

1 Introduction
Unsaturated amides serve as valuable building blocks for a vast array of organic compounds, which are commonly found in a wide range of bioactive natural products, functional materials, and pharmaceutical drugs.[1] For example, they often function as antitumor agents, anticancer agents, as well as histone deacetylase inhibitors.[2] Therefore, the exploration of effective approaches for synthesizing unsaturated amides remains a crucial research focus in the realm of organic chemistry. Approaches for the synthesis of α,β-unsaturated amides mainly involve direct amidation of cinnamic acids and amines,[3] C—H bond functionalizations,[4] decarboxylative acylation of cinnamic acids,[5] α,β-dehydrogenation of amides,[6] and others.[7] Nevertheless, the above-mentioned reactions suffer from multiple-step processes, complicated starting materials, and the use of strong oxidants or toxic reagents. Therefore, how to avoid these problems and develop new synthetic strategies is still challenging.
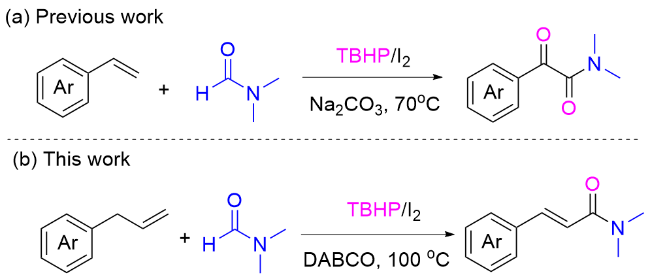
Alkenes, as abundant and simple chemical feedstocks, are highly appealing in organic synthesis due to their wide availability, simplicity, and low cost.[8] Moreover, approaches for their direct and selective functionalization are of great significance as they represent a vital means to fabricate more elaborate molecular structures. For example, Li and coworkers[9] disclosed an efficient intermolecular oxidative coupling reaction of alkenes and N,N-dialkylfor- mamides for the construction of α,β-unsaturated amides under FeCl3/DTBP catalytic system at a high reaction tem- perature. Recently, Cai and co-workers[10] reported a practical visible-light-enabled oxidative cross-coupling reaction of alkenes with dialkylformamides for the preparation of α,β-unsaturated amides. It is obvious that alkenes have been successfully applied and significant achievements have been made in the synthesis of α,β-unsaturated amides. However, it is still highly desirable to develop a facile, mild, and straightforward method for synthesizing α,β- unsaturated amide using simple substrate structures, with minimal use of transition-metal oxidants, and in a step- and atom-economical manner.
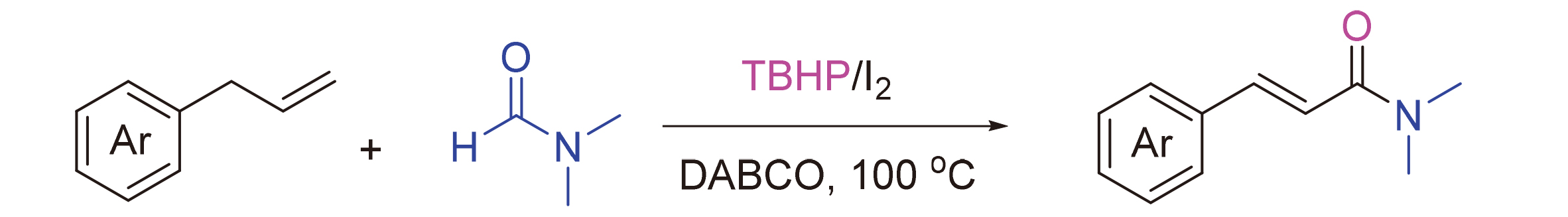
In previous work, our group successfully developed a series of t-butylhydroperoxide (TBHP)-mediated oxidative transformation reactions of alkenes for the construction of various complex molecules.[11-14] In 2022, we reported an I2/TBHP-promoted oxo-amidation of alkenes with N,N- dimethylformamide (DMF), in which DMF acted as both the solvent and the source of dimethylamine (Scheme 1, a).[13] This reaction achieved the efficient difunctionalization of substituted alkenes, enabling the facile and efficient synthesis of diverse amide derivatives. As a continuation of our interest in TBHP-promoted oxidation of alkenes, herein we report a facile one-pot synthesis of α,β-unsatu- rated amides from simple alkenes and dialkylformamides via a TBHP-mediated oxidation process, where dialkylfor- mamides serve as both solvent and reactant (Scheme 1, b).
2 Results and discussion
Our investigation began with the examination of the oxidative coupling of allylbenzene (1a) with DMF (2a) in the presence of TBHP (Table 1). A mixture of 1a (1 equiv.), TBHP (4 equiv.) and Na2CO3 (1 equiv.) in the presence of iodine (0.1 equiv.) and DMF (2a) reacted at 70 ℃ for 24 h. Gratifyingly, the target unsaturated amide 3a was obtained in 48% yield (Table 1, Entry 1). Then, a range of bases including CH3COONa, t-BuOK, 1,8-diazabicyclo- [5.4.0]undec-7-ene (DBU), N,N-dimethylpyridin-4-amine (DMAP), Et3N and 1,4-diazabicyclo[2.2.2]octane (DABCO) were screened, among which DABCO was identified as one of the best additives (Entries 2~7). However, increasing the amount of DABCO was not helpful for increasing the yield (Entries 9, 10). Both base and I2 are essential for this reaction. In the absence of base or I2, no or only a trace amount of 3a could be observed (Entries 8, 11). In the further investigation, when the amount of I₂ was increased from 0.1 equiv. to 0.5 equiv., the yield of 3a decreased obviously (Entry 12). Subsequently, the reaction conditions were optimized by adjusting the amount of TBHP. The results indicated that the optimal amount of TBHP was 4 equiv., and further increasing it to 6 equiv. did not improve the yield of 3a (Entries 13~15). Next, the effect of temperature on the reaction was also investigated. To our delight, the reaction was found to be more effective when the temperature was increased to 100 ℃ and the reaction time was shortened to 6 h, affording 3a in 75% yield (Entries 16, 17). When the temperature was further increased to 120 ℃, no significant increase in yield was observed (Entries 18). Finally, to clarify the role of DMF, control experiments were performed. When allylbenzene (1a) reacted with 2 equiv. of DMF in dimethyl sulfoxide (DMSO), PhCl or dioxane solvents in the presence of TBHP/I2, the major product 3a was obtained in 66%, 59%, and 63% yields, respectively (Entries 19~21). It suggests that DMF serves not only as a reactant but also as a solvent in this transformation.
Table 1 Optimization of reaction conditionsa |
| Entry | TBHB/equiv. | Base | Solvent | Yieldb/% |
|---|---|---|---|---|
| 1 | 4.0 | Na2CO3 | DMF | 48 |
| 2 | 4.0 | CH3COONa | DMF | 56 |
| 3 | 4.0 | t-BuOK | DMF | 70 |
| 4 | 4.0 | DBU | DMF | 36 |
| 5 | 4.0 | DMAP | DMF | 67 |
| 6 | 4.0 | Et3N | DMF | trace |
| 7 | 4.0 | DABCO | DMF | 71 |
| 8 | 4.0 | — | DMF | trace |
| 9 | 4.0 | DABCO (2 equiv.) | DMF | 70 |
| 10 | 4.0 | DABCO (5 equiv.) | DMF | 69 |
| 11c | 4.0 | DABCO | DMF | 0 |
| 12d | 4.0 | DABCO | DMF | 55 |
| 13 | 1.0 | DABCO | DMF | 33 |
| 14 | 2.0 | DABCO | DMF | 60 |
| 15 | 6.0 | DABCO | DMF | 65 |
| 16e | 4.0 | DABCO | DMF | 71 |
| 17f,g | 4.0 | DABCO | DMF | 75 |
| 18g,h | 4.0 | DABCO | DMF | 76 |
| 19f,g | 4.0 | DABCO | DMSO | 66 |
| 20f,g | 4.0 | DABCO | PhCl | 59 |
| 21f,g | 4.0 | DABCO | Dioxane | 63 |
a Reaction conditions: 1a (0.5 mmol), I2 (0.1 equiv.), TBHP (4.0 equiv.), solvent (1.0 mL), air, 70 ℃, 24 h; b GC yield; c In the absence of I2; d I2 (0.5 equiv.); e Temperature was r.t.; f Temperature was 100 ℃); g Time was 6 h; h Temperature was 120 ℃. |
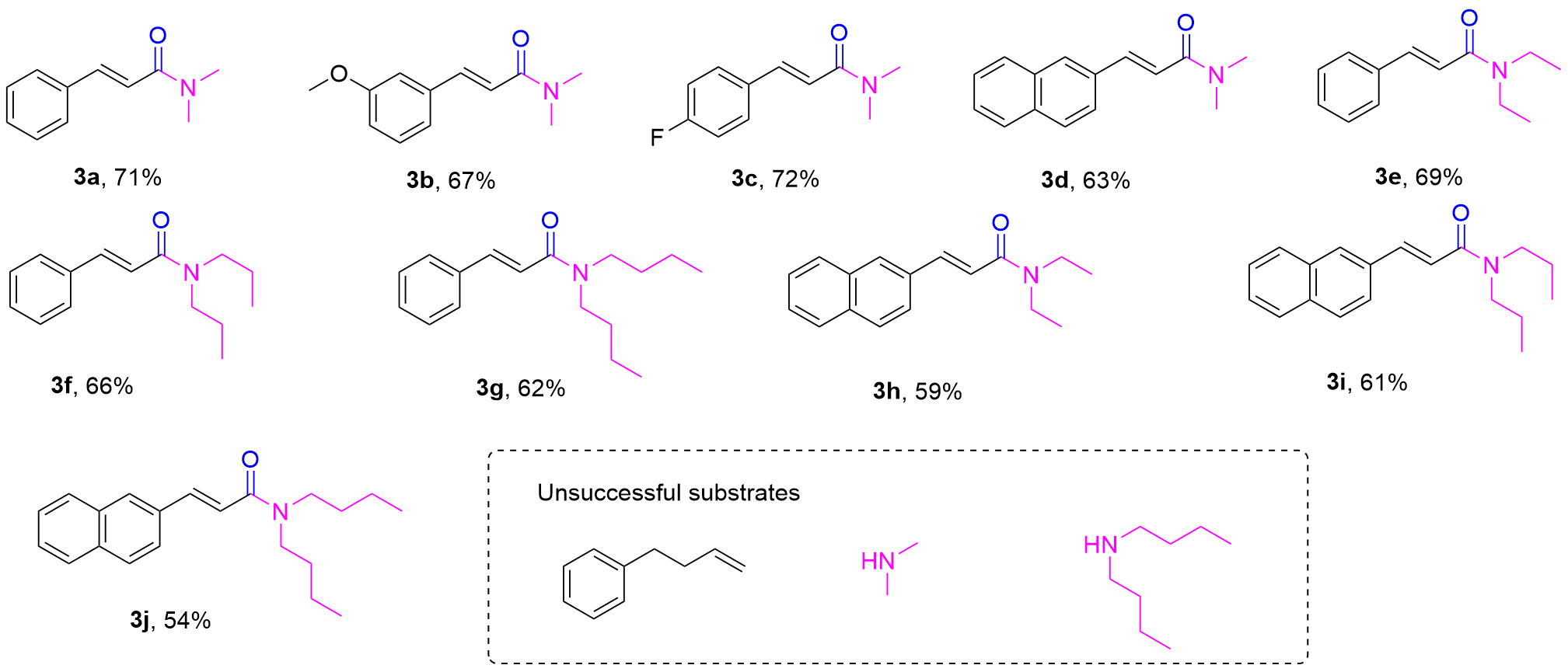
With the optimized conditions in hand, the scope of allylbenzenes 1 was subsequently explored (Table 2). The study revealed that allylbenzenes with electron-donating or electron-withdrawing groups proceeded well to yield desired products 3 in good yields (3b, 3c). Allylbenzene bearing an electron-withdrawing group (F) provided a higher yield than allylbenzene bearing an electron-dona- ting group (OCH3) (3b vs. 3c). Moreover, this method was proven to be efficient in converting 2-allylnaphthalene into the corresponding α,β-unsaturated amide with a satisfactory yield. Significantly, this TBHP-promoted reaction of allylbenzenes and formamides was successfully extended to other dialkylformamides 2 under the optimal reaction conditions. When 1a or 1d was used as substrates, formamides with ethyl, propyl, or butyl groups exhibited comparable reactivity (3e~3j), generating the corresponding α,β-unsaturated amides in 54%~69% yields. These results confirmed that the developed approach could be extensively applicable to compounds with diverse functional groups.
Table 2 Substrate scope for the synthesis of α,β-unsaturated amidesa,b |
 |
a Conditions: 1 (1.0 mmol), 2 (1.0 mL), TBHP (4.0 equiv.), I2 (10 mol%), DABCO (1.0 equiv.); b Isolated yields. |
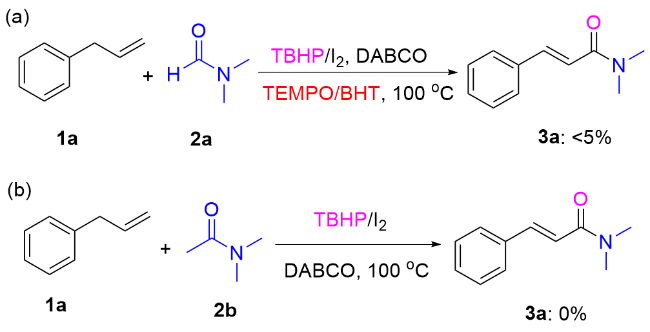
To understand the mechanism, control experiments were carried out as shown in Scheme 2. Initially, the addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butyla- ted hydroxytoluene (BHT) to the oxidative coupling reaction completely suppressed the process, thereby implicating a radical mechanism in this transformation (Scheme 2, a). Furthermore, when N,N-dimethylacetamide (2b) was employed instead of N,N-dimethylformamide (2a) to couple with allylbenzene 1a under standard reaction conditions, no anticipated product 3a could be observed at all (Scheme 2, b). It suggests that DMF serves not only as a solvent but also as a critical reagent, wherein its formyl group enables the formation of dimethylamine via decarbonylation, thereby promoting the formation of unsaturated amides.
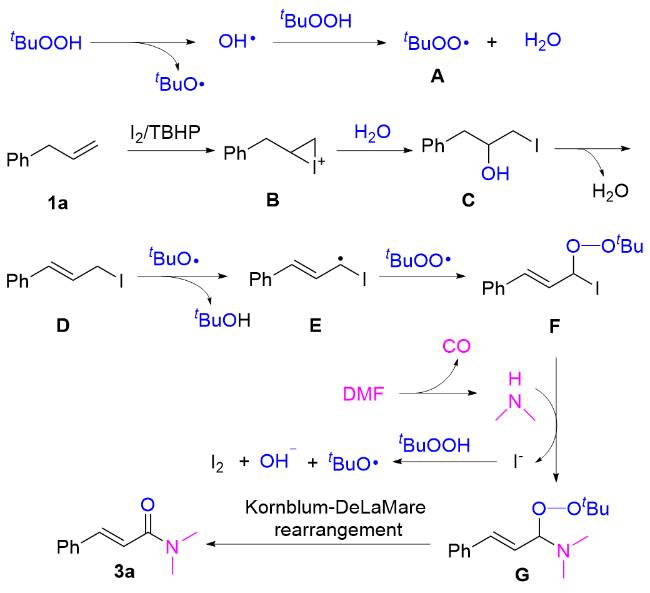
On the basis of the above observation and our previous publications,[11-14] a plausible mechanism was proposed to account for this reaction (Scheme 3). Initially, tert-butoxyl radicals and tert-butylperoxyl radicals were generated from TBHP under heating conditions. With TBHP involved, I2 reacted with allylbenzene to form an iodonium ion B. Subsequently, B underwent a nucleophilic attack by H2O, leading to the formation of intermediate C. After that, intermediate C dehydrated to produce intermediate D in the presence of base (DABCO). Intermediate D then combined with tBuO• radicals to generate species E. Reaction of E with tBuOO• radicals afforded intermediate F, which underwent nucleophilic substitution with in situ-formed dimethylamine from N,N-dimethylformamide (as established by Song, Wang, Hallberg et al.)[15-18] to produce intermediate G while generating iodide ions. The iodide ions were reoxidized to I2 by TBHP.[19] Finally, base-mediated Kornblum-DeLaMare rearrangement[20] of G in the presence of DABCO yielded product 3a.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In conclusion, a convenient and straightforward method for synthesizing α,β-unsaturated amides from simple alkenes and dialkylformamides via an I2/TBHP-mediated protocol was developed. A variety of allylbenzene and di- alkylformamide substrates are well-tolerated in this process. Notably, dialkylformamide serves not only as the solvent but also as the source of dialkylamine. This transformation is achieved in a one-pot procedure under mild conditions, highlighting its operational simplicity. Moreover, the protocol avoids the use of transition metals, thereby enhancing its practicality and environmental benignity. Given these advantages, we anticipate that this protocol will find numerous applications in pharmaceutical research.
4 Experimental section
4.1 General experimental information
Unless otherwise stated, all reagents (including 1 and 2) were purchased from commercial suppliers and utilized without additional purification. 1H NMR and 13C NMR spectra were measured using a Bruker model Bruker AV- 400 spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR) in solutions of CDCl3 with tetramethylsilane as the internal standard. Mass spectra were obtained from high resolution ESI mass spectrometer. HRMS was obtained on a Q-TOF micro spectrometer.
4.2 General procedure for the synthesis of α,β-un- saturated amides
A mixture of allylbenzene (1a, 118 mg, 1.0 mmol), I2 (26 mg, 0.1 mmol), DABCO (112 mg, 1.0 mmol), TBHP (750 mg, 6 mmol, 70% in water) and DMF (2a, 1.0 mL) was added successively in a round-bottom flask, and the resulting solution was stirred for 6 h at 100 ℃. The mixture was purified by column chromatography on silica gel to afford product 3a with petroleum ether (PE)/ethyl acetate (EA) (V∶V=20∶1) as the eluent. Compounds 3b~3j were synthesized via the same procedure. Compounds 3a~3j are all yellow oil.
(E)-N,N-Dimethylcinnamamide (3a):[7e] 71% yield. 1H NMR (400 MHz, CDCl3) δ: 7.69 (d, J=15.5 Hz, 1H), 7.58~7.50 (m, 2H), 7.41~7.33 (m, 3H), 6.91 (d, J=15.4 Hz, 1H), 3.19 (s, 3H), 3.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.7, 142.3, 135.4, 129.5, 128.8, 127.8, 117.5, 37.4, 35.9.
(E)-3-(3-Methoxyphenyl)-N,N-dimethylacrylamide (3b):[21] 67% yield. 1H NMR (400 MHz, CDCl3) δ: 7.64 (d, J=15.4 Hz, 1H), 7.35~7.25 (m, 1H), 7.14 (d, J=7.6 Hz, 1H), 7.06 (s, 1H), 6.90 (dd, J=15.0, 8.8 Hz, 2H), 3.85 (s, 3H), 3.18 (s, 3H), 3.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 159.8, 142.2, 136.8, 129.8, 120.3, 117.8, 115.0, 113.2, 55.3, 37.4, 35.9.
(E)-3-(4-Fluorophenyl)-N,N-dimethylacrylamide (3c):[7c] 72% yield. 1H NMR (400 MHz, CDCl3) δ: 7.65 (d, J=15.4 Hz, 1H), 7.53 (dd, J=8.6, 5.4 Hz, 2H), 7.08 (t, J=8.6 Hz, 2H), 6.83 (d, J=15.4 Hz, 1H), 3.19 (s, 3H), 3.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 162.2,141.1,131.6, 129.6, 117.1, 115.9, 37.4, 36.0.
(E)-N,N-Dimethyl-3-(naphthalen-1-yl)acrylamide (3d):[7e] 63% yield. 1H NMR (400 MHz, CDCl3) δ: 8.51 (d, J=15.2 Hz, 1H), 8.25 (d, J=8.2 Hz, 1H), 7.89 (d, J=8.4 Hz, 2H), 7.72 (s, 1H), 7.62~7.43 (m, 3H), 6.97 (d, J=15.2 Hz, 1H), 3.23 (s, 3H), 3.14 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 139.8, 133.6, 133.2, 131.5, 129.8, 128.6, 126.7, 126.2, 125.4, 124.5, 123.8, 120.5, 37.5, 36.0.
(E)-N,N-Diethylcinnamamide (3e):[7a] 69% yield. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J=15.4 Hz, 1H), 7.55 (dd, J=7.7, 1.4 Hz, 2H), 7.46~7.31 (m, 3H), 6.85 (d, J=15.4 Hz, 1H), 3.59~3.41 (m, 4H), 1.29 (t, J=6.9 Hz, 3H), 1.21 (dd, J=7.6, 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 142.3, 129.5, 128.8, 128.4, 127.8, 126.3, 117.8, 42.3, 41.1, 15.1, 13.2.
(E)-N,N-Dipropylcinnamamide (3f): 66% yield. 1H NMR (400 MHz, CDCl3) δ: 7.72 (d, J=15.4 Hz, 1H), 7.54 (dd, J=7.6, 1.6 Hz, 2H), 7.44~7.32 (m, 3H), 6.85 (d, J=15.4 Hz, 1H), 3.53~3.29 (m, 4H), 1.72~1.57 (m, 4H), 0.97 (dt, J=13.1, 7.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 166.2, 142.2, 135.6, 129.4, 128.8, 127.7, 117.9, 49.9, 48.6, 23.1, 21.2, 11.5, 11.4; HRMS calcd for C15H21NONa [M+Na]+ 232.1696, found 232.1688.
(E)-N,N-Dibutylcinnamamide (3g):[7b] 62% yield. 1H NMR (400 MHz, CDCl3) δ: 7.72 (d, J=15.4 Hz, 1H), 7.53 (d, J=6.7 Hz, 2H), 7.46~7.32 (m, 3H), 6.86 (d, J=15.4 Hz, 1H), 3.49~3.36 (m, 4H), 1.69~1.56 (m, 4H), 1.44~1.34 (m, 4H), 0.98 (dt, J=11.8, 7.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.0, 142.2, 135.6, 129.4, 128.8, 127.7, 117.9, 48.0, 46.7, 32.0, 30.1, 20.3, 20.1, 13.93, 13.86.
(E)-N,N-Diethyl-3-(naphthalen-1-yl)acrylamide (3h):[7c] 59% yield. 1H NMR (400 MHz, CDCl3) δ: 8.55 (d, J=15.1 Hz, 1H), 8.26 (dd, J=8.2, 1.0 Hz, 1H), 7.97~7.83 (m, 2H), 7.72 (d, J=7.2 Hz, 1H), 7.65~7.43 (m, 3H), 6.91 (d, J=15.1 Hz, 1H), 3.55 (dq, J=14.2, 7.1 Hz, 4H), 1.28 (dt, J=19.4, 7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 165.6, 139.7, 133.6, 133.4, 131.5, 129.7, 128.5, 126.6, 126.2, 125.4, 124.5, 123.9, 121.0, 42.4, 41.2, 15.1, 13.3.
(E)-3-(Naphthalen-1-yl)-N,N-dipropylacrylamide (3i): 61% yield. 1H NMR (400 MHz, CDCl3) δ: 8.53 (d, J=15.1 Hz, 1H), 8.26 (d, J=8.1 Hz, 1H), 7.89 (d, J=8.3 Hz, 2H), 7.71 (d, J=7.1 Hz, 1H), 7.60~7.47 (m, 3H), 6.91 (d, J=15.1 Hz, 1H), 3.51~3.37 (m, 4H), 1.73 (dd, J=9.4, 5.2 Hz, 4H), 0.99 (t, J=7.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 166.1, 139.6, 133.6, 133.4, 131.5, 129.6, 128.5, 126.6, 126.2, 125.4, 124.5, 123.9, 121.0, 50.0, 48.7, 23.1, 21.2, 11.5, 11.3l; HRMS calcd for C19H23NONa [M+Na]+ 281.1780, found 282.1852.
(E)-N,N-Dibutyl-3-(naphthalen-1-yl)acrylamide (3j): 54% yield. 1H NMR (400 MHz, CDCl3) δ: 8.54 (d, J=15.1 Hz, 1H), 8.26 (d, J=8.1 Hz, 1H), 7.93~7.82 (m, 2H), 7.71 (d, J=7.2 Hz, 1H), 7.64~7.42 (m, 3H), 6.91 (d, J=15.1 Hz, 1H), 3.54~3.39 (m, 4H), 1.45~1.37 (m, 4H), 0.98 (dd, J=14.0, 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 165.9, 139.6, 133.6, 133.4, 131.5, 129.6, 128.5, 126.6, 126.2, 125.4, 124.5, 123.9, 121.0, 48.1, 46.8, 32.0, 30.1, 20.4, 20.1, 14.0, 13.9l; HRMS calcd for C21H28NO [M+H]+ 310.2156, found 310.2156.
Supporting Information 1H NMR and 13C NMR spectra of compounds 3a~3j. The Supporting Information is available free of charge via the Internet at http://sioc- journal.cn.
(Zhao, C.)