

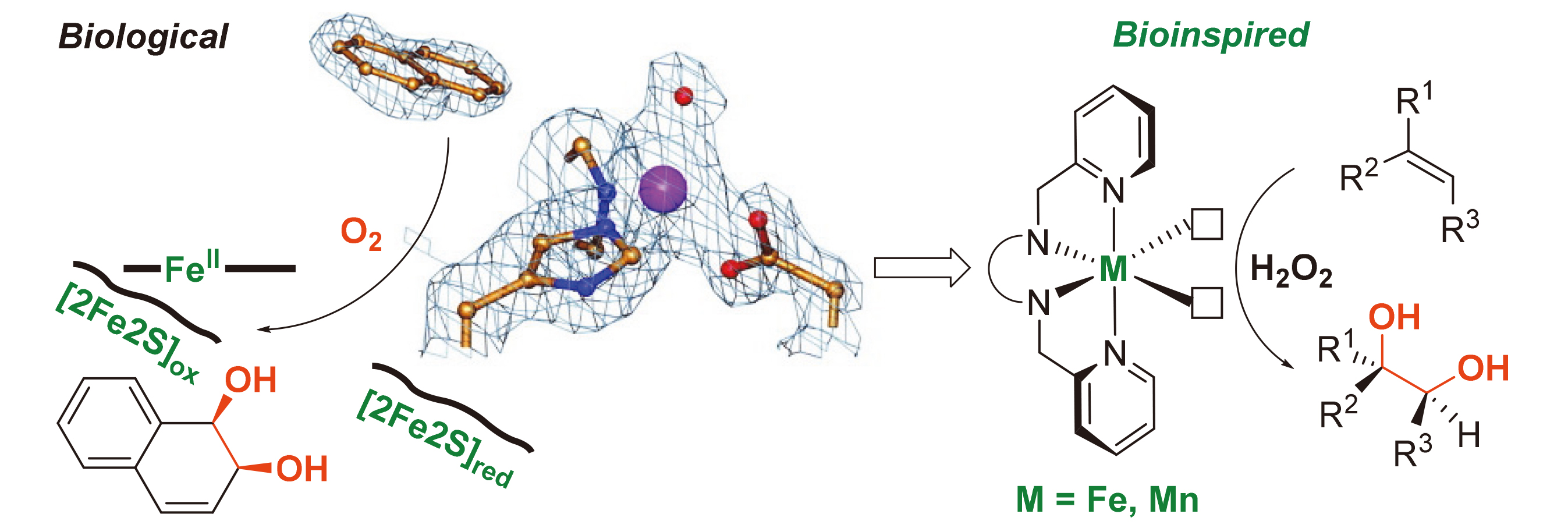
1 Rieske双加氧酶
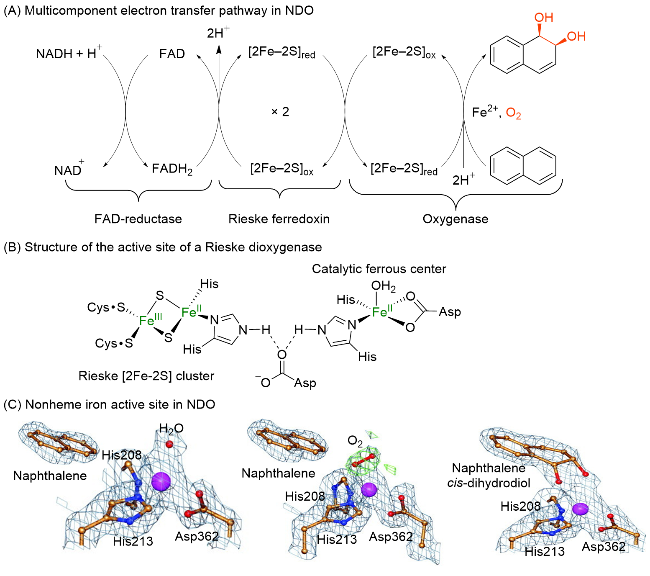
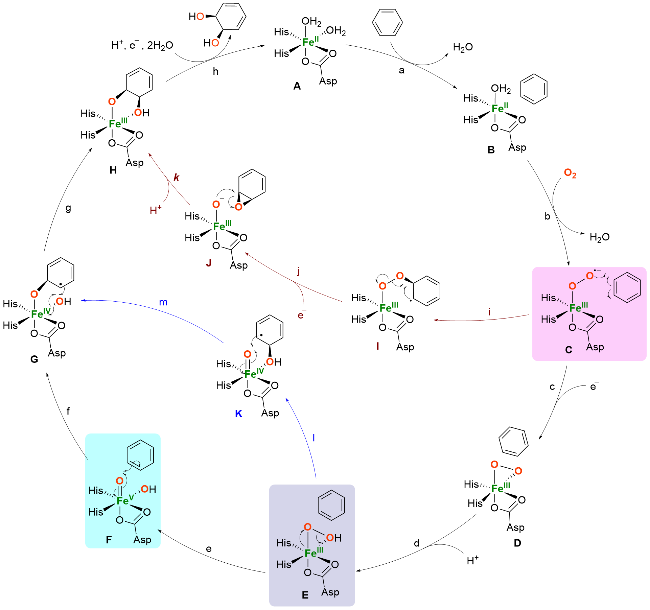
图2 (A)萘双加氧酶中的电子传输链、(B) Rieske双加氧酶活性位点结构示意图、(C)假单胞菌属萘双加氧酶铁活性位点的三维结构[从左到右依次为: 萘双加氧酶与底物(PDB ID: 1O7G)、萘双加氧酶与O2 (PDB ID: 1O7M)和萘双加氧酶与产物(PDB ID: 1O7P)的复合物, 原子颜色标注: 紫色为铁、蓝色为氮、红色为氧、金色为碳]Figure 2 (A) Electron transfer chain in NDO, (B) schematic structure of the active site of a Rieske dioxygenase, and (C) iron active site in NDO (Pseudomonas putida) during catalysis [left to right: Substrate-bound (PDB ID: 1O7G), O2-bound (PDB ID: 1O7M), and product-bound (PDB ID: 1O7P) states; Color code: violet=Fe, blue=N, red=O, gold=C] |
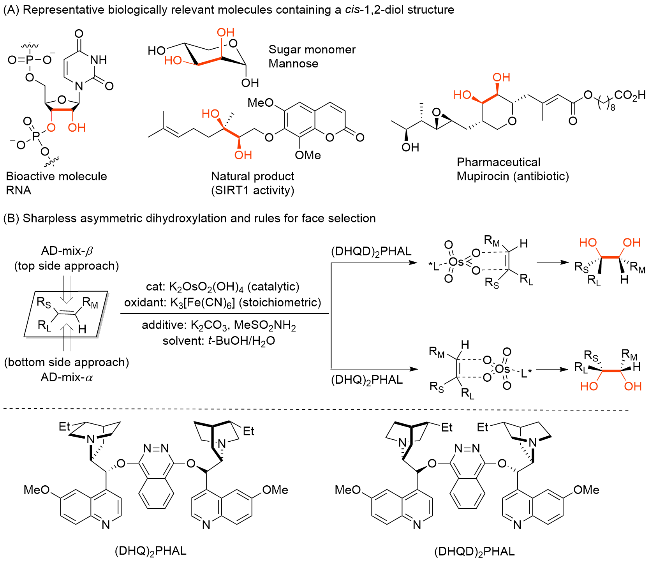
2 非血红素铁催化烯烃不对称双羟化反应
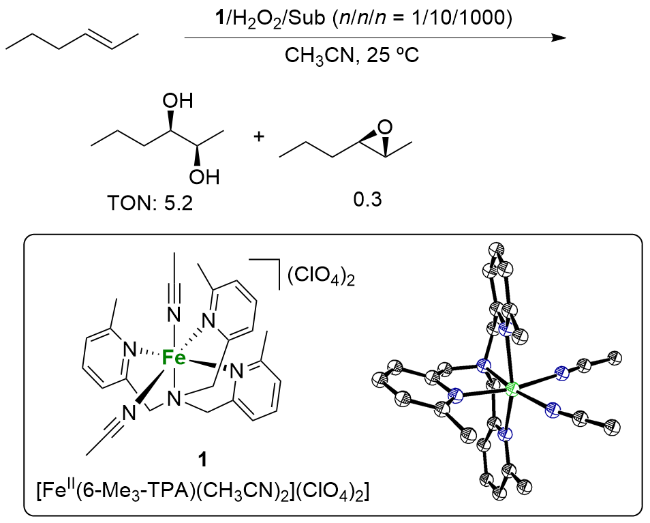
表1 底物限制量条件下配合物2催化各种烯烃氧化反应Table 1 Catalytic oxidation of various olefins by 2 under substrate-limiting conditions 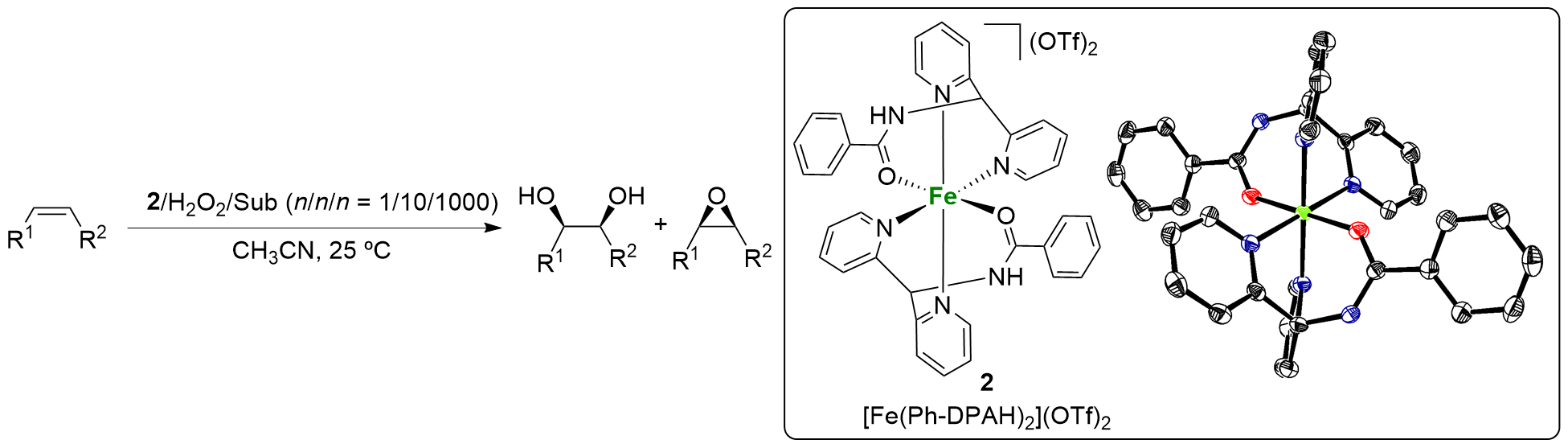 |
| Entry | Substrate | TONa | RCb/% | ||
|---|---|---|---|---|---|
| cis-Diol | Epoxide | cis-Diol/Epoxide | |||
| 1 | Styrene | 8.0 | 0.1 | 80 | — |
| 2 | 1-Octene | 7.6 | 0.1 | 76 | — |
| 3 | Cyclohexene | 6.2 | 0.7 | 9 | — |
| 4 | cis-2-Heptene | 4.9 | 0.7 | 7 | 99 |
| 5 | trans-2-Heptene | 4.9 | 0.5 | 10 | ˃ 99 |
| 6 | Ethyl trans-crotonate | 7.4 | — | ˃ 100 | — |
| 7 | tert-Butyl acrylate | 5.5 | — | ˃ 100 | — |
| 8 | Dimethyl fumarate | 5.3 | — | ˃ 100 | — |
a Yield expressed as turnover number (TON), mol product/mol catalyst. b The percentage of retention of configuration in the dihydroxylation of cis- and trans-2- heptene, expressed as the ratio of the diols: RC=(cis-trans)/(cis+trans)×100%. |
表2 底物限制量条件下配合物3催化各种烯烃氧化反应Table 2 Catalytic oxidation of various olefins by 3 under substrate-limiting conditions 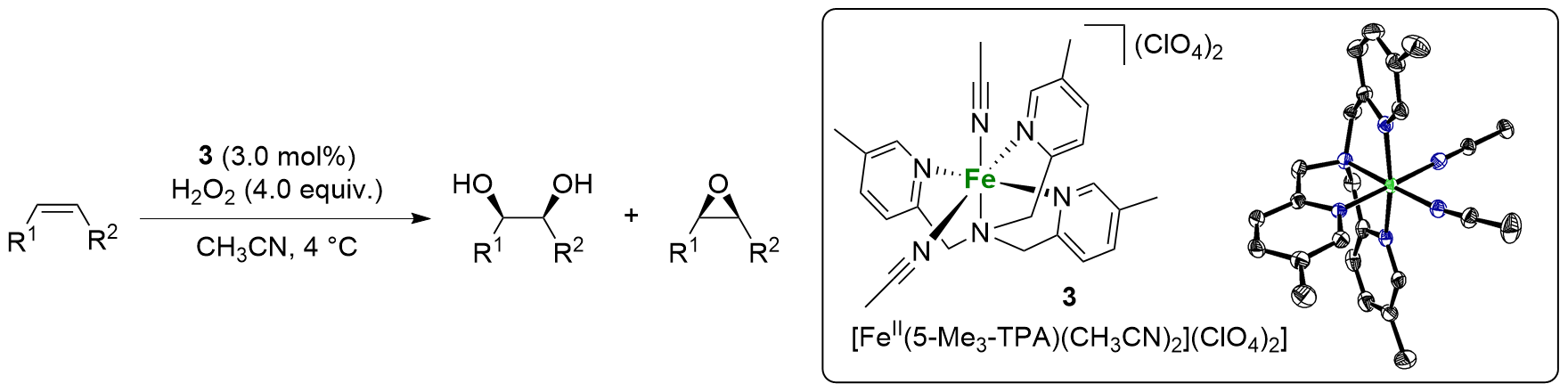 |
| Entry | Substrate | Yielda/% | RCb/% | ||
|---|---|---|---|---|---|
| cis-Diol | Epoxide | cis-Diol/Epoxide | |||
| 1 | 1-Octene | 67 | 20 | 3.4 | — |
| 2 | 1-Decene | 62 | 18 | 3.4 | — |
| 3 | Vinylcyclohexane | 57 | 16 | 3.6 | — |
| 4 | Cyclohexene | 45 | 30 | 1.5 | — |
| 5 | cis-2-Heptene | 60 | 20 | 3.0 | 97 |
| 6 | trans-2-Heptene | 61 | 14 | 4.4 | 99 |
a Conversion of substrate into product determined by GC analysis. b The percentage of retention of configuration in the dihydroxylation of cis- and trans-2- heptene, expressed as the ratio of the diols: RC=(cis-trans)/(cis+trans)×100%. |
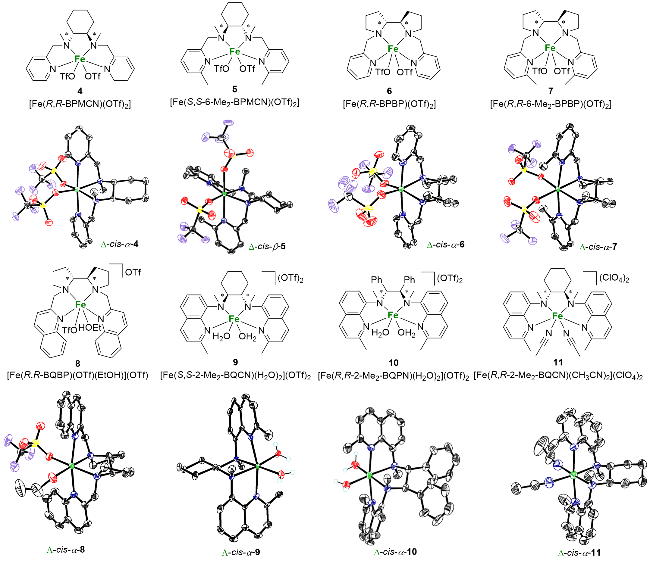
图3 用于催化烯烃不对称双羟化反应的非血红素铁配合物Figure 3 Nonheme iron complexes utilized in asymmetric cis-dihydroxylation of olefins |
表3 烯烃大大过量条件下的非血红素铁催化不对称双羟化Table 3 Nonheme iron-catalyzed asymmetric cis-dihydroxylation in the presence of a large excess of olefins |
| Entry | Complex | Labile site | Substrate | TONa | ee/% | ||
|---|---|---|---|---|---|---|---|
| cis-Diol | Epoxide | cis-Diol/Epoxide | |||||
| 1 | 4b | Two cis-α | trans-2-Heptene | 0.3 | 5.4 | 0.055 | 29 |
| 2 | 5c | Two cis-β | trans-2-Heptene | 7.5 | 2.4 | 3.1 | 79 |
| 3 | cis-2-Heptene | 7.8 | 4.5 | 1.7 | 9 | ||
| 4 | 1-Octene | 8.1 | 1.3 | 6.2 | 60 | ||
| 5 | tert-Butyl acrylate | 10 | 0.5 | 20 | 23 | ||
| 6 | 6d | Two cis-α | trans-2-Heptene | 1.1 | 5.1 | 0.22 | 38 |
| 7 | 1-Octene | 1.7 | 2.6 | 0.67 | 11 | ||
| 8 | tert-Butyl acrylate | 0.1 | 3.0 | 0.033 | — | ||
| 9 | 7d | Two cis-α | trans-2-Heptene | 5.2 | 0.2 | 26 | 97 |
| 10 | cis-2-Heptene | 3.4 | 0.6 | 5.7 | 11 | ||
| 11 | 1-Octene | 6.4 | 0.1 | 64 | 76 | ||
| 12 | Styrene | 6.5 | <0.1 | >65 | 15 | ||
| 13 | tert-Butyl acrylate | 4.0 | <0.1 | >40 | 68 | ||
| 14 | Ethyl trans-crotonate | 7.5 | 0.1 | >75 | 78 | ||
| 15 | 8d | Two cis-α | trans-2-Heptene | 3.6 | 0.9 | 4 | 78 |
| 16 | 1-Octene | 4.6 | 0.5 | 9 | 29 | ||
| 17 | tert-Butyl acrylate | 2.7 | <0.1 | >27 | 23 | ||
a Turnover numbers (TON), mol product/mol catalyst. b n(complex)∶n(H2O2)∶n(substrate)=1∶10∶1000. c n(complex)∶n(H2O2)∶n(substrate)=1∶20∶1000. d n(complex)∶n(H2O2)∶n(substrate)=1∶10∶500. |
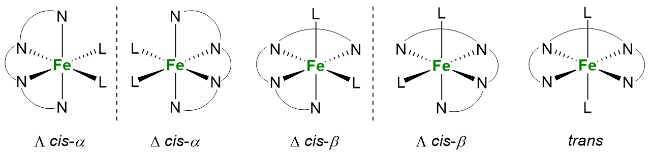
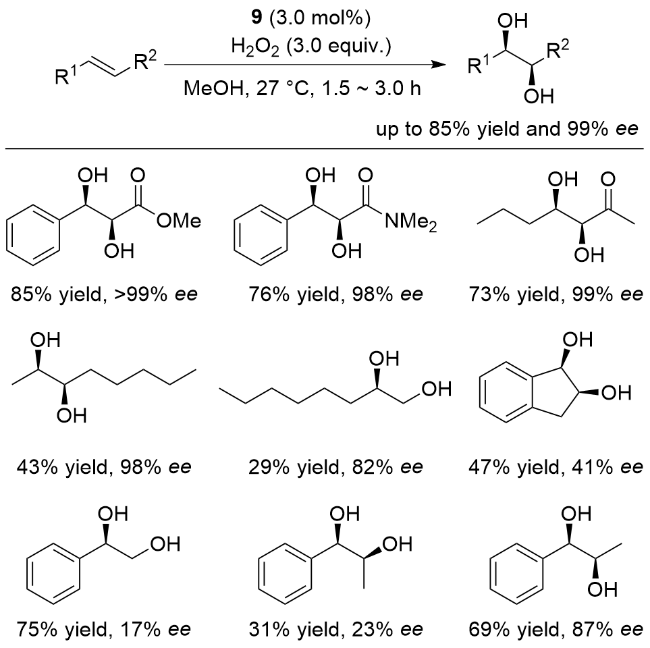
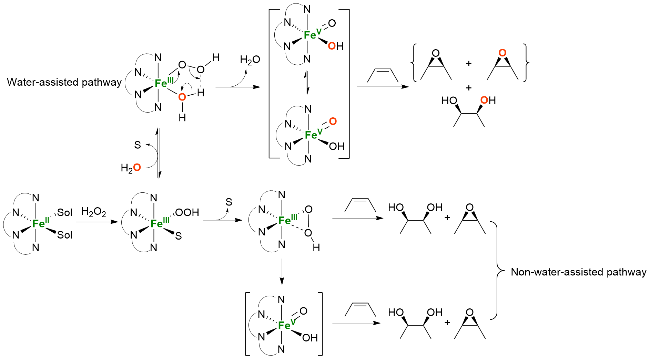
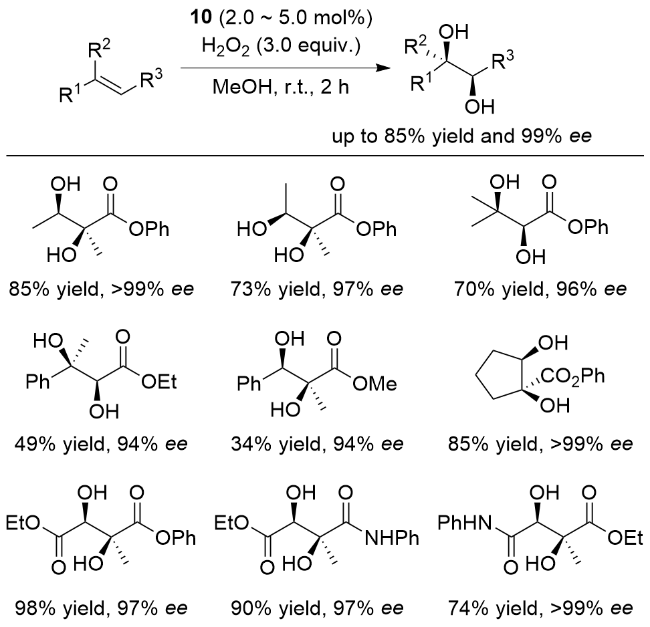
3 非血红素锰催化烯烃不对称双羟化反应
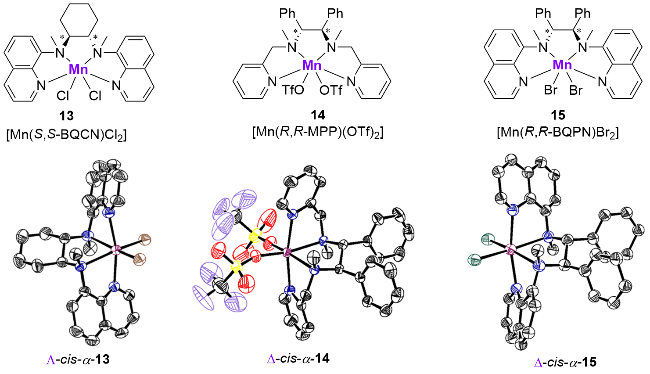
图5 用于催化烯烃不对称双羟化反应的非血红素锰配合物Figure 5 Nonheme manganese complexes utilized in asymmetric cis-dihydroxylation of olefins |
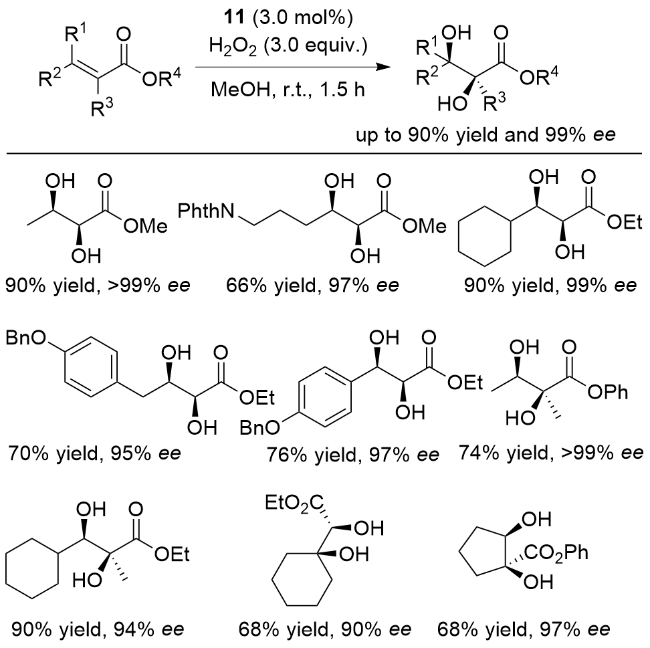
图式6 抗衡离子调控的非血红素铁催化烯烃不对称双羟化反应Scheme 6 Nonheme iron-catalyzed asymmetric cis-dihydroxy- lation by modulating the counterions of the ferrous complex |
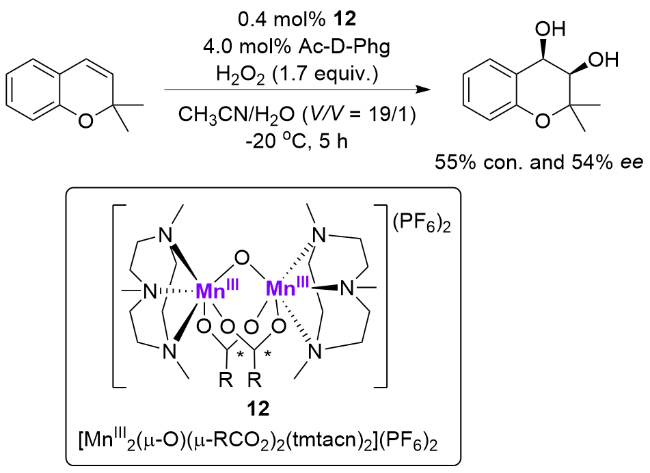
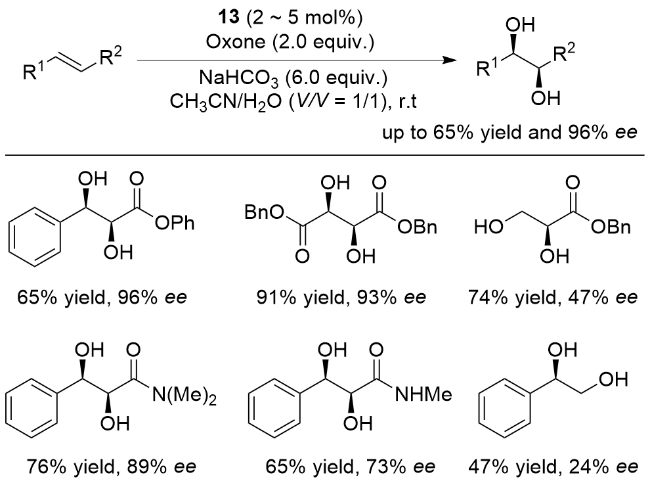
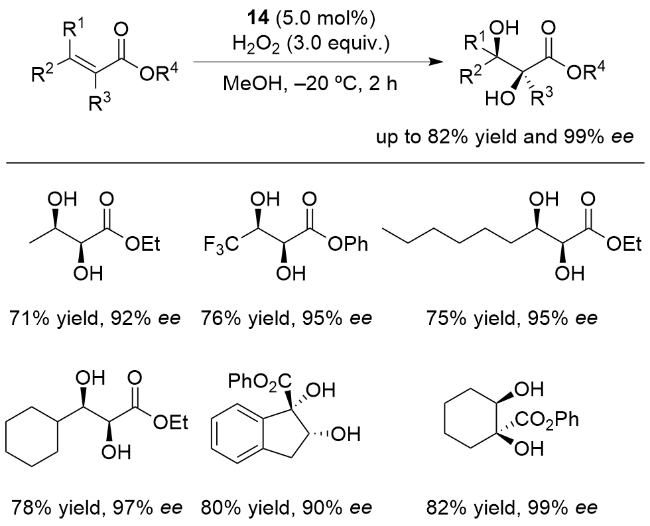
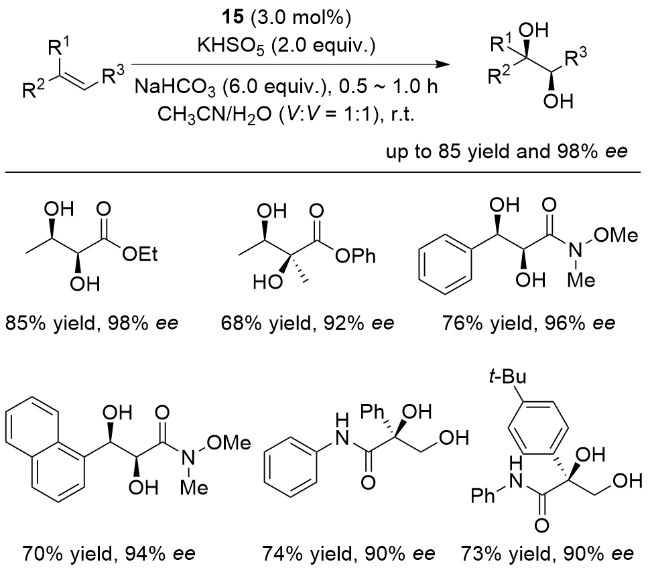
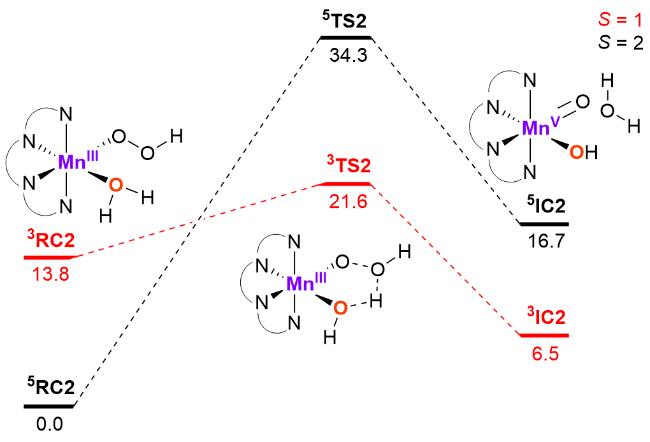
图6 MnIII(H2O)(OOH)经水分子辅助路径生成HO—MnV=O的DFT吉布斯能垒计算Figure 6 DFT-computed Gibbs energy profiles (in kcal mol−1) for water-assisted formation of HO—MnV=O from MnIII(H2O)- (OOH) |
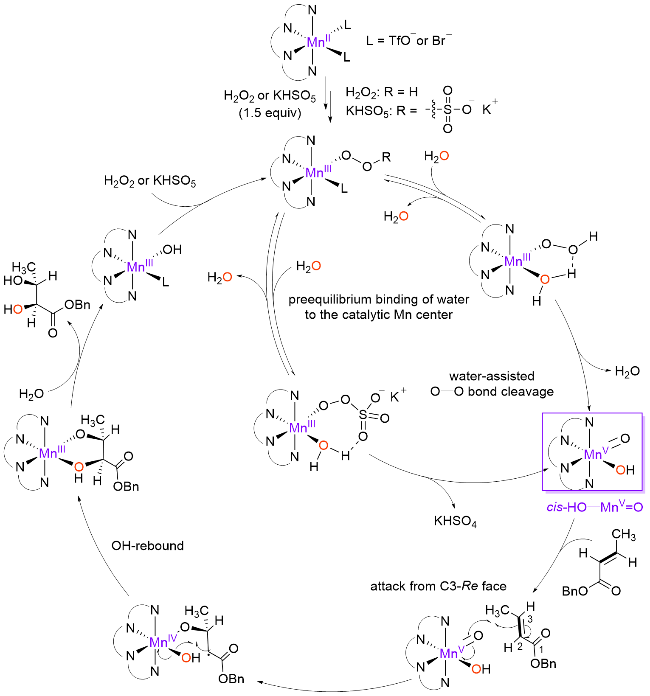
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}