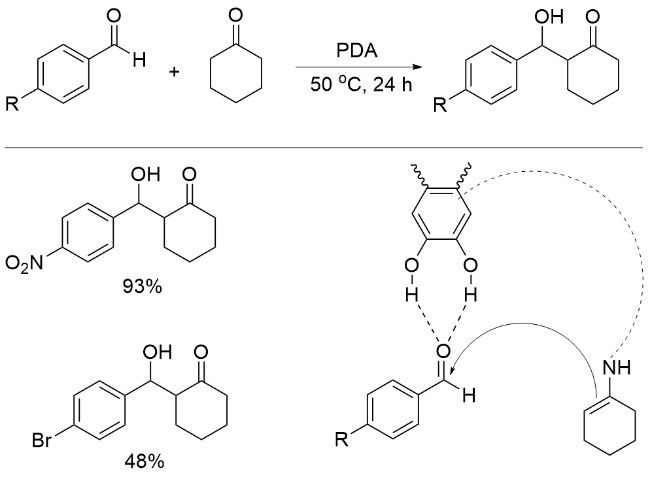
2014年, Liebscher课题组[69]报道了PDA催化的芳香醛与环己酮的Aldol反应, 再一次表明PDA不只是一种“惰性的”聚合物, 而是可以应用于有机转化的高效催化剂. PDA利用其酚羟基与氨基同时活化醛的羰基和酮的α-位, 使缺电子芳香醛顺利发生反应(Scheme 16). 但是PDA在循环一次后催化活性会明显下降; 将磁性Fe3O4与PDA结合, 回收后的催化剂同样失活. 基于以上两个实验, 作者提出了PDA失活的原因: 在反应过程中, PDA超分子结构中的低聚物从催化剂表面浸出, 剩余更稳定的PDA结构中含有的氨基较少, 催化活性降低; PDA中的氨基与芳香醛之间形成了亚胺, 阻断了碱性催化位点, 从而降低了催化活性.
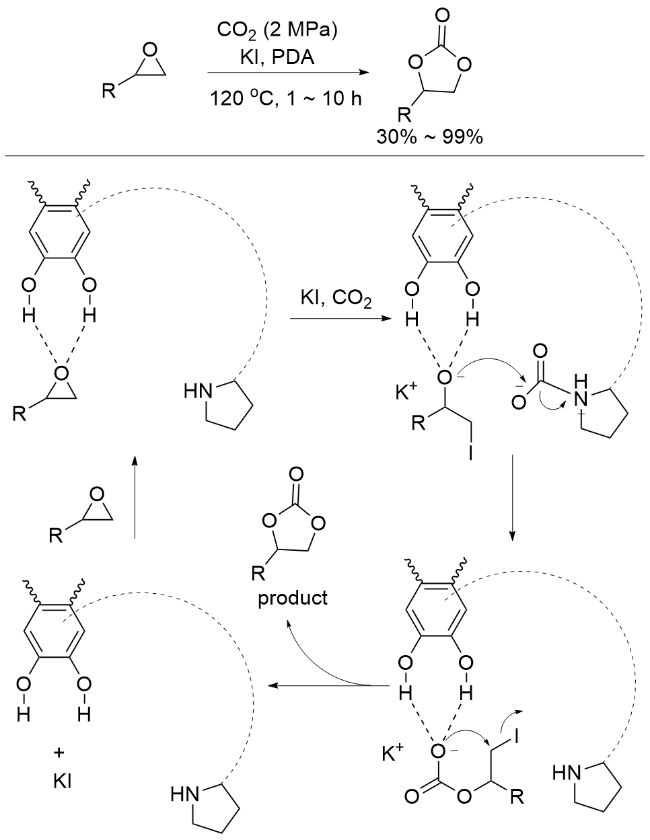
2.2 PDA催化的扩环反应
2.3 PDA催化的硫醇偶联反应
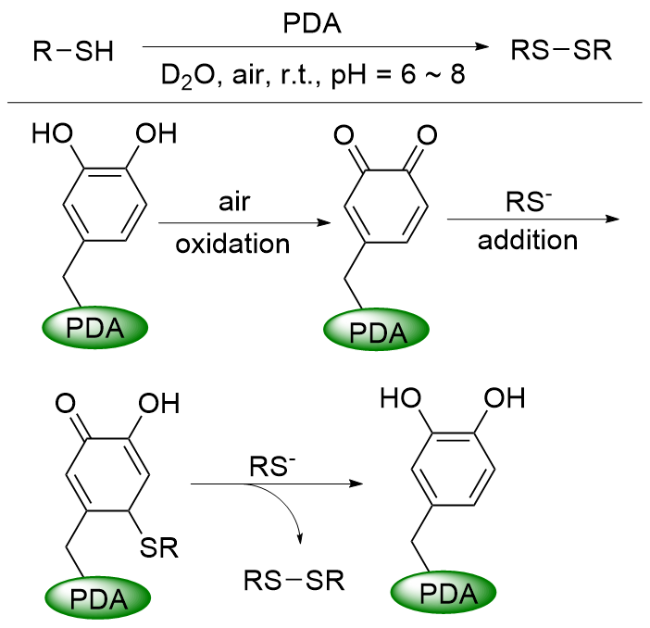
在前面的总结中, 可以看出M@PDA在偶联反应中具有优异的催化活性, 不仅如此, PDA在一定条件下也能够催化偶联反应. 2015年, 徐志康课题组[71]报道了PDA催化的硫醇在室温下水溶液中的偶联. 与上述PDA作为酸-碱协同催化剂不同, 在该反应中, PDA中的醌结构单元起到了至关重要的作用(Scheme 18). 首先, RS-基团对醌结构进行亲核进攻, 经迈克尔加成反应生成相应的硫醚产物, 随后, 生成的硫醚继续与RS-反应生成二硫醚产物, 并伴随具有儿茶酚结构的PDA催化剂生成, 当其暴露在空气中时, 儿茶酚结构又被重新氧化到醌态, 完成催化循环. 该反应条件兼容性好, 在中性、弱碱性或弱酸性环境中都能有效地进行; PDA催化剂可回收性高, 重复使用五次活性未见明显下降.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 总结与展望
综上所述, 无论是M@PDA还是PDA自身都可以作为异相催化剂, 应用于C—C键、C—N键及S—S键的偶联, 双键、硝基和羰基的还原, Aldol反应, 串联反应或环氧乙烷的扩环反应中, 具有应用范围广、反应条件温和、催化剂可回收再利用、廉价无毒及绿色环保等诸多优点. 但是目前, 基于PDA在异相催化领域的研究仍处于初级发展阶段, 还存在很多挑战: (1) PDA在其它类型的有机反应中的应用仍需拓展, 特别是基于PDA的捕光性质, 其在光催化的有机转化领域的应用需要深入探索. (2)受限于当前的催化理论与表征技术, 目前对于PDA负载金属时, 金属纳米颗粒的尺寸、形貌及金属离子的价态对催化活性影响的探索并不深入, 催化位点与催化活性之间具体的构效关系仍有待进一步研究. (3) PDA结构中包含大量羟基、氨基及共轭双键等活性基团, 通过对PDA修饰来调控材料结构, 对于提高PDA或M@PDA的催化性能至关重要, 目前此类研究鲜有报道, 在未来将拥有广阔的发展空间. (4) PDA催化剂在绿色化工领域的应用亟待探究, 以提升此类仿生材料的实际应用价值. 总之, PDA材料在有机合成领域的利用至今只进行了大约十年, 基于PDA的绿色、经济性, 值得开发其在更多反应中的应用.
(Zhao, C.)