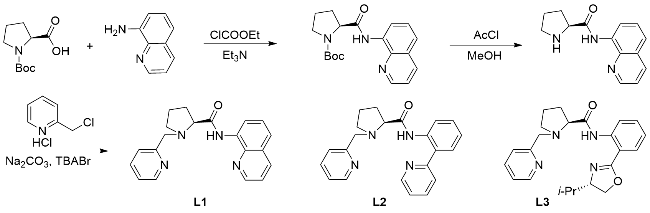
The synthesis of ligand
L1 was accomplished in three steps according to
Scheme 1. The detailed procedure for
L1 is described below, while
L2 and
L3 were prepared analogously by replacing 8-aminoquinoline used in
L1 synthesis with 2-(pyridin-2-yl)aniline
[17] (for
L2) or (
S)-2- (4-isopropyl-4,5-dihydrooxazol-2-yl)aniline
[18] (for
L3), respectively.
Step 1: A mixture of Boc-
L-proline (1.1 g, 5.0 mmol), anhydrous tetrahydrofuran (THF, 20 mL), and triethyl- amine (0.51 mg, 700 μL, 5.0 mmol) was added into a 100 mL flask. The flask was immersed in a low-temperature cooling circulator to maintain an internal temperature of -10 ℃. Ethyl chloroformate (0.54 g, 5.0 mmol) was then added dropwise over 30 min using a pressure-equalizing dropping funnel while keeping the temperature at -10 ℃. Upon completion of the addition, a significant amount of white solid precipitated, indicating the reaction was complete. The triethylamine hydrochloride byproduct was removed by filtration, and the resulting filtrate was re- cooled to -10 ℃. To the cooled filtrate, quinolin-8-amine (0.72 g, 5.0 mmol) was added, and the mixture was stirred at -10 ℃ for 30 min before being allowed to warm to room temperature and stirred overnight. The solvent was then removed under reduced pressure using a rotary evaporator. The crude residue was dissolved in a saturated aqueous NH
4Cl solution and extracted with dichloro- methane (CH
2Cl
2, 20 mL×3). The combined organic layers were dried over with anhydrous sodium sulfate (Na
2SO
4), filtered, and concentrated under vacuum. The product was purified by flash column chromatography on silica gel using a petroleum ether/ethyl acetate gradient (
V∶
V=10∶1) to afford (
S)-
tert-butyl 2-(quinolin-8-yl- carbamoyl)pyrrolidine-1-carboxylate
[19a] as a white solid (1.25 g, 3.75 mmol, 75% yield).
Rf=0.20 (
V(petroleum ether)∶
V(ethyl acetate)=20∶1). m.p. 136.1~137.8 ℃;
1H NMR (400 MHz, CDCl
3) 10.37 (s, 1H), 8.82~8.75 (m, 2H), 8.15 (s, 1H), 7.57~7.48 (m, 3H), 4.54~4.33 (m, 1H), 3.69~3.56 (m, 2H), 2.34~2.27 (m, 1H), 2.05~1.90 (m, 3H), 1.35 (s, 9H). HRMS (ESI) calcd for C
19H
24N
3O
3 [M+H]
+ 342.1818, found 342.1811.
Step 2: (
S)-
tert-Butyl 2-(quinolin-8-ylcarbamoyl)pyr- rolidine-1-carboxylate (1.0 g, 3.0 mmol) and anhydrous methanol (25 mL) were combined in a 100 mL flask. The flask was immersed in a low-temperature cooling circulator maintained at 0 ℃. Acetyl chloride (1.4 g, 18 mmol) was added dropwise via a pressure-equalizing dropping funnel over 30 min while keeping the temperature at 0 ℃. The reaction mixture was stirred at 0 ℃ for 30 min, then allowed to warm to room temperature and stirred overnight. Saturated aqueous sodium carbonate (Na
2CO
3) solution was added to the reaction mixture, which was then extracted with dichloromethane (CH
2Cl
2, 20 mL×3). The combined organic layers were dried over anhydrous sodium sulfate (Na
2SO
4), filtered, and concentrated under reduced pressure using a rotary evaporator. The crude product was purified by flash column chromatography on silica gel with a petroleum ether/ethyl acetate gradient (
V∶
V=3∶1) to afford (
S)-
N-(quinolin-8-yl)pyrrolidine-2-carboxamide
[19b] as a white solid (0.69 g, 2.88 mmol, 96% yield).
Rf=0.20 (
V(petroleum ether)∶
V(ethyl acetate)=1∶1). m.p. 203.2~204.9 ℃;
1H NMR (600 MHz, CDCl
3)
δ: 10.34 (s, 1H), 8.71 (d,
J=3.7 Hz, 1H), 8.60 (dd,
J=6.5, 2.5 Hz, 1H), 8.02 (d,
J=8.2 Hz, 1H), 7.43~7.29 (t,
J=6.3 Hz, 3H), 4.74 (t,
J=7.7 Hz, 1H), 3.63~3.43 (m, 2H), 2.65~2.48 (m, 1H), 2.29~2.19 (m, 1H), 2.15~2.01 (m, 2H). HRMS (ESI) calcd for C
14H
16N
3O [M+H]
+ 242.1293, found 242.1298.
Step 3: Under an argon atmosphere, 2-chloromethylpyri-dine hydrochloride (0.59 g, 3.6 mmol), (S)-N-(quinolin-8-yl)pyrrolidine-2-carboxamide (0.72 g, 3.0 mmol), and anhydrous acetonitrile (55 mL) were combined in a 100 mL flask. Anhydrous sodium carbonate (Na2CO3, 1.59 g, 15 mmol) and tetrabutylammonium bromide (TBABr, 30 mg) were added directly to the mixture as solids. The reaction was heated under reflux for 4 h. After cooling to room temperature, the resulting yellow suspension was filtered, and the filter cake was washed with dichloro- methane (CH2Cl2). The combined filtrates were concen- trated under reduced pressure using a rotary evaporator. The crude residue was dissolved in aqueous NaOH solution (1.0 mol/L, 25 mL) and extracted with CH2Cl2 (20 mL×3). The organic layers were combined, dried over anhydrous sodium sulfate (Na2SO4), filtered, and concentrated under vacuum. The product was purified by flash column chromatography on silica gel using a petro- leum ether/ethyl acetate gradient (V∶V=3∶1) to yield (S)-1-(pyridin-2-ylmethyl)-N-(quinolin-8-yl)pyrrolidine-2-carboxamide (L1) as a yellow oil (0.63 g, 1.89 mmol, 63% yield). Rf=0.23 (V(petroleum ether)∶V(ethyl acetate)= 10∶1); ${[\alpha ]}_{\text{D}}^{\text{25}}$+99.29 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 11.65 (s, 1H), 8.88~8.80 (m, 2H), 8.49 (dd, J=4.9, 0.9 Hz, 1H), 8.23~8.11 (m, 2H), 7.73 (td, J=7.7, 1.9 Hz, 1H), 7.58~7.49 (m, 2H), 7.46 (dd, J=8.3, 4.2 Hz, 1H), 7.16 (dd, J=7.0, 5.4 Hz, 1H), 4.26 (d, J=13.8 Hz, 1H), 3.80 (d, J=13.8 Hz, 1H), 3.53 (dd, J=10.0, 5.4 Hz, 1H), 3.24 (ddd, J=9.4, 6.2, 3.1 Hz, 1H), 2.52 (td, J=9.3, 7.4 Hz, 1H), 2.46~2.33 (m, 1H), 2.21~2.07 (m, 1H), 1.94~1.84 (m, 2H); 13C NMR (151 MHz, CDCl3) δ: 173.5, 159.0, 148.8, 148.2, 139.1, 136.5, 136.3, 134.5, 128.1, 127.4, 123.5, 122.2, 121.7, 121.6, 116.6, 69.0, 62.0, 54.0, 31.0, 24.4. HRMS (ESI) calcd for C20H21N4O [M+H]+ 333.1715, found 333.1719.
(S)-tert-Butyl 2-((2-(pyridin-2-yl)phenyl)carbamoyl)-pyrrolidine-1-carboxylate: White solid, 75% yield. Rf= 0.20 (V(petroleum ether)∶V(ethyl acetate)=20∶1); m.p. 135.1~136.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 12.83 (s, 1H), 8.81~8.70 (m, 2H), 7.85~7.64 (m, 3H), 7.45~7.38 (m, 1H), 7.28~7.23 (m, 1H), 7.18 (t, J=7.6 Hz, 1H), 4.32 (dd, J=8.7, 4.0 Hz, 1H), 3.66~3.53 (m, 2H), 2.37~2.22 (m, 1H), 2.22~2.08 (m, 1H), 1.91~1.79 (m, 2H), 1.21 (s, 9H); 13C NMR (151 MHz, CDCl3) δ: 172.2, 157.9, 154.5, 148.2, 137.6, 130.1, 128.8, 125.5, 123.5, 122.5, 121.8, 121.2, 81.7, 63.9, 46.9, 31.7, 28.0, 23.8. HRMS (ESI) calcd for C21H26N3O3 [M+H]+ 368.1974, found 368.1966.
(S)-N-(2-(Pyridin-2-yl)phenyl)pyrrolidine-2-carbox-amide: Yellow oil, 96% yield. Rf=0.20 (V(petroleum ether)∶V(ethyl acetate)=1∶1); 1H NMR (600 MHz, CDCl3) δ: 12.34 (s, 1H), 8.67 (d, J=6.4 Hz, 1H), 8.53 (d, J=8.3 Hz, 1H), 7.81~7.78 (m, 1H), 7.60 (d, J=8.0 Hz, 1H), 7.54 (dd, J=7.7, 1.6 Hz, 1H), 7.41~7.37 (m, 1H), 7.27~7.23(m, 1H), 7.17~7.13 (m, 1H), 3.84 (dd, J=9.2, 5.1 Hz, 1H), 3.03~2.98 (m, 1H), 2.90~2.86 (m, 1H), 2.18~2.13 (m, 1H), 1.98~1.91 (m, 2H), 1.70~1.59(m, 2H); 13C NMR (151 MHz, CDCl3) δ: 174.7, 158.2, 148.0, 137.4, 136.8, 129.8, 129.2, 128.1, 123.6, 123.4, 121.8, 61.7, 47.3, 31.2, 26.2. HRMS (ESI) calcd for C16H18N3O [M+H]+ 268.1450, found 268.1443.
(S)-N-(2-(Pyridin-2-yl)phenyl)-1-(pyridin-2-ylmethyl)-pyrrolidine-2-carboxamide (L2): Yellow oil, 60% yield. Rf=0.23 (V(petroleum ether)∶V(ethyl acetate)=10∶1); ${[\alpha ]}_{\text{D}}^{\text{25}}$+121.23 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 12.33 (s, 1H), 8.54~8.40 (m, 3H), 7.81 (td, J=7.7, 1.8 Hz, 1H), 7.65 (d, J=8.0 Hz, 1H), 7.57 (dd, J=7.8, 1.6 Hz, 1H), 7.51~7.44 (m, 1H), 7.44~7.37 (m, 2H), 7.24~7.14 (m, 2H), 7.13~7.06 (m, 1H), 4.08 (d, J=13.4 Hz, 1H), 3.75 (d, J=13.4 Hz, 1H), 3.44 (dd, J=10.2, 4.4 Hz, 1H), 3.14~2.96 (m, 1H), 2.55 (td, J=9.5, 6.2 Hz, 1H), 2.29~2.15 (m, 1H), 1.93~1.81 (m, 4.0 Hz, 1H), 1.79~1.66 (m, 1H), 1.65~1.52 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 174.2, 158.7, 158.4, 148.9, 147.8, 137.4, 136.8, 136.5, 129.9, 129.4, 128.1, 123.8, 123.6, 123.4, 122.2, 121.9, 68.3, 61.3, 53.0, 31.1, 24.5. HRMS (ESI) calcd for C22H23N4O [M+H]+ 359.1872, found 359.1873.
(S)-tert-Butyl 2-((2-((S)-4-isopropyl-4,5-dihydrooxazol-2-yl)phenyl)carbamoyl)pyrrolidine-1-carboxylate: White solid, 75% yield. Rf=0.20 (V(petroleum ether)∶V(ethyl acetate)=20∶1); m.p. 119.7~120.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.79 (d, J=8.4 Hz, 1H), 7.86 (d, J=6.2 Hz, 1H), 7.47 (t, J=8.1 Hz, 1H), 7.12~7.05 (m, 1H), 4.34~4.22 (m, 3H), 4.18~4.10 (m, 1H), 3.69~3.60 (m, 1H), 3.58~3.50 (m, 1H), 2.32~2.22 (m, 1H), 2.15~2.02 (m, 2H), 2.01~1.94 (m, 1H), 1.91~1.83 (m, 1H), 1.31 (s, 9H), 1.02~0.97 (m, 3H), 0.85 (d, J=6.7 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 172.8, 163.3, 154.2, 139.8, 132.6, 129.3, 122.5, 119.7, 113.5, 80.1, 72.5, 67.9, 62.7, 47.0, 31.8, 28.4, 24.0, 19.6, 17.0. HRMS (ESI) calcd for C22H32N3O4 [M+H]+ 402.2393, found 402.2385.
(S)-N-(2-((S)-4-Isopropyl-4,5-dihydrooxazol-2-yl)phen- yl)pyrrolidine-2-carboxamide: White solid, 97% yield. Rf=0.20 (V(petroleum ether)∶V(ethyl acetate)=1∶1); m.p. 111.2~112.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 12.65 (s, 1H), 8.78 (d, J=9.6 Hz, 1H), 7.83 (dd, J=7.9, 1.7 Hz, 1H), 7.51~7.37 (m, 1H), 7.16~6.93 (m, 1H), 4.45~4.32 (m, 1H), 4.24~4.12 (m, 1H), 4.04 (t, J=8.2 Hz, 1H), 3.88 (dd, J=8.9, 5.5 Hz, 1H), 3.17~3.05 (m, 1H), 3.03~2.93 (m, 1H), 2.24~2.15 (m, 2H), 1.99 (dt,J=12.4, 6.2 Hz, 1H), 1.87~1.71 (m, 3H), 1.07 (d, J=6.7 Hz, 3H), 0.98 (d, J=6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 175.4, 162.9, 139.7, 132.3, 129.3, 122.4, 120.0, 114.1, 73.2, 69.2, 62.3, 47.5, 33.2, 31.7, 26.1, 18.8. HRMS (ESI) calcd for C17H24N3O2 [M+H]+ 302.1869, found 302.1861.
(S)-N-(2-((S)-4-Isopropyl-4,5-dihydrooxazol-2-yl)-phenyl)-1-(pyridin-2-ylmethyl)pyrrolidine-2-carboxamide (L3): White solid, 72% yield. Rf=0.25 (V(petroleum ether)∶V(ethyl acetate)=20∶1); m.p. 108.9~110.2 ℃; ${[\alpha ]}_{\text{D}}^{\text{25}}$+40.38 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 12.26 (s, 1H), 8.64 (d, J=9.7 Hz, 1H), 8.49 (d, J=6.7 Hz, 1H), 7.85 (dd, J=7.9, 1.7 Hz, 1H), 7.58~7.49 (m, 1H), 7.49~7.38 (m, 2H), 7.14~7.02 (m, 2H), 4.33 (dd, J=9.4, 7.9 Hz, 1H), 4.22~4.10 (m, 1H), 4.10~4.01 (m, 2H), 3.83 (d, J=13.5 Hz, 1H), 3.52~3.31 (m, 1H), 3.23~3.07 (m, 1H), 2.65~2.53 (m, 1H), 2.32~2.13 (m, 1H), 1.98~1.79 (m, 4H), 0.99 (d, J=6.7 Hz, 3H), 0.91(d, J=6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 174.7, 164.2, 158.6, 149.0, 139.5, 136.3, 132.2, 129.4, 123.9, 122.5, 122.1, 120.6, 114.4, 73.2, 68.7, 60.8, 54.0, 32.7, 31.0, 24.0, 19.0, 17.7. HRMS (ESI) calcd for C23H29N4O2 [M+H]+ 393.2291, found 393.2289.



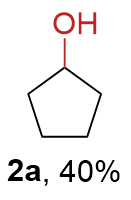
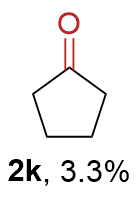
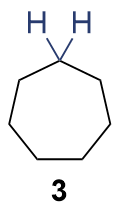
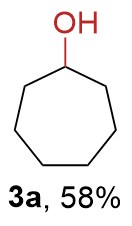
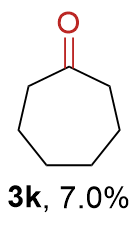

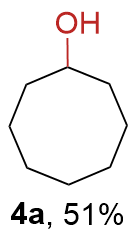
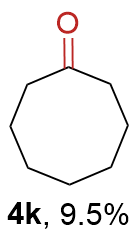
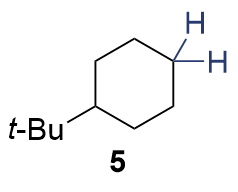
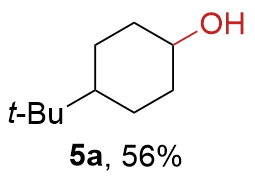
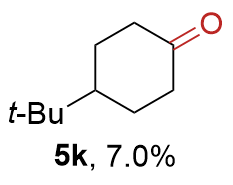
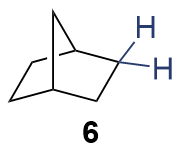
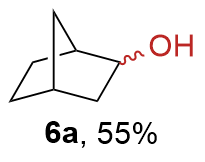
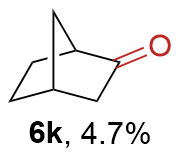
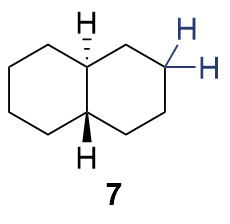
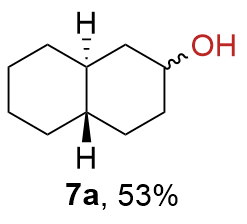
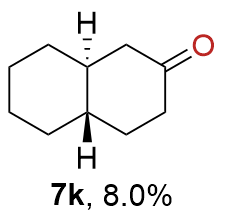
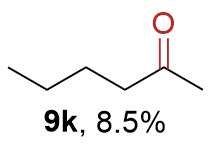
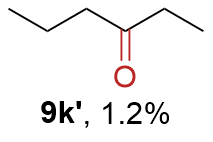
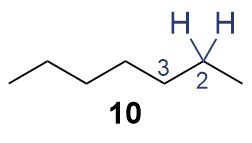
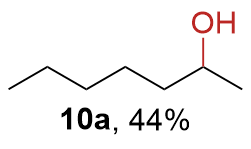
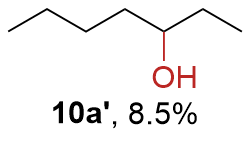
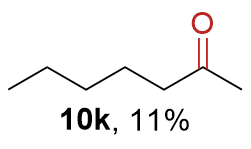
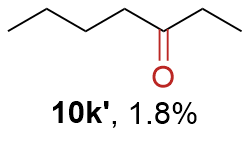
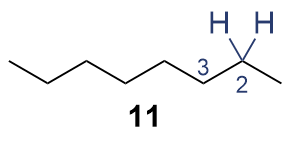

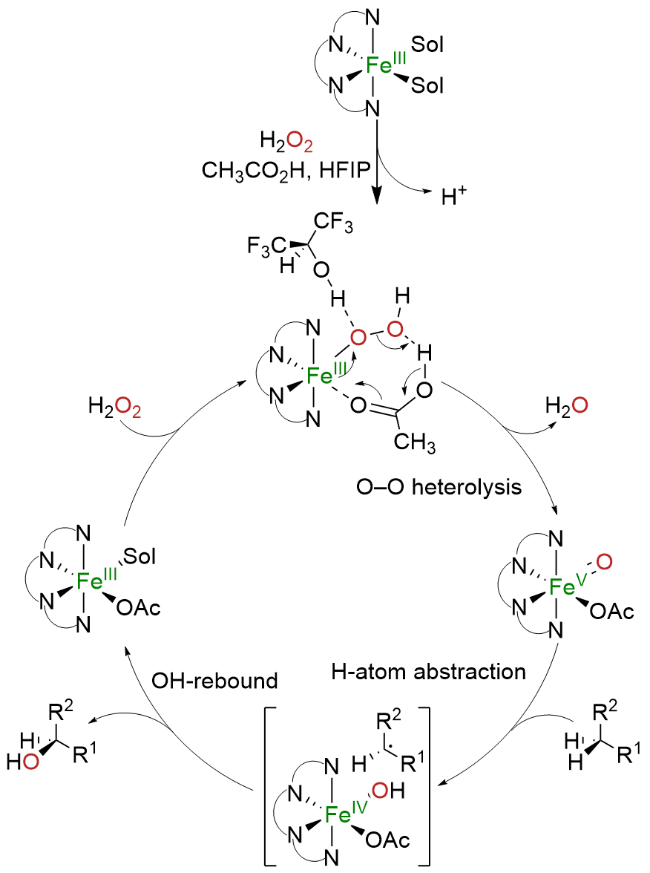
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}