


1 Introduction
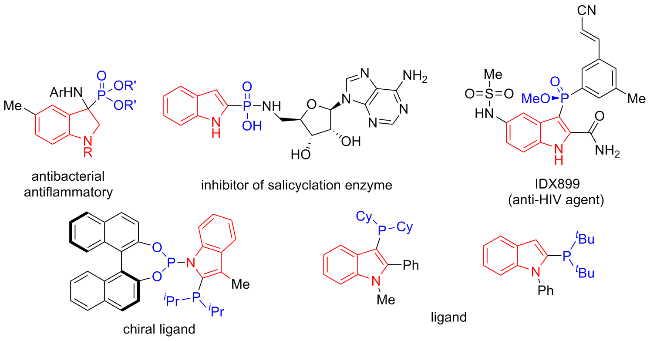
Organophosphorus compounds is recognized as one valuable functional scaffold in organic chemistry,[1] which have excellent application prospects as materials,[2] pharmaceuticals,[3-4] and ligands.[5] Among these compounds, phosphinoylindoles are basic elementary components of active natural products and materials in pharmaceuticals, demonstrating promising application prospects in functional molecules design (Figure 1).[6] Therefore, the synthetic methodology of phosphinoylindoles has attracted considerable research attention in the scientific community.[7] Despite these remarkable advancements, the development of novel and efficient synthetic methodologies for constructing phosphorylated indoles remains imperative.
Over the last few decades, numerous synthetic strategies have been developed for efficient synthesis of phosphonylated heterocycles. These approaches primarily encompass direct phosphonylation of heterocycles catalyzed by transition metals, visible light or electricity, and addition/ coupling reactions of heterocyclic compounds (Scheme 1, a).[8] Furthermore, owing to their unique reactivity, isocyanides serve as highly efficient synthons for constructing heterocyclic frameworks.[9] Hence, the cascade cyclization reactions involving isocyanides and phosphoryl groups have been widely applied to the synthesis of heterocyclic compounds containing phosphorus skeletons (Scheme 1b, path 1).[10] In 2018, Yang’s group[10g] have demonstrated a new, mild approach for the synthesis of 2-phosphinoyl- indoles through photoredox catalysis without the use of ex- ternal oxidant. In 2021, Yu’s group[10j] developed a visible light-induced proton-coupled electron transfer strategy for the generation of phosphorus-centered radicals, via which a wide range of phosphorylated heterocycles were constructed. In 2022, Wu’s group[10k] have successfully used single cobaloxime photocatalysis for the synthesis of phosphorylated heteroaromatics under mild redox-neutral conditions, and a variety of phosphine oxides directly added to isocyanides via P-radical addition and β-H elimination. In 2024, Studer’s group[10m] presented a radical cascade addition cyclization sequence to access quinoline- based benzophosphole oxides from ortho-alkynylated aromatic phosphine oxides using various aryl isonitriles as radical acceptors. However, these reactions mainly proceeded through a radical mechanism, while the use of phosphorus central ions for the construction of phosphonylated heterocycles have been rarely reported.[[8c-8f] In 2024, Xu’s group[11] reported a silver catalyzed P-centered anion-initiated cascade reaction of functionalized arylisocyanides with H-phosphorus oxides (Scheme 1b, path 2). Although this approach has efficiently achieved the construction of phosphonated indoles, the reaction conditions remain suboptimal due to the reliance on noble metal catalysts and thermal activation.

Inspired by our research group’s interest in isocyanide chemistry and green synthesis,[12] and aiming to expand the cascade cyclization reactions involving phosphoryl anions and isocyanides, we developed a metal-free annulation reaction that efficiently converts readily available 2-iso- cyanobenzaldehydes and aryl phosphine oxides into 2- phosphinoylindoles under mild conditions (Scheme 1c). This conversion pathway provides a new strategy for the synthesis of highly valuable phosphorylated heterocycle compounds.
2 Results and discussion
Initially, the cyclization reaction of 2-isocyanobenzalde- hyde 1a and diphenyl phosphine oxide 2a was chosen as a model to screen the reaction conditions. We firstly examined the solvents and it was found that MeCN was used to achieve high yield (Table 1, Entries 1~4). When MeCN was used as the solvent, a series of bases including 1,4- diazabicyclo[2.2.2]octane (DABCO), t-BuOK, tetramethyl- guanidine (TMG) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were tested (Table 1, Entries 4~8), and the results showed that the desired product 3aa was obtained in 73% yield in the presence of DBU (Table 1, Entry 4). To further optimize the reaction, the temperature was screened, and the yield of 3aa was found to decrease at both elevated and reduced temperature. Furthermore, after determining the optimal base loading to be 2.5 equiv., the influence of substrate equivalents on the reaction efficiency was subsequently investigated. Notably, when substrate 2a was employed at 2.5 equiv., the reaction demonstrated enhanced productivity, achieving a product yield of up to 85% (Table 1, Entries 10~13). Finally, a series of control experiments were conducted to investigate the effects of oxygen and water on the reaction. The results indicated that water exhibited no significant effect on the reaction process, whereas the presence of oxygen decreased the product yield. Therefore, the reaction conditions in Entry 13 was proved to be the optimal.
Table 1 Optimization of the reaction conditionsa |

| Entry | Base | Solvent | Yieldb/% |
|---|---|---|---|
| 1 | DBU | CH2Cl2 | 55 |
| 2 | DBU | DMSO | 67 |
| 3 | DBU | Toluene | 31 |
| 4 | DBU | MeCN | 73 |
| 5 | DABCO | MeCN | N.D. |
| 6 | TMG | MeCN | 69 |
| 7 | t-BuOK | MeCN | 51 |
| 8 | Na2CO3 | MeCN | N.D. |
| 9c | DBU | MeCN | 76 |
| 10c, d | DBU | MeCN | N.D. |
| 11c, e | DBU | MeCN | 85 |
| 12c, f | DBU | MeCN | 74 |
| 13c, e, g | DBU | MeCN | 84 (87)h |
a Unless otherwise noted, the reactions were carried out with 1a (0.15 mmol), 2a (2.0 equiv.), base (2.0 equiv.), solvent (1 mL), under N2, 25 ℃, 12 h; b HPLC yields; c base (2.5 equiv.); d 1a∶2a=2.0∶1.0, 2a (0.15 mmol); e 1a∶2a=1.0∶2.5; f 1a∶2a=1.0∶4.0; g 1a (0.2 mmol); h Isolated yield. ND=not detected. |
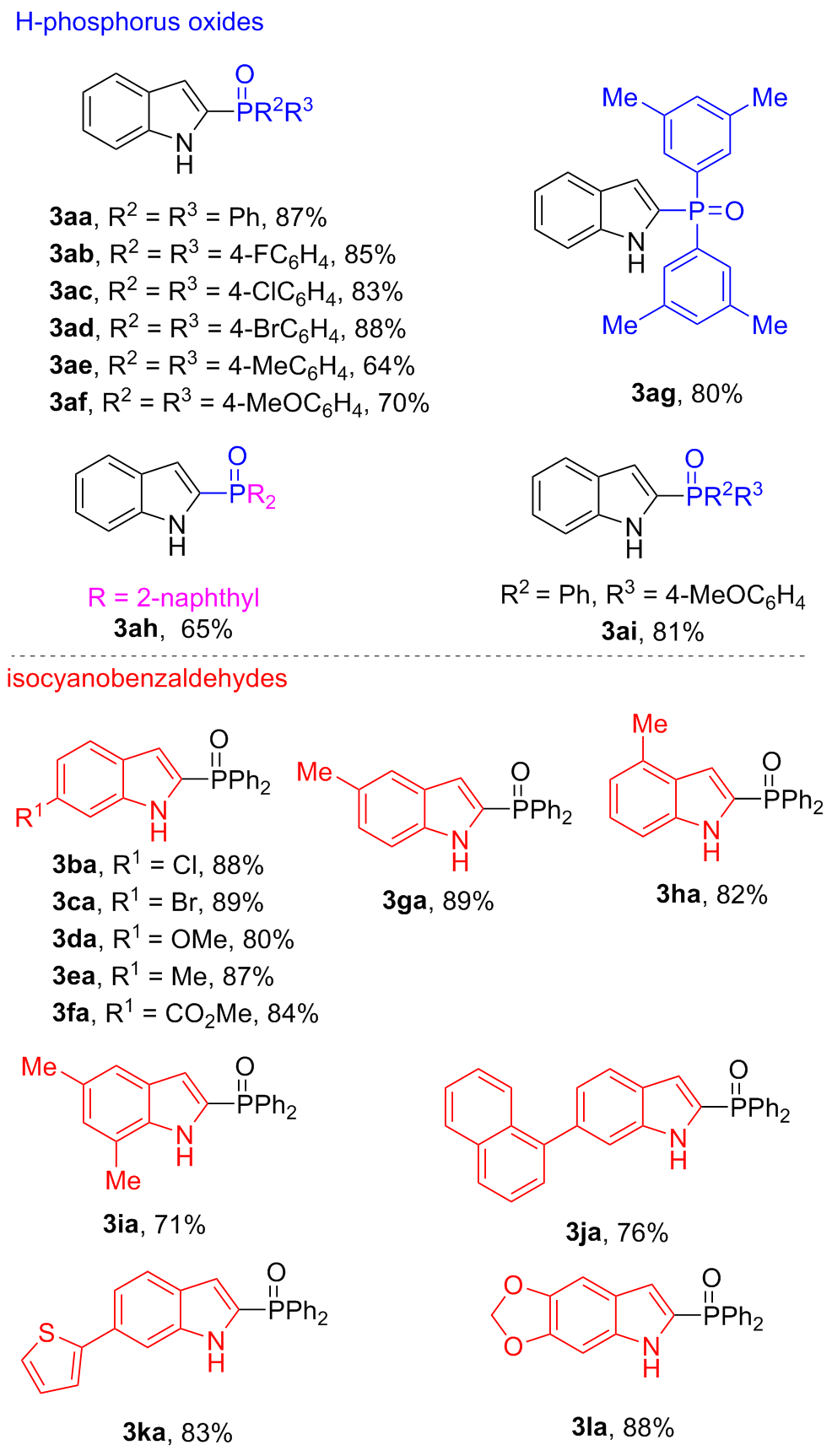
With the optimized reaction conditions, we firstly examined the substrate scope of the H-phosphorus oxides (Table 2). When para-substituted aryl phosphine oxides bearing electron-withdrawing groups were employed, the corresponding 2-phosphinoyl indoles were obtained in 83%~88% yields (3ab~3ad). In comparison, substrates with electron-donating groups (OMe, Me) afforded products 3ae~3af in slightly lower yields. In addition, the scope could be extended to 3,5-dimethyl-substituted aryl phosphine oxide (3ag). The product 3ah containing 2-naphthyl was synthesized in moderate yield. However, alkyl phosphine oxides failed to generate the desired products under the same conditions, indicating the necessity of aromatic substrates for this transformation. Furthermore, the target product 3ai was also obtained in 81% yield when the R2 and R3 groups of the H-phosphorus oxide 2i were distinct. Subsequently, the substrate scope of 2-isocyano- benzaldehydes 1 were investigated (Table 2). Substrates with both electron-withdrawing and electron-donating substituents on the aromatic ring afford the corresponding products in excellent yields (3ba~3fa). Additionally, when the position and number of substituents on the aryl ring of the 2-isocyanobenzaldehydes were systematically varied, the corresponding products were obtained in good yields (Table 2, 3ga~3ia). Finally, when the para-position of the aryl ring in substrate 1a was substituted with a 1-naphthyl or 2-thienyl group, the corresponding products were obtained in 76%~83% yields (Table 2, 3ja~3ka). Notably, product 3la, containing a 1,3-benzodioxole motif, was obtained in 88% yield.
Table 2 Substrate scope of 2-phosphonyl indolesa,b |
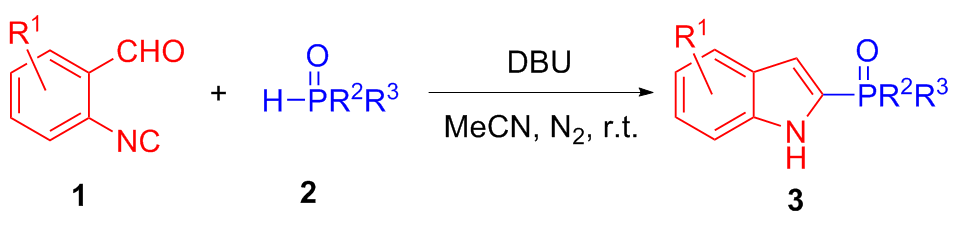
 |
a General conditions: 1 (0.2 mmol), 2 (0.5 mmol), DBU (0.5 mmol), MeCN (1 mL), under N2, at 25 ℃, 12 h. b Isolated yields. |
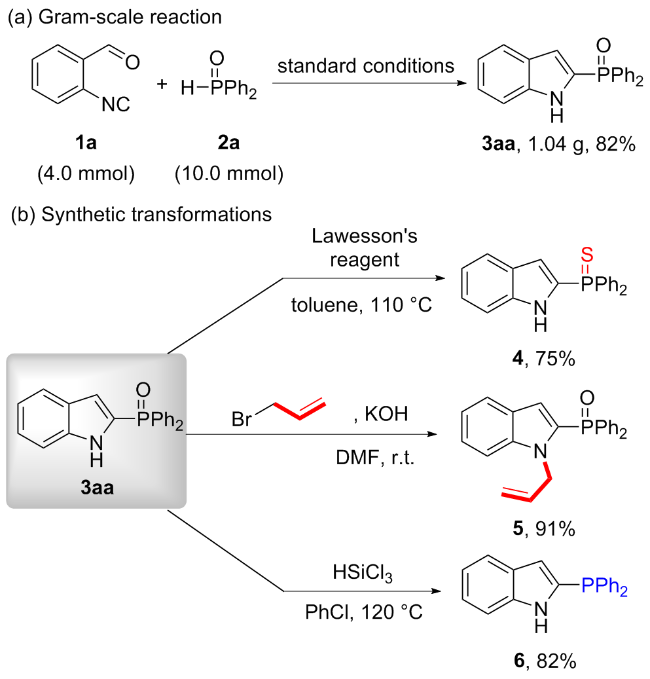
To evaluate the synthetic applicability of this cascade reaction, gram-scale syntheses of 3aa was performed. Under the standard conditions, 1.04 g of product 3aa was obtained in 82% yield (Scheme 2a). Subsequently, several synthetic transformations were carried out. At first, The thiolation of P=O bond of 3aa was conveniently realized by using Lawessonʼs reagent, affording corresponding pro- duct 4 yield of 75%.[13] In the presence of potassium hydroxide, compound 3aa underwent nucleophilic substitution with allyl bromide in N,N-dimethylformamide (DMF), affording the allylation product 5 in 91% yield.[14] Finally, the reduction of phosphinoylindole 3aa using HSiCl3 afforded 6 in 82% yield (Scheme 2b).[7d]
Subsequently, a series of control experiments were carried out to gain insights into the reaction mechanism. No desired product 3aa was observed when the cyclization reaction was treated in the absence of base. When 2,2,6,6- tetramethylpiperidine-1-oxyl (TEMPO, 5.0 equiv.) and 2,6-di-tert-butyl-4-methylphenol (BHT, 5.0 equiv.) were added as radical inhibitors under standard conditions, they could not markedly influence the yield of 3aa (Table 3). These results indicate that a P-centered radical pathway could most likely be ruled out in this transformation. When H-phosphorus oxide 2j containing a large site-blocking group was used as a substrate, bisphosphonylaminomethane 7 was obtained in 70% yield, while the corresponding 2-phosphonyl indole product could not be detected (Scheme 3). These results indicate that under steric hindrance, iso-cyanide 1a undergoes anionic 1,1-bisphosphination[16] competing with cyclization, further confirming the absence of a radical pathway in this synthetic strategy.
Table 3 Control experimenta |

| Entry | Variation of reaction condition | Yieldb/% |
|---|---|---|
| 1 | — | 87 |
| 2 | w/o base | 0 |
| 3 | TEMPO (5.0 equiv.) | 84 |
| 4 | BHT (5.0 equiv.) | 82 |
a General conditions: 1a (0.2 mmol), 2a (0.5 mmol), DBU (2.5 equiv.), MeCN (1 mL), under N2, 25 ℃, 12 h; b Isolated yields. |
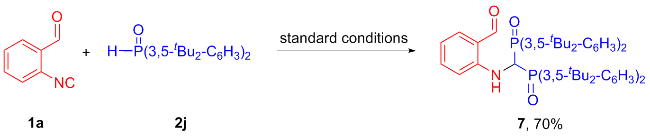
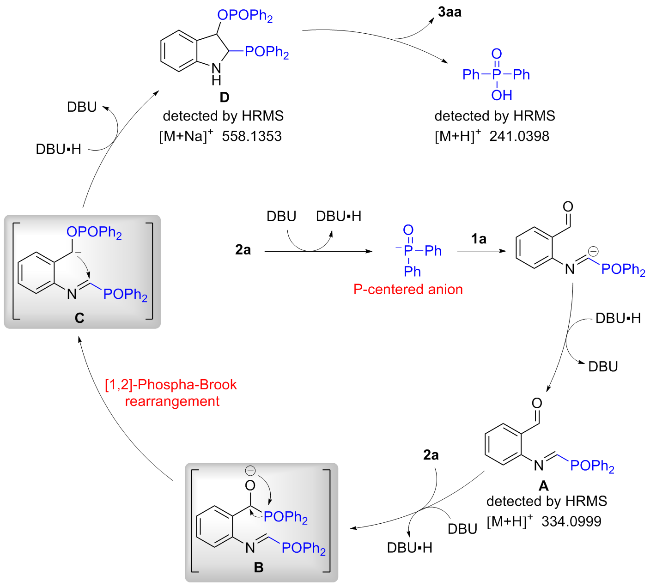
Based on the above results and previous reports in the literature,[15b,16] a plausible reaction mechanism is proposed (Scheme 4). Initially, the presence of DBU facilitates the deprotonation process of compound 2a, producing a P- center anion. Then, the nucleophilic addition of isocyanobenzaldehyde 1a leads to the imine intermediate A. Subsequently, intermediate A would be attacked by another H-phosphorus oxide 2a to form intermediate B, followed by a [1,2]-Phospha-Brook rearrangement to generate intermediate C. Intermediate C then undergoes cyclization and protonation to afford intermediate D. Finally, elimination of a molecule of diphenylphosphinic acid from intermediate D yields the final product 3aa. The molecular weight of the intermediates A, D and diphenylphosphinic acid were detected by HRMS.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In conclusion, we have established a practical and scala-ble synthetic strategy for constructing 2-phosphinoylindoles through base-promoted cascade reactions of isocyanobenzaldehydes with phosphorus oxides. This protocol features mild conditions, excellent functional group tolerance, and scalability. This approach represents a green and sustainable alternative to metal-catalyzed, photochemical, or electrochemical phosphorylation strategies. Extensive experimental validation confirms the versatility of this method, which offers significant advantages in phosphine ligand synthesis, biorthogonal chemistry, and drug discovery.
4 Experimental section
4.1 General information
Reactions were monitored by analytical thin-layer chro- matography (TLC) on silica gel plates (GF254). The TLC plates were isualized by shortwave (254 nm) or longwave (365 nm) UV light. Column chromatography was carried out using silica gel (200~300 mesh) to purify the products. 1H NMR (400 MHz), 13C NMR (101 MHz), 19F NMR (376 MHz) and 31P NMR (162 MHz) spectra were recorded in CDCl3 or DMSO-d6 on Bruker AVANCE III 400 MHz spectrometers using TMS as the internal standard (CDCl3 δH 7.26, δC 77.16; DMSO-d6 δH 2.50, δC 39.52). The high- resolution mass spectra (HRESIMS) were acquired using the water G2-Xs qtof mass spectrometer. Measured values are reported to 4 decimal places of the calculated value. The calculated values are based on the most abundant isotope. All reagents and solvents were purchased from commercial sources and used without further purification.
4.2 Experimental method
4.2.1 Typical procedure for synthesis of compounds 3
In a flame-dried 10.0 mL Schlenk tube equipped with a magnetic stir bar was charged sequentially with 1a (0.2 mmol, 1.0 equiv.) and 2a (0.5 mmol, 2.5 equiv.), followed by the addition of anhydrous MeCN (1.0 mL). To the resulting mixture was added DBU (0.5 mmol, 2.5 equiv.) under nitrogen atmosphere. Then the mixture was stirred at room temperature for 12 h under nitrogen atmosphere until the reaction was completed, as monitored by TLC analysis. The residue was purified by silica gel chromatography with petroleum ether/ethyl acetate (V∶V=1∶1) to afford 2-phosphinoyl indoles 3aa (87% yield). Compounds 3ab~3la were prepared through the same procedure.
(1H-Indol-2-yl)diphenylphosphine oxide (3aa): White solid, 87% yield. m.p. 193~193.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.87 (s, 1H), 7.71~7.40 (m, 12H), 7.25 (t, J=6.7 Hz, 1H), 7.09 (t, J=6.9 Hz, 1H), 6.54 (s, 1H); 13C NMR (101 MHz,CDCl3) δ: 139.8~139.6 (m), 132.20 (d, J=2.6 Hz), 132.19 (d, J=110.0 Hz), 131.98 (d, J=10.5 Hz), 128.6 (d, J=12.6 Hz), 128.3 (d, J=124.5 Hz), 127.4 (d, J=12.6 Hz), 124.2, 121.5, 120.3, 113.2 (d, J=16.0 Hz), 112.9~112.8 (m); 31P NMR (162 MHz, CDCl3) δ: 22.38; HRMS (ESI) calcd for C20H16NOPNa [M+Na]+ 340.0862, found 340.0870.
Bis(4-fluorophenyl)(1H-indol-2-yl)phosphine oxide (3ab): White solid, 85% yield. m.p. 238.4~238.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.87 (s, 1H), 7.67~7.56 (m, 6H), 7.30~7.25 (m, 1H), 7.13~7.09 (m, 5H), 6.49 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 166.7 (d, JC-F=3.0 Hz), 164.1 (d, JC-F=3.3 Hz), 139.8 (d, J=9.8 Hz), 134.4 (dd, J=12.1, 8.9 Hz), 128.5~128.2 (m), 127.4~127.0 (m), 124.6, 121.6, 120.6, 116.1 (dd, J=21.4, 13.9 Hz), 113.6 (d, J=16.4 Hz), 112.8; 19F NMR (376 MHz, CDCl3) δ: 105.61; 31P NMR (162 MHz, CDCl3) δ: 21.06; HRMS (ESI) calcd for C20H14F2NOPNa [M+Na]+ 376.0673, found 376.0682.
Bis(4-chlorophenyl)(1H-indol-2-yl)phosphine oxide (3ac): White solid, 83% yield. m.p. 278.8~279.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.84 (s, 1H), 7.60~7.53 (m, 6H), 7.40 (d, J=6.7 Hz, 4H), 7.31~7.25 (m, 1H), 7.12 (t, J=7.4 Hz, 1H), 6.51 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 139.9 (d, J=9.7 Hz), 139.2 (d, J=3.4 Hz), 133.3 (d, J=11.5 Hz), 130.3 (d, J=112.2 Hz), 129.1 (d, J=13.3 Hz), 127.2 (d, J=12.8 Hz), 127.0 (d, J=127.4 Hz), 124.8, 121.7, 120.7, 113.8 (d, J=16.5 Hz), 112.8; 31P NMR (162 MHz, CDCl3) δ: 20.91; HRMS (ESI) calcd for C20H14Cl2- NOPNa [M+Na]+ 408.0082, found 408.0089.
Bis(4-bromophenyl)(1H-indol-2-yl)phosphine oxide (3ad): White solid, 88% yield. m.p.>288 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 11.97 (s, 1H), 7.80 (d, J=7.0 Hz, 4H), 7.64~7.59 (m, 5H), 7.44 (d, J=8.1 Hz, 1H), 7.24 (t, J=7.4 Hz, 1H), 7.07 (t, J=7.3 Hz, 1H), 6.68 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ: 139.3 (d, J=9.8 Hz), 133.3 (d, J=11.1 Hz), 132.0 (d, J=12.8 Hz), 131.0, 128.7, 127.0 (d, J=93.6 Hz), 126.7 (d, J=3.1 Hz), 124.2, 121.6, 120.1, 113.0 (d, J=15.8 Hz), 112.4; 31P NMR (162 MHz, DMSO- d6) δ: 16.89. HRMS (ESI) calcd for C20H14Br2- NOPNa [M+Na]+ 495.9072, found 495.9078.
(1H-Indol-2-yl)di-p-tolylphosphine oxide (3ae): White solid, 64% yield. m.p. 260.1~260.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.31 (s, 1H), 7.59~7.50 (m, 6H), 7.25~7.20 (m, 5H), 7.09 (t, J=7.2 Hz, 1H), 6.54~6.53 (m, 1H), 2.39 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 142.7 (d, J=2.8 Hz), 139.4 (d, J=9.4 Hz), 132.0 (d, J=10.9 Hz), 129.3 (d, J=13.0 Hz), 129.1 (d, J=113.1 Hz), 129.0 (d, J=124.2 Hz), 127.4 (d, J=12.4 Hz), 124.1, 121.5, 120.3, 112.9 (d, J=15.2 Hz), 112.7, 21.8; 31P NMR (162 MHz, CDCl3) δ: 22.51; HRMS (ESI) calcd for C22H20NOPNa [M+Na]+ 368.1175, found 368.1183.
(1H-Indol-2-yl)bis(4-methoxyphenyl)phosphine oxide (3af): White solid, 70% yield. m.p. 264.3~264.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.76 (s, 1H), 7.62~7.57 (m, 5H), 7.51 (d, J=8.1 Hz, 1H), 7.26~7.24 (m, 1H), 7.11 (t, J=7.3 Hz, 1H), 6.92 (d, J=7.2 Hz, 4H), 6.55 (s, 1H), 3.84 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 162.7 (d, J=2.6 Hz), 139.2 (d, J=9.0 Hz), 133.9 (d, J=11.9 Hz), 129.4 (d, J=124.4 Hz), 127.5 (d, J=12.2 Hz), 124.2, 123.1, 121.6, 120.4, 114.2 (d, J=13.1 Hz), 112.9 (d, J=15.3 Hz), 112.6, 55.5; 31P NMR (162 MHz, CDCl3) δ: 21.90; HRMS (ESI) calcd for C22H20NO3PNa [M+Na]+ 400.1073, found 400.1082.
Bis(3,5-dimethylphenyl)(1H-indol-2-yl)phosphine oxide (3ag): White solid, 80% yield. m.p. 283.0~283.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.08 (s, 1H), 7.62 (q, J=8.0 Hz, 2H), 7.36 (d, J=13.0 Hz, 4H), 7.29 (t, J=7.6 Hz, 1H), 7.18~7.12 (m, 3H), 6.62 (s, 1H), 2.31 (s, 12H); 13C NMR (101 MHz, CDCl3) δ: 139.3 (d, J=9.1 Hz), 138.2 (d, J=13.3 Hz), 134.0 (d, J=2.8 Hz), 132.8 (d, J=109.1 Hz), 129.5 (d, J=10.5 Hz), 128.2 (d, J=4.1 Hz), 127.6 (d, J=12.3 Hz), 124.0, 121.5, 120.3, 112.9, 112.7, 21.4; 31P NMR (162 MHz, CDCl3) δ: 22.78; HRMS (ESI) calcd for C24H24NOPNa [M+Na]+ 396.1488, found 396.1497.
(1H-Indol-2-yl)di(naphthalen-2-yl)phosphine oxide (3ah): White solid, 64% yield. m.p.>288 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 11.99 (s, 1H), 8.45 (d, J=14.2 Hz, 2H), 8.11~8.07 (m, 5H), 8.02 (d, J=8.1 Hz, 2H), 7.78~7.73 (m, 2H), 7.70~7.60 (m, 5H), 7.45 (d, J=8.2 Hz, 1H), 7.23 (t, J=7.4 Hz, 1H), 7.07 (t, J=7.5 Hz, 1H), 6.76 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ: 139.3 (d, J=9.6 Hz), 134.4 (d, J=2.3 Hz), 133.0 (d, J=9.8 Hz), 132.1 (d, J=13.4 Hz), 130.5, 130.0, 129.4, 128.8 (d, J=49.6 Hz), 128.7, 128.4, 127.5 (d, J=63.2 Hz), 126.7, 126.5 (d, J=11.0 Hz), 124.0, 121.6, 120.0, 112.7 (d, J=15.2 Hz), 112.4; 31P NMR (162 MHz, DMSO-d6) δ: 17.58; HRMS (ESI) calcd for C28H20- NOPNa [M+Na]+ 440.1175, found 440.1184.
(1H-Indol-2-yl)(4-methoxyphenyl)(phenyl)phosphine oxide (3ai): White solid, 81% yield. m.p. 190~190.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.28 (s, 1H), 7.63~7.58 (m, 2H), 7.54~7.42 (m, 5H), 7.33 (td, J=7.6, 2.9 Hz, 2H), 7.17 (t, J=7.3 Hz, 1H), 7.02 (t, J=7.3 Hz, 1H), 6.83 (dd, J=8.8, 2.1 Hz, 2H), 6.46~6.45 (m, 1H), 3.75 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 162.8 (d, J=3.0 Hz), 139.5 (d, J=9.4 Hz), 133.9 (d, J=11.9 Hz), 133.1, 132.1 (d, J=2.7 Hz), 132.0 (d, J=10.6 Hz), 128.8 (d, J=124.4 Hz), 128.5 (d, J=12.6 Hz), 127.4 (d, J=12.4 Hz), 124.2, 123.2 (d, J=116.6 Hz), 121.5, 120.3, 114.2 (d, J=13.7 Hz), 113.0 (d, J=15.8 Hz), 112.7, 55.5; 31P NMR (162 MHz, CDCl3) δ: 22.23; HRMS (ESI) calcd for C21H18NO2PNa [M+Na]+ 370.0967, found 370.0975.
(6-Chloro-1H-indol-2-yl)diphenylphosphine oxide (3ba): White solid, 88% yield. m.p. 284.7~284.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 12.17 (s, 1H), 7.65 (dd, J=12.5, 7.7 Hz, 4H), 7.58~7.50 (m, 3H), 7.48~7.42 (m, 5H), 7.05 (d, J=8.4 Hz, 1H), 6.48 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 140.0 (d, J=9.8 Hz), 132.4 (d, J=3.0 Hz), 131.9 (d, J=10.6 Hz), 131.8 (d, J=111.1 Hz), 130.2, 129.8, 128.7 (d, J=12.7 Hz), 125.9 (d, J=12.6 Hz), 122.4, 121.3, 113.1 (d, J=16.0 Hz), 112.6; 31P NMR (162 MHz, CDCl3) δ: 22.51; HRMS (ESI) calcd for C20H15ClNOPNa [M+Na]+ 374.0472, found 374.0480.
(6-Bromo-1H-indol-2-yl)diphenylphosphine oxide (3ca): White solid, 89% yield. m.p. 283.4~283.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.94 (s, 1H), 7.71 (s, 1H), 7.65 (dd, J=12.6, 7.4 Hz, 4H), 7.55 (t, J=7.1 Hz, 2H), 7.45~7.42 (m, 5H), 7.19 (dd, J=8.5, 1.1 Hz, 1H), 6.49 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 140.2 (d, J=9.1 Hz), 132.5 (d, J=2.7 Hz), 132.0 (d, J=10.6 Hz), 131.8 (d, J=110.8 Hz), 129.8, 128.8 (d, J=13.1 Hz), 126.2 (d, J=12.5 Hz), 123.9, 122.8, 118.1, 115.6, 113.2 (d, J=15.8 Hz); 31P NMR (162 MHz, CDCl3) δ: 22.37; HRMS (ESI) m/z calcd for C20H15BrNOPNa [M+Na]+ 417.9967, found 417.9977.
(6-Methoxy-1H-indol-2-yl)diphenylphosphine oxide (3da): White solid, 80% yield. m.p. 267.8~268.0 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.47 (s, 1H), 7.68 (dd, J=12.5, 7.5 Hz, 4H), 7.53 (t, J=7.2 Hz, 2H), 7.44~7.42 (m, 5H), 6.99 (s, 1H), 6.77 (d, J=8.7 Hz, 1H), 6.46 (s, 1H), 3.80 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 158.2, 140.6 (d, J=9.5 Hz), 132.3 (d, J=110.3 Hz), 132.3~131.9 (m), 130.8 (d, J=11.5 Hz), 129.1 (d, J=12.1 Hz), 128.6 (d, J=12.6 Hz), 122.3, 121.8 (d, J=12.7 Hz), 113.7 (d, J=15.9 Hz), 112.0, 94.5, 55.6; 31P NMR (162 MHz, CDCl3) δ: 22.10; HRMS (ESI) m/z calcd for C21H18NO2PNa [M+Na]+ 370.0967, found 370.0977.
(6-Methyl-1H-indol-2-yl)diphenylphosphine oxide (3ea): White solid, 87% yield. m.p. 275.2~275.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.95 (s, 1H), 7.68 (dd, J=11.9, 7.7 Hz, 4H), 7.53~7.41 (m, 7H), 7.31 (s, 1H), 6.94 (d, J=7.9 Hz, 1H), 6.51 (s, 1H), 2.44 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 139.9 (d, J=10.1 Hz), 134.5, 132.8, 132.2~131.9 (m), 131.7, 128.6 (d, J=12.7 Hz), 127.4 (d, J=124.9 Hz), 125.4 (d, J=11.8 Hz), 122.6, 121.2, 113.3 (d, J=15.6 Hz), 112.3, 22.0; 31P NMR (162 MHz, CDCl3) δ: 22.25; HRMS (ESI) calcd for C21H18NOPNa [M+Na]+ 354.1018, found 354.1026.
2-(Diphenylphosphoryl)-1H-indol-6-yl acetate (3fa): White solid, 84% yield. m.p.>288 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.38 (s, 1H), 8.12 (s, 1H), 7.74~7.57 (m, 12H), 6.73 (s, 1H), 3.86 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 166.8, 138.3 (d, J=9.7 Hz), 133.2 (d, J=118.3 Hz), 132.67, 132.4 (d, J=2.5 Hz), 131.6, 131.3 (d, J=10.3 Hz), 130.0 (d, J=11.7 Hz), 128.8 (d, J=12.3 Hz), 124.8, 121.5, 120.2, 114.3, 112.1 (d, J=15.1 Hz), 52.0; 31P NMR (162 MHz, DMSO-d6) δ: 17.81; HRMS (ESI) calcd for C22H18NO3PNa [M+H]+ 376.1097, found 376.1103.
(5-Methyl-1H-indol-2-yl)diphenylphosphine oxide (3ga): White solid, 89% yield. m.p. 242.2~242.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.64 (s, 1H), 7.69~7.64 (m, 4H), 7.53~7.46 (m, 3H), 7.39 (td, J=7.6, 2.9 Hz, 4H), 7.34 (s, 1H), 7.09 (d, J=8.4 Hz, 1H), 6.45~6.44 (m, 1H), 2.41 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 138.1 (d, J=9.7 Hz), 132.2 (d, J=110.1 Hz), 132.4~131.8 (m), 129.5, 128.7, 128.5 (d, J=12.6 Hz), 127.6, 127.5 (d, J=2.1 Hz), 126.2, 120.7, 112.6 (d, J=9.1 Hz), 112.5, 21.9; 31P NMR (162 MHz, CDCl3) δ: 22.42; HRMS (ESI) calcd for C21H18NOPNa [M+Na]+ 354.1018, found 354.1026.
(4-Methyl-1H-indol-2-yl)diphenylphosphine oxide (3ha): White solid, 82% yield. m.p. 264.6~264.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.67 (s, 1H), 7.69 (dd, J=11.1, 7.6 Hz, 4H), 7.52~7.41 (m, 7H), 7.16 (t, J=7.2 Hz, 1H), 6.89 (d, J=6.3 Hz, 1H), 6.55 (s, 1H), 2.47 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 139.4 (d, J=9.4 Hz), 132.8, 131.7, 132.1 (dd, J=21.0, 6.5 Hz), 131.2, 128.6 (d, J=12.6 Hz), 127.6 (d, J=12.4 Hz), 127.5 (d, J=124.8 Hz), 124.4, 120.3, 111.6 (d, J=15.7 Hz), 110.3, 18.9; 31P NMR (162 MHz, CDCl3) δ: 22.39; HRMS (ESI) calcd for C21H18- NOPNa [M+Na]+ 354.1018, found 354.1028.
(5,7-Dimethyl-1H-indol-2-yl)diphenylphosphine oxide (3ia): White solid, 71% yield. m.p. 235.6~235.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 10.57 (s, 1H), 7.68~7.63 (m, 4H), 7.53 (td, J=7.4, 1.3 Hz, 2H), 7.42 (td, J=7.7, 2.9 Hz, 4H), 7.20 (s, 1H), 6.91 (s, 1H), 6.47 (dd, J=3.7, 2.0 Hz, 1H), 2.52 (s, 3H), 2.39 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 137.6 (d, J=8.9 Hz), 132.3 (d, J=109.9 Hz), 132.2 (d, J=2.8 Hz), 132.0 (d, J=10.6 Hz), 130.1, 128.6 (d, J=12.6 Hz), 127.9 (d, J=97.3 Hz), 127.2 (d, J=14.8 Hz), 126.9, 121.8, 118.4, 113.4 (d, J=15.8 Hz), 21.5, 17.2; 31P NMR (162 MHz, CDCl3) δ: 22.25; HRMS (ESI) calcd for C22H20NOPNa [M+Na]+ 368.1175, found 368.1184.
(6-(Naphthalen-1-yl)-1H-indol-2-yl)diphenylphosphine oxide (3ja): White solid, 75% yield. m.p. 201~202 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.07 (s, 1H), 7.97 (dd, J=21.4, 8.0 Hz, 2H), 7.85 (d, J=8.2 Hz, 1H), 7.77~7.72 (m, 5H), 7.66~7.46 (m, 11H), 7.18 (d, J=8.0 Hz, 1H), 6.75 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ: 140.2, 139.3 (d, J=9.5 Hz), 135.8, 133.4, 133.1, 132.3 (d, J=2.0 Hz), 132.1, 131.3 (d, J=10.2 Hz), 131.1, 131.0 (d, J=121.3 Hz), 128.8 (d, J=12.2 Hz), 128.3, 127.4, 127.0, 126.2, 126.1, 125.9 (d, J=6.1 Hz), 125.6 (d, J=2.9 Hz), 122.5, 121.3, 113.2, 112.4 (d, J=15.1 Hz); 31P NMR (162 MHz, DMSO-d6) δ: 17.78; HRMS (ESI) calcd for C30H22NOPNa [M+Na]+ 466.1331, found 466.1336.
Diphenyl(6-(thiophen-2-yl)-1H-indol-2-yl)phosphine oxide (3ka): White solid, 83% yield. m.p. 275~275.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 11.49 (s, 1H), 7.71 (s, 1H), 7.65 (dd, J=12.6, 7.5 Hz, 4H), 7.49~7.45 (m, 3H), 7.38~7.31 (m, 5H), 7.23 (d, J=3.1 Hz, 1H), 7.17 (d, J=5.0 Hz, 1H), 7.00 (t, J=3.8 Hz, 1H), 6.46 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 145.5, 139.8 (d, J=9.4 Hz), 132.4 (d, J=2.7 Hz), 132.0 (d, J=10.6 Hz), 131.2 (d, J=47.0 Hz), 128.7 (d, J=12.7 Hz), 128.1, 123.8 (d, J=149.3 Hz), 122.0, 119.7, 113.3 (d, J=15.6 Hz), 109.6; 31P NMR (162 MHz, CDCl3) δ: 22.14; HRMS (ESI) calcd for C24H18NOPSNa [M+Na]+ 422.0739, found 422.0746.
(5H-[1,3]Dioxolo[5-f]indol-6-yl)diphenylphosphine oxide (3la): White solid, 88% yield. m.p.>288 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 11.76 (s, 1H), 7.70~7.55 (m, 10H), 7.05 (s, 1H), 6.89 (s, 1H), 6.47 (s, 1H), 5.98 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 146.4, 143.2, 134.9 (d, J=9.7 Hz), 132.9 (d, J=107.5 Hz), 132.1 (d, J=2.0 Hz), 131.3 (d, J=10.1 Hz), 128.7 (d, J=12.1 Hz), 127.1 (d, J=125.5 Hz), 120.8 (d, J=12.3 Hz), 112.9 (d, J=15.7 Hz), 100.7, 99.0, 92.1; 31P NMR (162 MHz, DMSO-d6) δ: 17.16; HRMS (ESI) calcd for C21H16NO3PNa [M+Na]+ 384.0760, found 384.0767.
4.2.2 Synthesis of 1H-indol-2-yl)diphenylphosphine sulfide (4)
To a 10 mL Schlenk tube were added phosphinoylindole 3aa (0.2 mmol, 63.4 mg), Lawesson’s reagent (2.0 equiv., 162 mg) and toluene (2 mL). Then the reaction mixture was stirred for 2 h at 110 ℃. Upon completion, the solvent was removed in vacuo and the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, V∶V=10∶1) to give the product 4. Yellow solid, 75% yield. m.p. 145.4~146.0 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.14 (s, 1H), 7.79 (dd, J=13.6, 7.4 Hz, 4H), 7.61~7.44 (m, 8H), 7.29 (t, J=7.5 Hz, 1H), 7.14 (t, J=7.3 Hz, 1H), 6.63 (s, 1H); 13C NMR (101 MHz, CDCl3) δ: 138.4 (d, J=9.1 Hz), 133.0, 132.1 (d, J=3.0 Hz), 132.0 (d, J=11.5 Hz), 128.8 (d, J=13.1 Hz), 128.4 (d, J=11.6 Hz), 127.3 (d, J=105.5 Hz), 124.7, 121.8, 121.0, 112.3 (d, J=12.2 Hz), 112.1 (d, J=1.0 Hz); 31P NMR (162 MHz, CDCl3) δ: 31.49; HRMS (ESI) calcd for C20H17NPS [M+H]+ 334.0814, found 334.0823.
4.2.3 Synthesis of (1-allyl-1H-indol-2-yl)diphenylpho- sphine oxide (5)
In a 10 mL Schlenk tube, phosphinoyl indole 3aa (0.2 mmol, 1.0 equiv.) was diluted with dry DMF (2 mL) and KOH (0.22 mmol, 1.1 equiv.) was added at room temperature. Allyl bromide (0.28 mmol, 1.4 equiv.) was added to the resulting stirred solution dropwise. The reaction mixture was checked by TLC until completion (12 h). The mixture was diluted with EtOAc (10 mL) and washed with brine (10 mL×3). The organic phase was dried with Na2SO4 and concentrated under reduced pressure. The crude product 5 was loaded in a silica gel column and purified by flash chromatography using petroleum ether/ethyl acetate (V∶ V=10∶1) as eluents. Colourless oil, 91% yield. 1H NMR (400 MHz, CDCl3) δ: 7.76~7.71 (m, 4H), 7.59~7.55 (m, 3H), 7.50~7.46 (m, 4H), 7.36~7.27 (m, 2H), 7.14~7.10 (m, 1H), 6.37 (d, J=4.1 Hz, 1H), 5.69~5.60 (m, 1H), 5.06 (d, J=5.5 Hz, 2H), 4.93~4.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 140.2 (d, J=7.8 Hz), 132.9, 132.3 (d, J=2.8 Hz), 132.17 (d, J=10.2 Hz), 132.16 (d, J=109.8 Hz), 130.6 (d, J=120.6 Hz), 128.6 (d, J=12.5 Hz), 126.8 (d, J=13.0 Hz), 124.5, 122.1, 120.6, 117.1, 115.1 (d, J=16.2 Hz), 111.0, 48.6; 31P NMR (162 MHz, CDCl3) δ: 21.21; HRMS (ESI) calcd for C23H20NOPNa [M+Na]+ 380.1175, found 380.1184.
4.2.4 Synthesis of 2-(diphenylphosphanyl)-1H-indole (6)
To a 10 mL Schlenk tube were added phosphinoyl indole 3aa (0.2 mmol, 63.4 mg), HSiCl3 (10.0 equiv., 270.9 mg) and PhCl (2 mL). The flask was placed in a metal bath and heated at 120 ℃ for 24 h until the complete consumption of 3aa as monitored by TLC. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (petroleum ether/ethyl acetate, V∶V=20∶1) to give product 6. Colorless oil, 82% yield. 1H NMR (400 MHz, CDCl3) δ: 7.87 (s, 1H), 7.50 (d, J=7.3 Hz, 1H), 7.30~7.23 (m, 10H), 7.17 (dd, J=8.1, 0.7 Hz, 1H), 7.08 (td, J=7.0, 1.2 Hz, 1H), 7.02~6.98 (m, 1H), 6.65~6.63 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 138.8 (d, J=2.4 Hz), 136.2 (d, J=8.1 Hz), 133.4, 133.2, 132.6 (d, J=14.5 Hz), 129.1, 128.8 (d, J=6.9 Hz), 123.0, 120.9, 120.1, 113.2 (d, J=22.0 Hz), 111.1; 31P NMR (162 MHz, CDCl3) δ: 25.01; HRMS (ESI) calcd for C20H17NP [M+H]+ 302.1093, found 302.1100.
4.2.5 Mechanism experiment
In a flame-dried 10.0 mL Schlenk tube equipped with a magnetic stir bar was charged sequentially with 1a (0.2 mmol) and 2j (0.5 mmol, 2.5 equiv.), followed by the addition of anhydrous MeCN (1.0 mL). To the resulting mixture was added DBU (0.5 mmol, 2.5 equiv.) under nitrogen atmosphere. Then the mixture was stirred at room temperature for 12 h under nitrogen atmosphere until the reaction was completed as monitored by TLC analysis. The residue was purified by silica gel chromatography with petroleum ether and ethyl acetate (V∶V=5∶1) to afford bisphosphonylaminomethane 7.
2-((Bis(bis(3,5-di-tert-butylphenyl)phosphoryl)methyl)-amino)benzaldehyde (7): Yellow solid, 70%, yield. m.p. 244~244.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.60 (s, 1H), 9.22 (d, J=10.4 Hz, 1H), 7.75~7.68 (m, 8H), 7.41 (d, J=11.8 Hz, 4H), 7.15 (dd, J=7.6, 1.3 Hz, 1H), 6.85 (t, J=7.4 Hz, 1H), 6.45 (t, J=7.4 Hz, 1H), 6.03 (d, J=8.6 Hz, 1H), 5.16 (q, J=13.8 Hz, 1H),1.21 (d, J=2.8 Hz, 72H); 13C NMR (101 MHz, CDCl3) δ: 193.2, 150.4 (dt, J=12.0, 5.9 Hz), 148.8 (d, J=2.3 Hz), 136.1, 135.0, 130.8 (d, J=40.1 Hz), 129.8 (t, J=39.0 Hz), 126.4 (t, J=5.1 Hz), 126.1 (t, J=5.1 Hz), 119.3, 116.0, 110.6, 35.1, 31.4 (d, J=4.0 Hz); 31P NMR (162 MHz, CDCl3) δ: 32.15; HRMS (ESI) calcd for C64H91NO3P2K [M+K]+ 1022.6106, found 1022.6097.
Supporting Information Optimization of reaction conditions, mechanism verification experiments, and 1H NMR, 13C NMR, 19F NMR and 31P NMR spectra of compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Zhao, C.)