


杂环骨架广泛存在于天然产物、药物及生物活性分子中, 是许多常见药物的重要组成部分, 例如抗癌药物、抗病毒药物、激素、核苷、维生素等[1]. 其中氟烷基取代的杂环化合物对药物化学的发展做出了重大贡献, 因此关于氟烷基修饰的杂环化合物的开发和生物活性评价引起了科研工作者的极大兴趣[2-6].
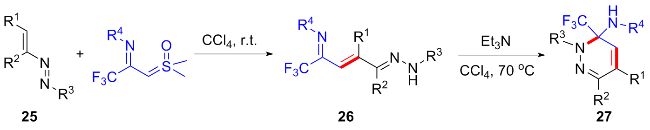
作为重氮化合物的替代物, 亚砜叶立德相较于重氮化合物具有更易制备、更安全等优点, 且表现出于重氮化合物相似的反应性, 被广泛应用于杂环类化合物的合成中[7-8]. 2021年, 程国林课题组[9]首次报道了一种新型的亚砜叶立德CF3-亚胺酰基亚砜叶立德(TFISYs) (Scheme 1), 随后研究发现, CF3-亚胺酰基亚砜叶立德与酰基亚砜叶立德性质相似, 其参与的环加成反应为氟烷基修饰的杂环化合物的合成提供了新的途径. 最近, CF3-亚胺酰基亚砜叶立德在杂环化合物的构建中引起了越来越多的关注. CF3-亚胺酰基亚砜叶立德不仅可以作为卡宾前体参与金属催化的环化反应, 还可以在不添加金属催化剂的条件下, 作为偶极子经亲核取代反应串联环化过程构建杂环化合物. 刘占祥、张玉红[10]及张建涛[11]分别总结了亚砜叶立德参与的碳氢键活化/环化反应及亚砜叶立德在合成五元/六元氮杂环中的应用进展, 但关于CF3-亚胺酰基亚砜叶立德参与的环化反应暂未有综述报道. 因此, 本文将结合近年来关于CF3-亚胺酰基亚砜叶立德的研究报道, 分别按其作为偶极子及卡宾前体, 对其参与合成氟烷基修饰的杂环骨架的环化反应研究进展进行归纳总结.
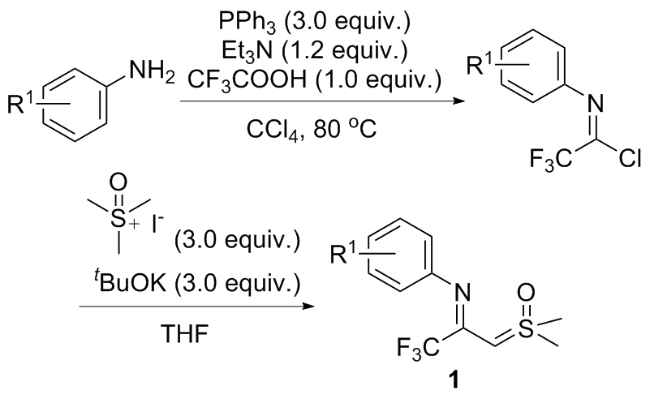
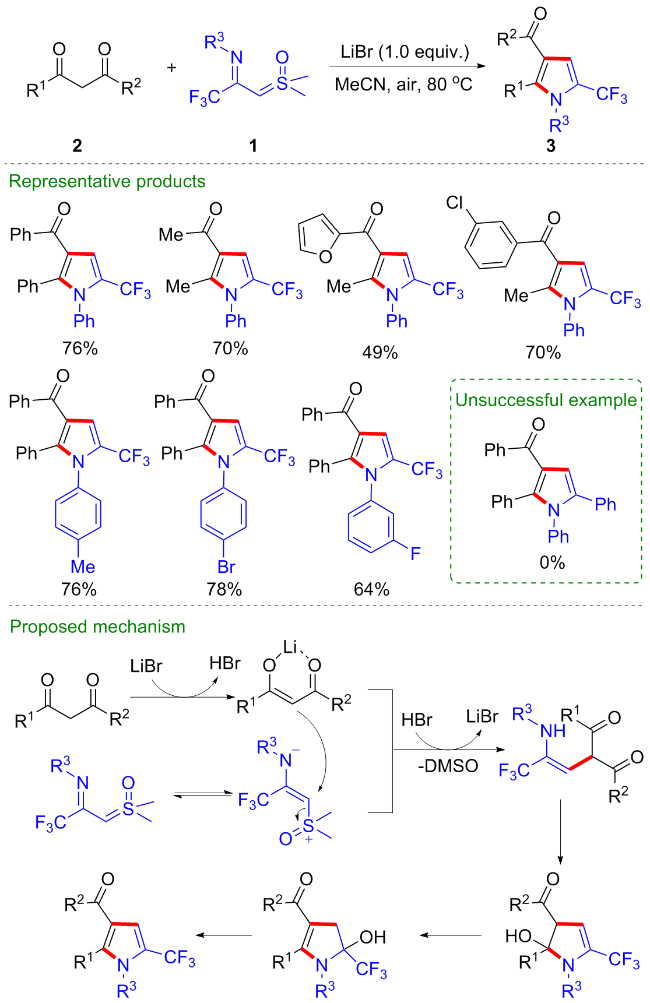
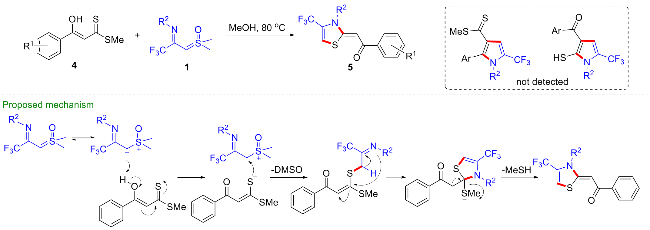
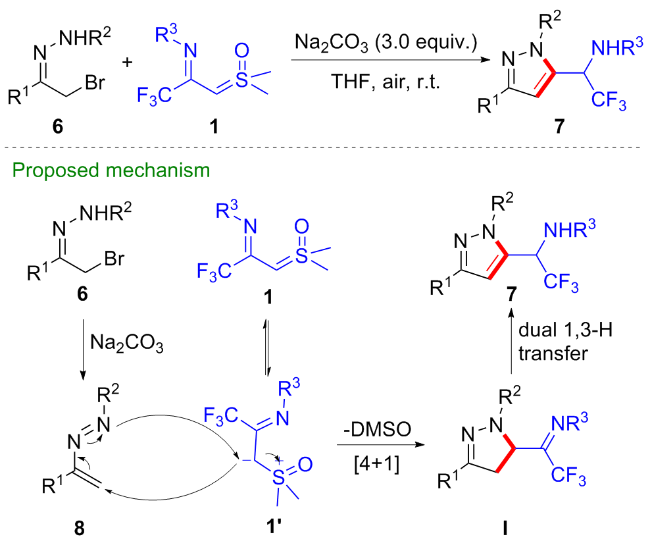
1 作为偶极子参与的环化反应
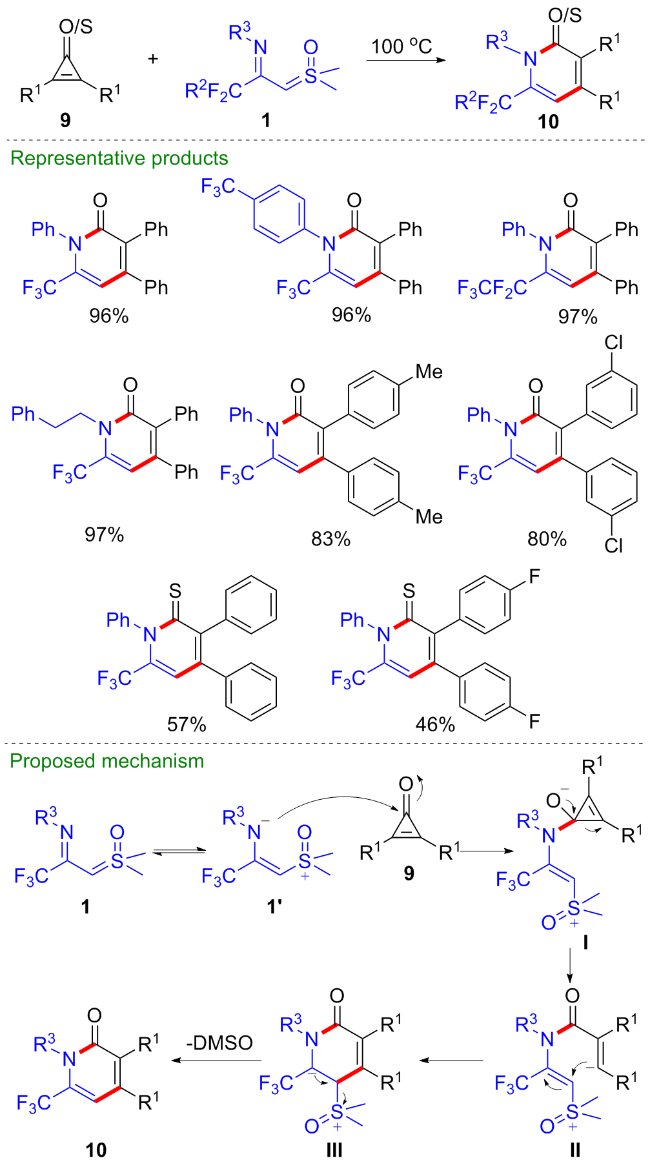
2022年, 程国林等[14]报道了CF3-亚胺酰基亚砜叶立德与环丙烯酮9的[3+3]环化反应, 为多取代的三氟甲基哌啶酮类化合物的合成提供了新的策略(Scheme 5). 该反应不需要添加任何金属催化剂、添加剂, 在无溶剂条件下直接加热到100 ℃, 便可以以优秀的产率得到一系列氟烷基取代的哌啶酮类化合物10. 值得一提的是, 当使用环丙烯硫酮底物时, 反应可以顺利地得到对应的哌啶硫酮类产物. 作者提出了可能的反应机理, CF3-亚胺酰基亚砜叶立德共振为偶极子中间体1', 对环丙烯酮进行亲核加成得到环丙烯中间体I, 随后经开环反应、分子内亲核加成得到中间体III, 最后离去一分子二甲基亚砜得到三氟甲基哌啶酮产物10.
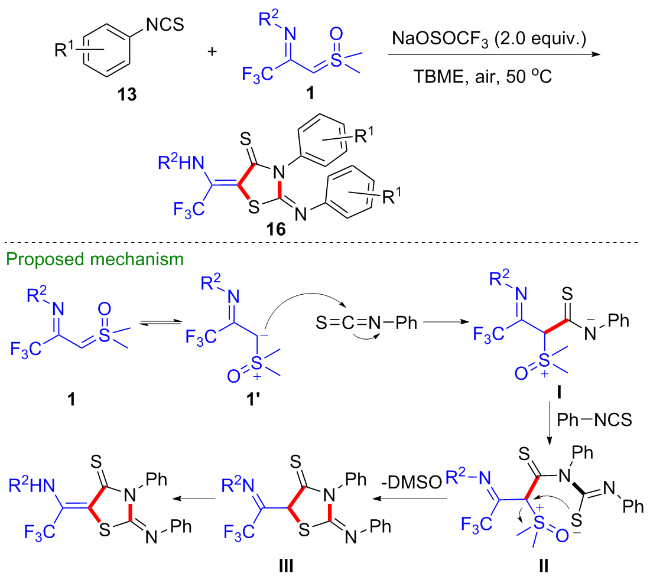
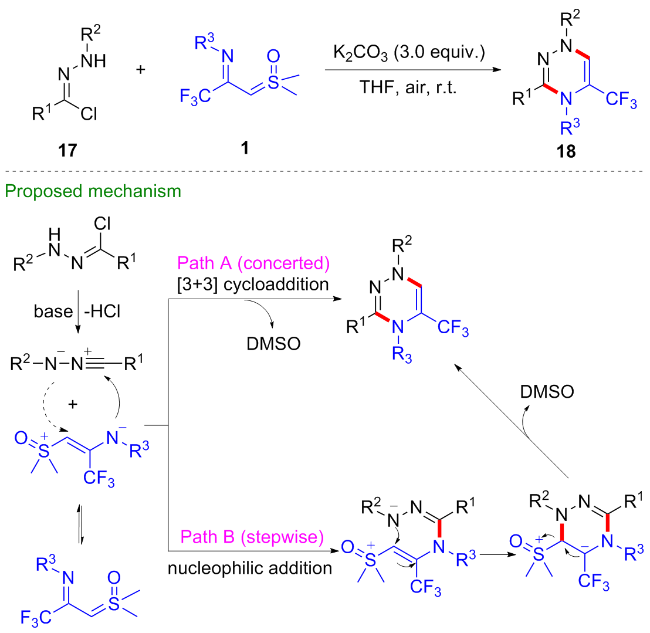
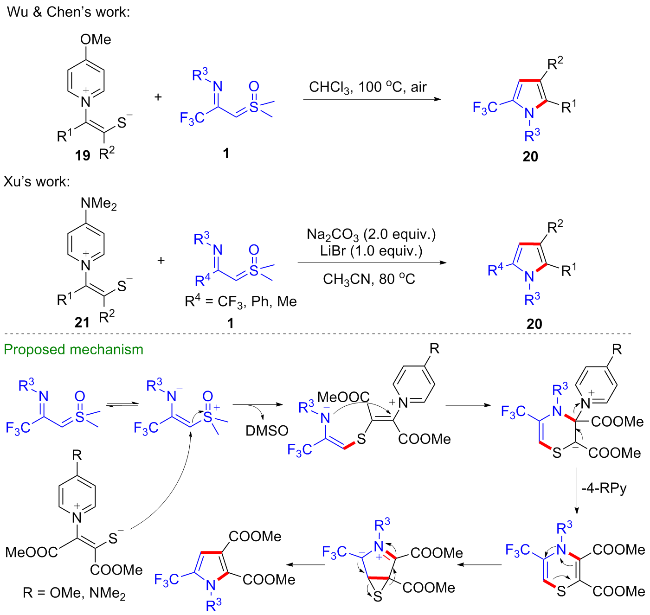
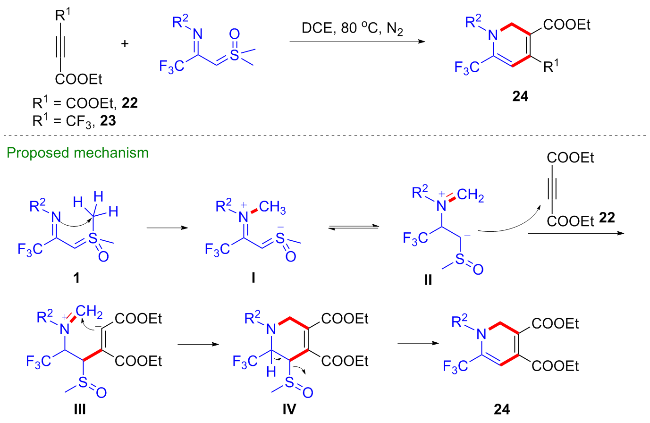
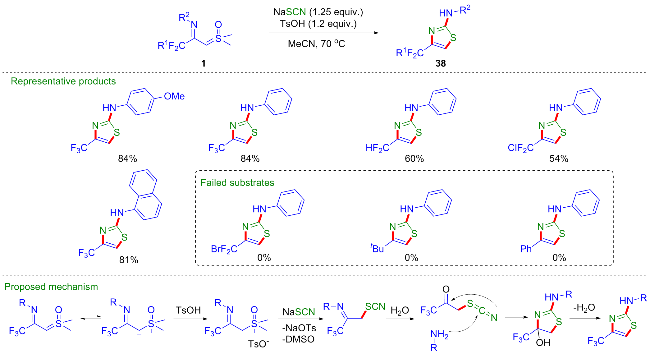
考虑到1,4-吡啶硫内鎓盐19也是一种具有多个反应位点且反应性能优异的偶极子[20-22], 吴晓峰、陈铮凯课题组[23]和胥波课题组[24]先后研究了CF3-亚胺酰基亚砜叶立德与1,4-吡啶硫内鎓盐19的环化反应(Scheme 11). 吴晓峰课题组研究发现, 在不需要添加任何催化剂的条件下, 两者可以顺利地发生[(3+3)-1]环化反应,高效地合成了一系列三氟甲基取代的吡咯类化合物20. 在该反应中, 1,4-吡啶硫内鎓盐中的吡啶片段作为离去基团参与反应, 而更富电子的吡啶片段对反应产率有益, 他们选择了对甲氧基取代的1,4-吡啶硫内鎓盐作为最优底物. 胥波等选择了对二甲氨基取代的1,4-吡啶硫内鎓盐21作为最优底物, 同时添加碳酸钠和溴化锂作为添加剂, 得到了类似的反应结果, 值得一提的是在该反应条件下烷基取代的亚胺酰基亚砜叶立德也能顺利参与反应. 此外, 胥波等还研究了CF3-亚胺酰基亚砜叶立德与丁炔酸二酯22、三氟甲基丙炔酸酯23的形式 [4+2]环化反应(Scheme 12), 在氘代实验的基础上, 作者推测在反应过程中, CF3-亚胺酰基亚砜叶立德首先发生甲基迁移生成亚胺偶极子中间体I, 该中间体作为四元合成子与丁炔酸二酯发生[4+2]环化反应, 最终消除一分子CH3SOH得到二氢吡啶产物24.
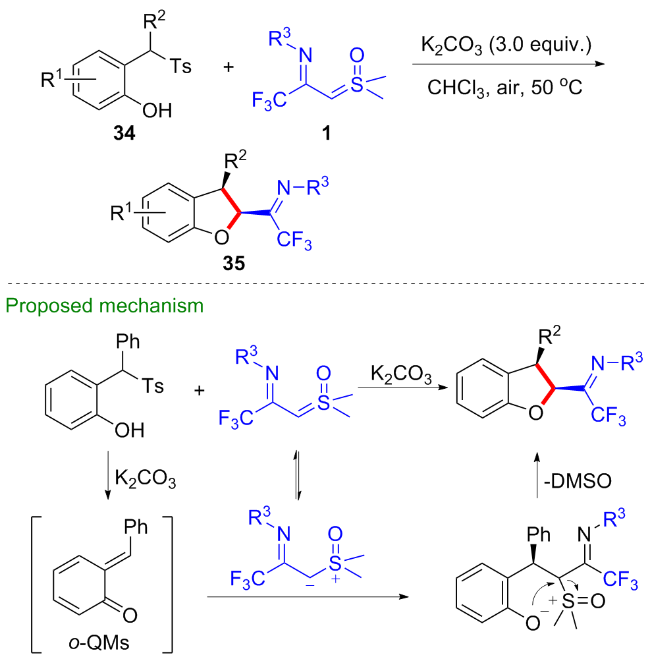
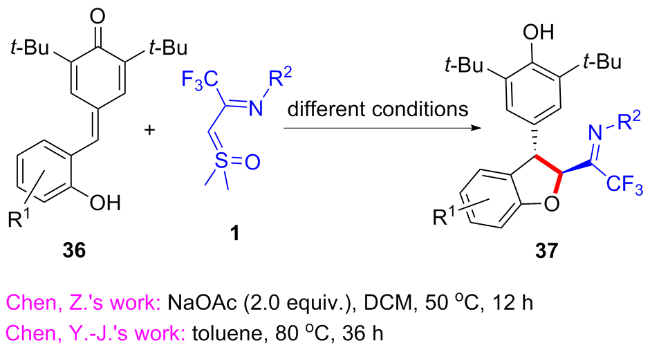
邻羟苯基甲亚基对苯醌36常作为氧杂四元合成子参与[4+m]环化反应, 为多种杂环化合物的构建提供了高效的策略[29-30]. 2024年, 陈铮凯课题组[31]与陈亚静课题组[32]分别独立报道了CF3-亚胺酰基亚砜叶立德与邻羟苯基甲亚基对苯醌36的[4+1]环化反应(Scheme 17). 陈铮凯课题组添加醋酸钠为碱, 以二氯甲烷为溶剂, 在50 ℃下反应以优秀的收率和非对映选择性得到一系列反式1,2-二氢苯并呋喃类化合物37. 陈亚静等发现当将温度提高至80 ℃, 无需添加任何添加剂, 仅通过延长反应时间, 两者就可以顺利地发生该[4+1]环化反应, 而对反应的产率和非对映选择性几乎没有影响.
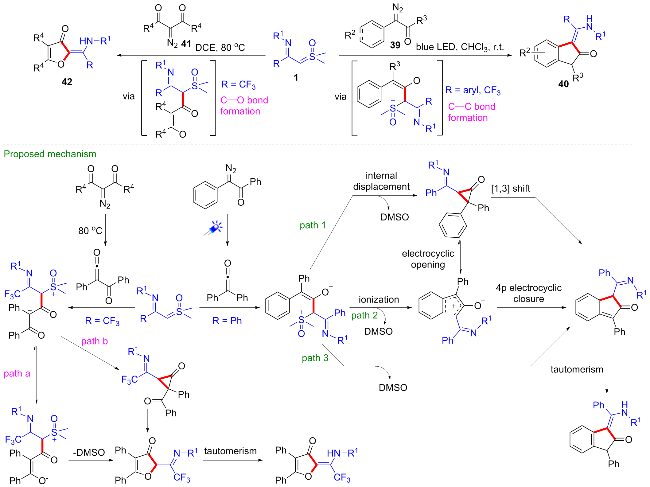
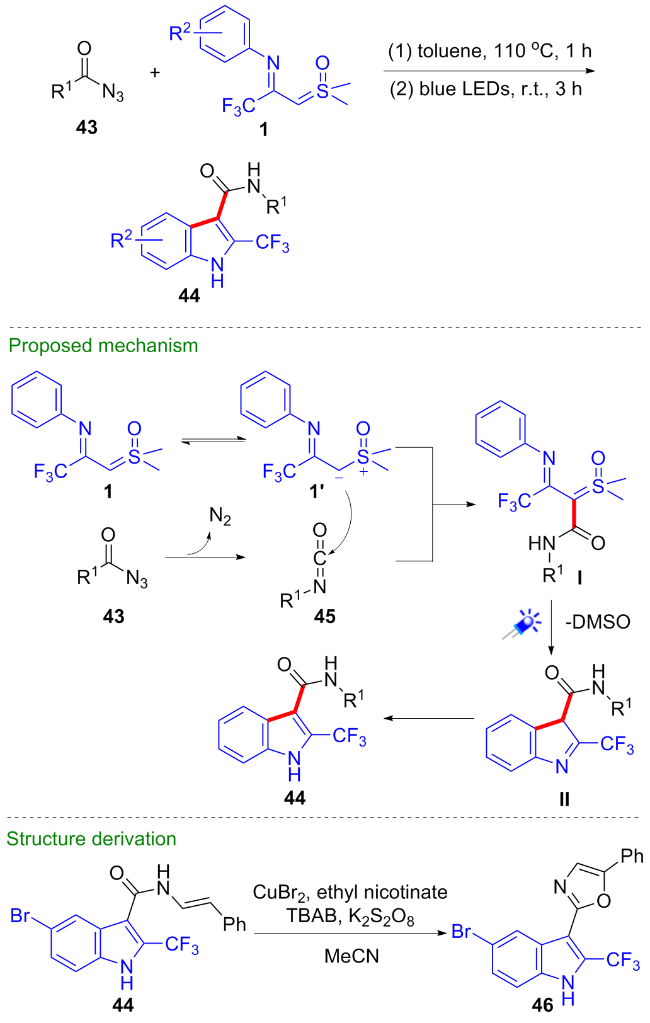
2024年, 刘培念课题组[34]报道了一种新颖的底物调节的亚胺酰基亚砜叶立德与重氮化合物参与的[4+1]环化反应(Scheme 19). 该反应策略不需要添加任何催化剂, 当使用α-芳基重氮酮39作为底物时, 反应在光照条件下进行, 芳基亚胺酰基亚砜叶立德与CF3-亚胺酰基亚砜叶立德均可顺利发生该环化反应, 得到2-茚酮类化合物40, 但CF3-亚胺酰基亚砜叶立德产率略低; 当使用2-重氮-1,3-二酮类化合物41为底物时, 反应在加热条件下进行, 仅CF3-亚胺酰基亚砜叶立德可顺利发生环化反应, 得到2(2H)-呋喃酮类化合物42. 在反应中, 重氮化合物在光照或加热条件下首先发生重排原位生成烯酮化合物, 与亚胺酰基亚砜叶立德加成得到活性偶极中间体, 随后选择性地生成碳碳键或碳氧键构建五元环体系.
2 作为卡宾前体参与的环化反应
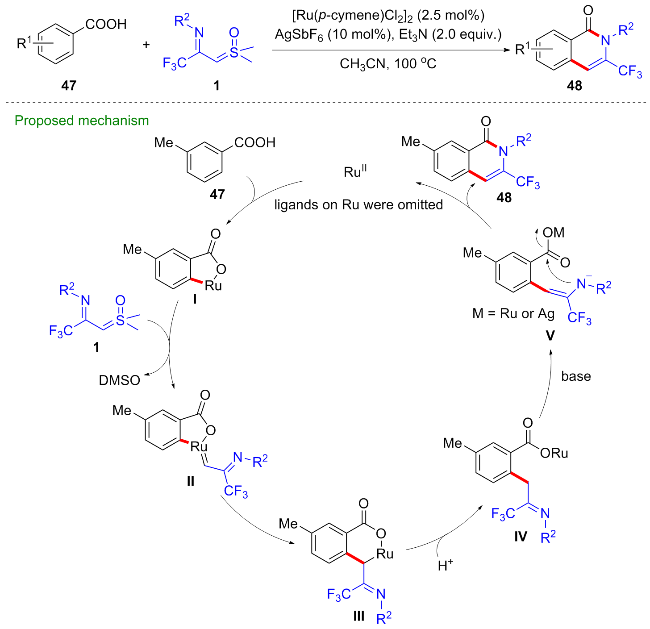
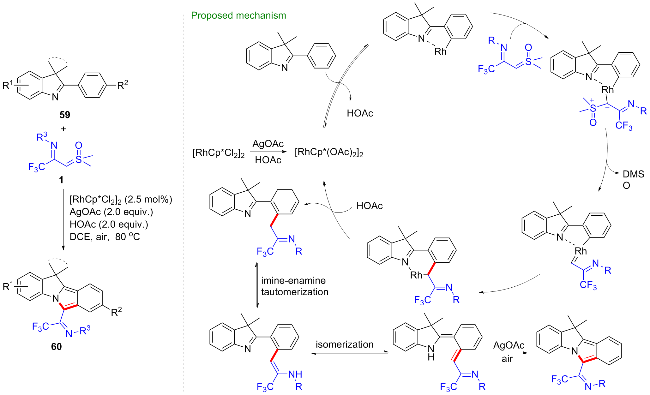
过渡金属催化的导向碳氢键活化反应具有反应效率高、选择性好、原子经济等优点, 已经发展成为高效构筑碳碳键和碳杂键的新方法[36]. 其中, 卡宾前体参与的碳氢键活化反应已经发展为一种新的碳氢键转化类型和方法[37]. 近年来, 国内外科研工作者利用亚砜叶立德作为卡宾前体, 成功合成了一系列在药物活性分子中普遍存在的杂环骨架[38]. 2022年, 程国林等[39]利用CF3-亚胺酰基亚砜叶立德作为卡宾前体, 实现了钌催化的苯甲酸47与CF3-亚胺酰基亚砜叶立德的碳氢键活化/环化反应, 以中等到良好的收率得到了一系列的3-三氟甲基异喹诺啉酮类化合物48 (Scheme 21). 其中苯甲酸中的羧基作为弱共价导向基团, 促进了邻位碳氢键活化的进行. 首先苯甲酸与钌催化剂发生弱的氧配位, 随后发生邻位碳氢键活化得到五元环钌中间体I, 该中间体捕获CF3-亚胺酰基亚砜叶立德生成钌卡宾II, 经迁移插入反应得到六元环钌中间体III, 质子化得到烷基化中间体IV, 在碱作用下经烯胺化、分子内酰胺化最终得到异喹诺啉酮产物48.
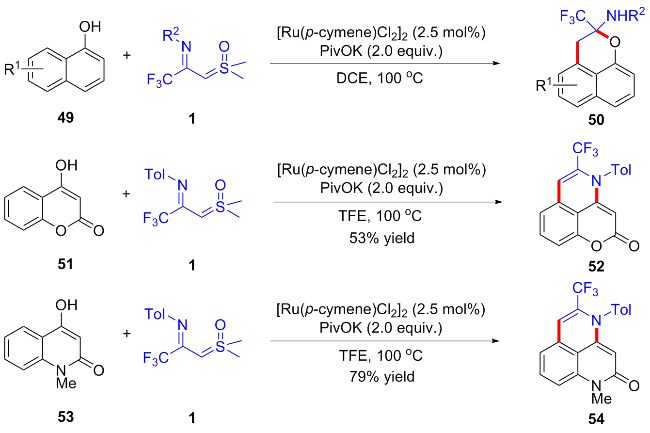
随后, 吴晓峰与陈铮凯课题组[40]利用CF3-亚胺酰基亚砜叶立德作为卡宾前体, 实现了钌催化的α-萘酚49与CF3-亚胺酰基亚砜叶立德的碳氢键活化/环化反应, 得到了一系列2-三氟甲基-2,3-二氢苯并[de]色烯-2-胺类化合物50 (Scheme 22). 其中酚羟基作为弱共价导向基团, 实现α-萘酚peri位选择性碳氢键活化, 随后发生亲核加成反应得到目标产物. 有趣的是, 当使用4-羟基香豆素51或4-羟基-1-甲基喹诺啉酮53作为底物时, 反应能顺利发生peri位碳氢键活化, 但随后经分子内脱水分别得到吡喃酮[2,3,4-ij]异喹啉-2(4H)-酮(52)和苯并[de][1,6]萘吡啶-2(4H)-酮(54).
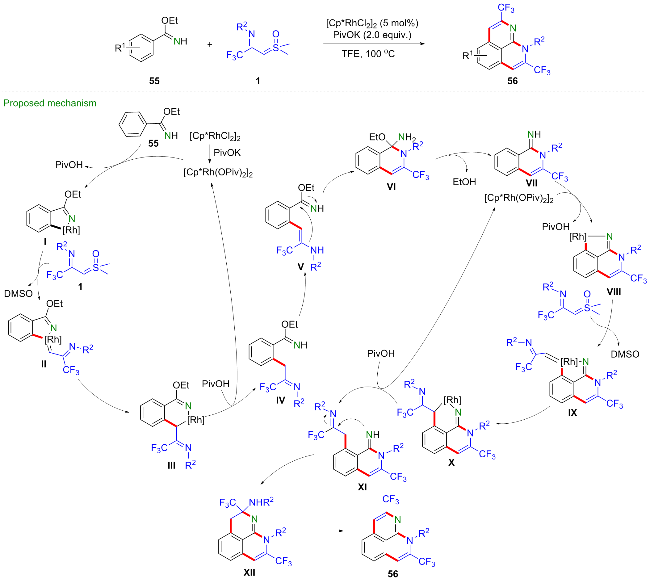
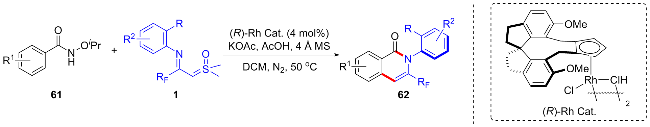
考虑到苯甲亚胺酸酯55既可以作为导向基团又可以参与环化反应, 吴晓峰与陈铮凯等[41]实现了铑催化的苯甲亚胺酸酯与CF3-亚胺酰基亚砜叶立德的双碳氢键活化串联环化反应, 合成得到了一系列三氟甲基取代的苯并[de][1,8]萘吡啶类化合物56 (Scheme 23). 该新颖的合成策略在高效地生成四根新的化学键的同时构建了两个氮杂环, 此外, 作者还研究了稠环芳香族产物的光电学性质. 2024年, 程国林课题组[42]利用[Cp*RhCl2]2作为催化剂, 醋酸钠作为碱, 六氟锑酸银作为添加剂,甲苯中100 ℃下反应, 也得到了类似的反应结果. 苯甲亚胺酸酯与铑催化剂发生环金属化得到五元环铑中间体I, 该中间体捕获CF3-亚胺酰基亚砜叶立德得到铑卡宾中间体II, 经迁移插入反应、质子化得到烷基化中间体IV, 然后经烯胺化、分子内环化、消除反应得到中间体VII, 在亚胺键的配位作用下经相同过程再次发生碳氢键活化/串联环化反应得到中间体XII, 最后发生消除反应脱去一分子胺得到目标产物56.
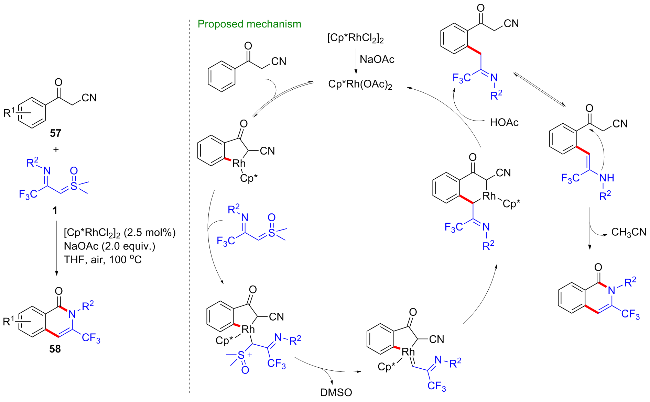
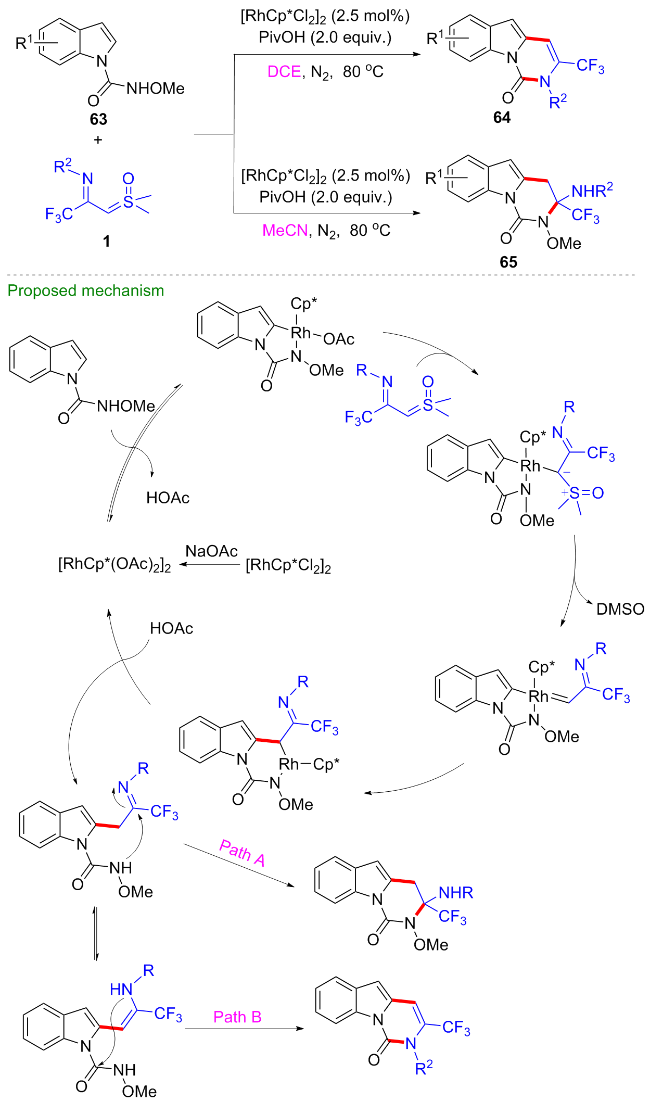
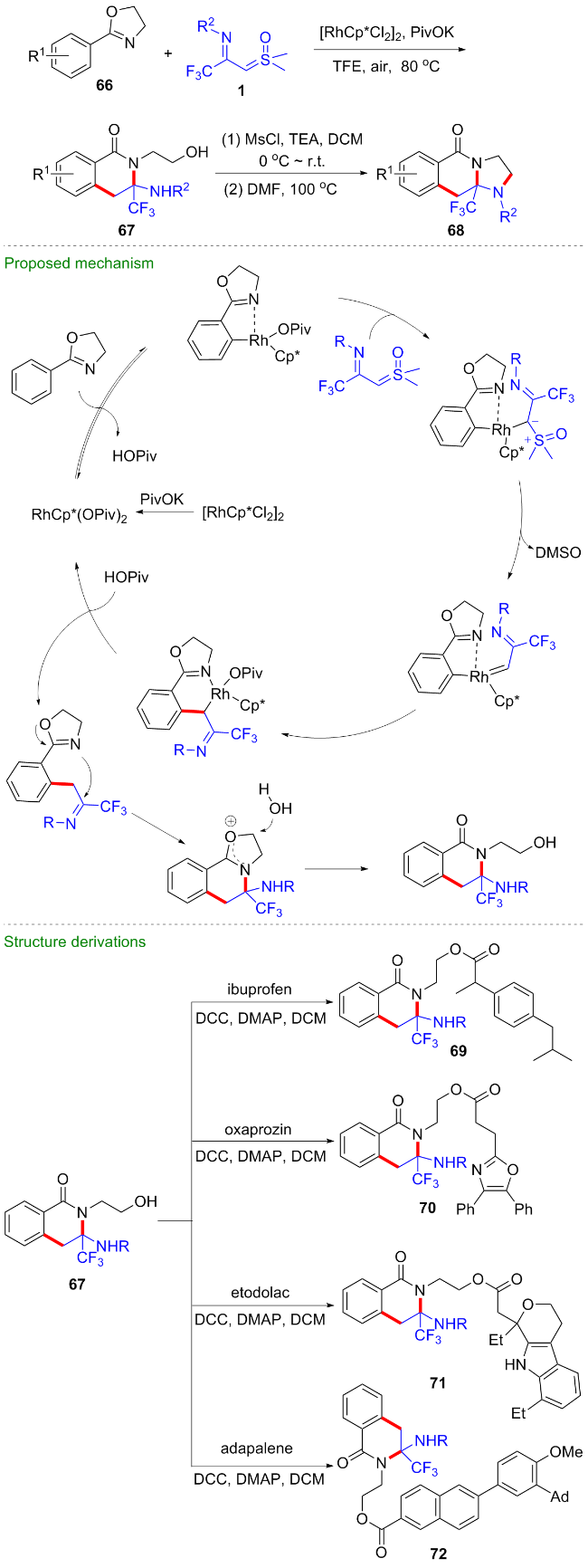
2024年, 范学森课题组[47]利用噁唑啉为导向基团, 开发了CF3-亚胺酰基亚砜叶立德参与的形式[4+2]环加成反应, 在铑(III)催化剂的作用下, 反应经过噁唑啉辅助的芳基烷基化、分子内缩合、水促进的噁唑啉开环等过程, 高效地构建了N-羟乙基取代的异喹啉酮67 (Scheme 28). 值得一提的是, 由于产物的高度官能团化, 产物经甲磺酰化后, 在碱性条件下可顺利地转化为咪唑并-CF3-异喹啉酮类化合物68. 此外, 产物还可以与布洛芬、奥沙普秦、依托度酸、阿达帕林等临床药物在N,N'-二环己基碳二亚胺(DCC)的作用下发生缩合反应, 得到一系列药物衍生物69~72, 这对新药研发具有潜在的重要用途.
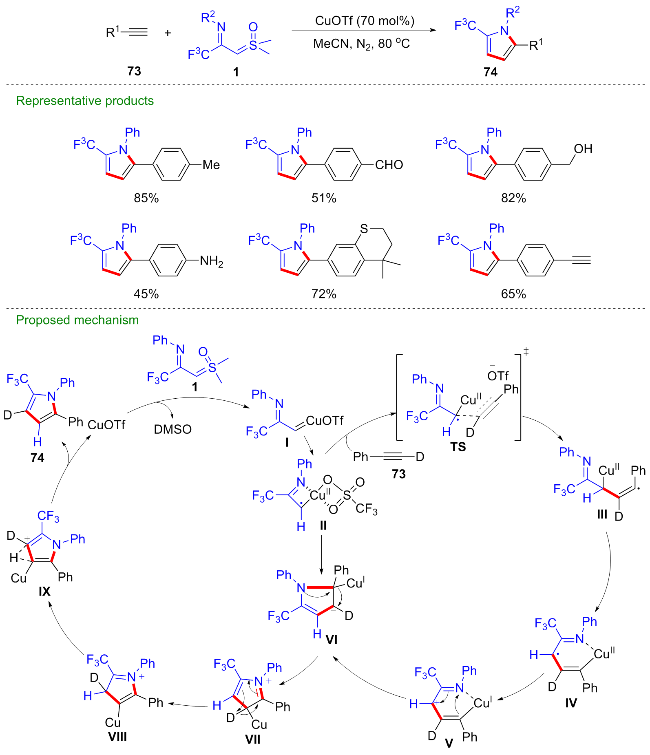
2024年, 熊彪课题组[48]报道了三氟甲磺酸铜催化的CF3-亚胺酰基亚砜叶立德与末端炔烃73的[3+2]环加成反应, 能以优秀的产率得到5-三氟甲基吡咯类化合物74, 为该类化合物的合成提供了实用性方法(Scheme 29). 该反应具有底物适用性广及良好的官能团兼容性等优点, 醛基、羰基、羟基、胺基、酯基等活性基团均能兼容. 作者通过控制实验及密度泛函理论(DFT)推测反应过程包含铜卡宾自由基对末端炔烃的加成及氢转移等步骤. 首先CF3-亚胺酰基亚砜叶立德与三氟甲磺酸铜反应得到铜卡宾I, 随后发生单电子转移过程得到铜卡宾自由基II, 进攻炔键得到烯基自由基III, 经1,3-金属迁移得到自由基IV, 再次发生单电子转移得到阳离子中间体V, 分子内环化得到中间体VI, 最后经异构化、氢迁移等过程得到最终产物.
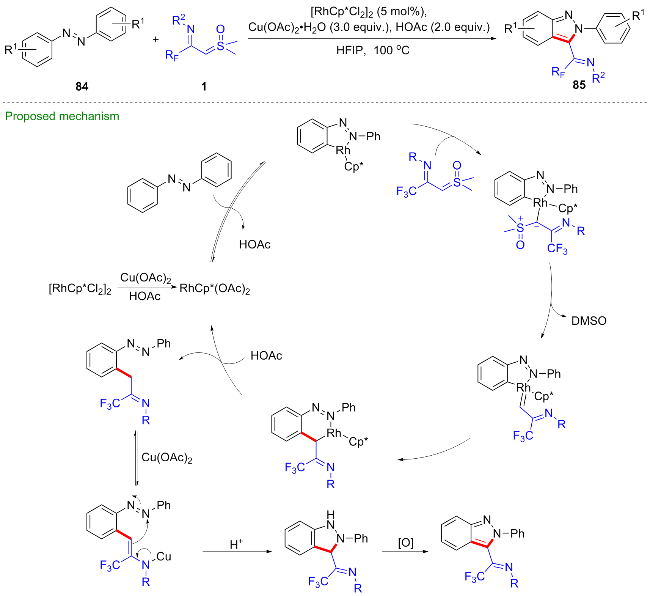
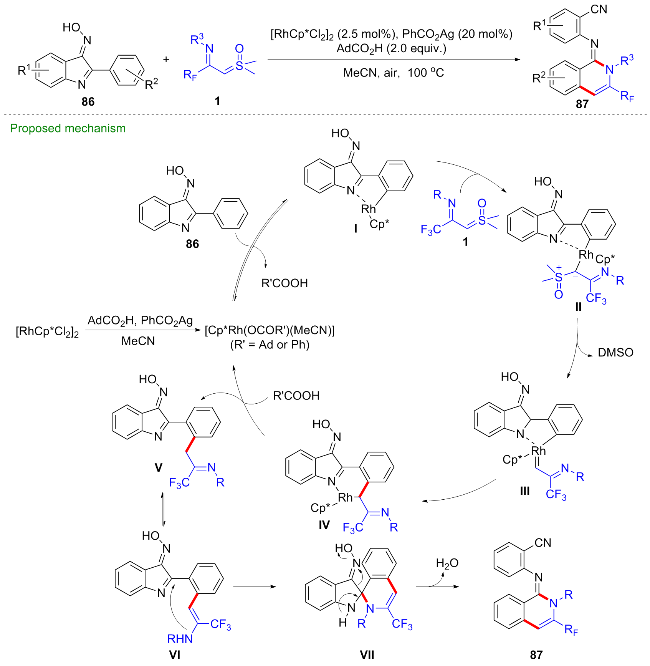
最近, 陈铮凯课题组[53]发展了一种铑(III)催化的芳基肟86与CF3-亚胺酰基亚砜叶立德的碳氢键活化/串联环化反应, 得到了一系列N-(2-氰芳基)-3-三氟甲基异喹啉-1(2H)-亚胺类化合物87 (Scheme 34). 作者依据控制实验结果, 推测反应过程包含了连续的碳氢键活化、分子内螺环化、碳碳键活化及氮氧键断裂等步骤, 最终以优异的产率得到目标产物. 首先, [Cp*RhCl2]2与1-金刚烷甲酸、苯甲酸银及乙腈作用, 生成活性铑(III)催化剂, 然后经碳氢键活化得到环铑中间体I, 与CF3-亚胺酰基亚砜叶立德配位得到铑烷基配合物II, 经α-消除离去一分子二甲基亚砜(DMSO)得到铑卡宾中间体III, 该中间体发生迁移插入得到六元环铑中间体IV, 质子化后得到亚胺中间体V, 并异构化为中间体VI, 在催化剂Rh/Ag的作用下发生分子内亲核加成得到螺环中间体VII, 最终经过协同的氮氧键及碳碳键断裂得到产物.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论与展望
综上所述, 近年来关于CF3-亚胺酰基亚砜叶立德参与的环化反应研究取得了较大的进展, 这些方法丰富了氟烷基取代的杂环化合物的合成途径, 而且在药物化学等领域也有较广泛的应用. 尽管如此, 对于CF3-亚胺酰基亚砜叶立德的研究仍具有较大的研究潜力和发展空间. 在已报道的CF3-亚胺酰基亚砜叶立德作为卡宾前体参与的环化反应研究中, 大部分反应都需要使用昂贵的Rh催化剂, 使用廉价金属催化剂的反应仍待开发. 另外, CF3-亚胺酰基亚砜叶立德参与的不对称催化反应报道较少, 随着光催化、电催化等绿色合成方法的不断完善, 相信可以实现对CF3-亚胺酰基亚砜叶立德参与的环化反应的立体选择性控制, 为氟烷基修饰的杂环化合物的不对称合成提供新的策略. 我们相信随着科技的进步以及广大科研工作者的努力, 未来将会开发出更多CF3-亚胺酰基亚砜叶立德参与的反应类型, 拓展其应用范围, 从而进一步推动合成化学、药物化学及材料化学等领域的发展.
(Cheng, F.)