


1 Introduction
Cyclic aldimine derivatives are significant intermediates, attributed to their diverse biological effects and their function as essential reagents or intermediates in organic synthesis.[1] As a result, cyclic aldimines have been extensively modified through diverse chemical transformations in previous studies.[2] Given that the C=N double bonds in imines are the primary reactive sites, the reported methodologies mainly focus on nucleophilic addition,[3] ring- expansion,[4] and annulation reactions.[5] In contrast, the unsaturated C=N bond in cyclic aldimines must be maintained for the synthesis of many desired molecules, which is typically achieved through C—H functionalization. However, research in this area is constrained due to the inherent stability of the C—H bond (Scheme 1, a).[6] The radical- mediated three-component C—H functionalization remains underexplored and has not been thoroughly investigated.
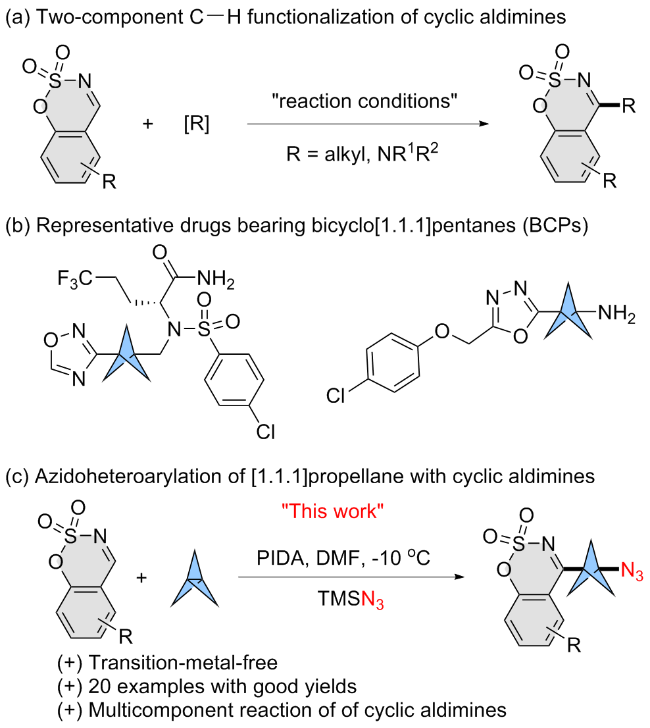
Strained small sp3-rich rings are frequently employed as unique bioisosteres in modern medicinal chemistry, mainly because their small size and structural rigidity often lead to enhanced pharmacological properties compared to their parent molecules, such as aqueous solubility and metabolic stability.[7] For instance, 1,3-disubstituted bicyclo[1.1.1]- pentanes (BCPs) are frequently used as bioisosteres for alkynes and para-substituted aromatic rings.[8] Likewise, mono-substituted bicyclo[1.1.1]pentanes (BCPs) are sought after as replacements for tertiary butyl and phenyl groups.[9] Consequently, significant efforts have been conducted to develop versatile and effective strategies for synthesizing the strained bicyclo[1.1.1]pentanes (BCPs) rings.
Recently, the development of radical-mediated multicomponent reaction has received considerable attention due to their multifaceted technical advantages, significantly simplifying the cumbersome purification steps inherent in traditional multi-step synthesis.[10-11] On the other hand, the hypervalent iodine is commonly employed in these systems, which eliminates the need for transition metal catalysts, simultaneously resolving heavy metal contamination concerns.[12] These multicomponent reactions also have emer- ged as promising approaches for the rapid synthesis of 1,3-disubstituted bicyclo[1.1.1]pentanes (BCPs).[13-15]
To address the research shortcomings and to continue our work on the functionalization of [1.1.1]propellane and green synthesis,[16] herein, we disclose a practical method for direct 1,3-azidoheteroarylation of [1.1.1]propellane with cyclic aldimines and azidotrimethylsilane (TMSN3) under mild conditions. Importantly, the resulting functionalized strained rings act as synthetically flexible building blocks for a variety of transformations, making this method particularly appealing for constructing complex medicinal skeletons.
2 Results and discussion
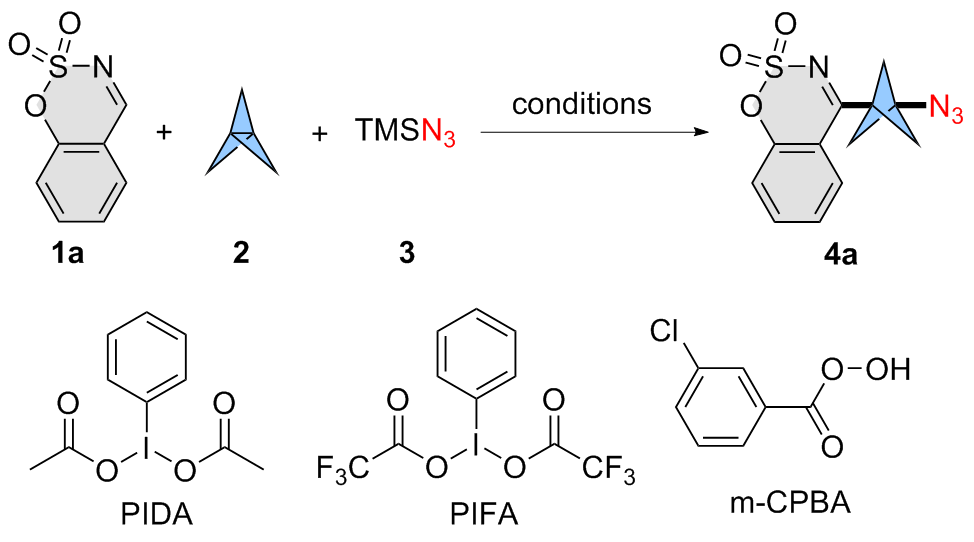
To achieve the transformation, N-sulfonyl ketimine (1a), [1.1.1]propellane (2), and azidotrimethylsilane (TMSN3) (3) were selected as the initial reactants. The optimal reaction conditions were determined through a systematic evaluation involving the selection of an appropriate oxidant, solvent, reaction atmosphere and temperature (Table 1). When the reaction was conducted in MeCN under a nitrogen atmosphere with (diacetoxyiodo)benzene (PIDA) serving as the oxidant, the target 1-azido-3-heteroaryl bicyclo[1.1.1]pentane (BCP) (4a) was synthesized with a yield of 40%. Alternative oxidant, including bis(trifluoro- acetoxyiodo)benzene (PIFA), 3-chloroperbenzoic acid (m-CPBA), and hydrogen peroxide (H2O2), demonstrated negligible catalytic activity in the reactions (Table 1, Entries 2~4). Control experiment showed that the presence of an oxidant was a crucial and essential element for the successful execution of this transformation. To enhance the yield of corresponding product, a wide range of organic solvents including dimethylsulfoxide (DMSO), N-methyl- pyrrolidone (NMP), dimethyl formamide (DMF), ethyl acetate (EA), methyl alcohol (MeOH), and dichloromethane (DCM) were explored (Table 1, Entries 6~11), and it was found that dimethyl formamide (DMF) was the optimal solvent for 1,3-azidoheteroarylation of [1.1.1]propellane.
Table 1 Screening of reaction conditionsa |
| Entry | Oxidant | Solvent | Yieldb/% |
|---|---|---|---|
| 1 | PIDA | MeCN | 40 |
| 2 | PIFA | MeCN | 18 |
| 3 | m-CPBA | MeCN | 0 |
| 4 | H2O2 | MeCN | 0 |
| 5 | — | MeCN | 0 |
| 6 | PIDA | DMSO | 67 |
| 7 | PIDA | NMP | 36 |
| 8 | PIDA | DMF | 72 |
| 9 | PIDA | EA | 20 |
| 10 | PIDA | MeOH | Trace |
| 11 | PIDA | DCM | 25 |
| 12c | PIDA | DMF | 70 |
| 13d | PIDA | DMF | 38 |
| 14e | PIDA | DMF | 0 |
| 14f | PIDA | DMF | 61 |
| 15g,h,i | PIDA | DMF | 60, 62, 65 |
a Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), 3 (0.3 mmol), oxidant (0.3 mmol), solvent (1 mL), 10 ℃, N2, 6 h. b Isolated yield. c 3 (0.4 mmol). d 3 (0.1 mmol). e Without 3. f Under air. g At room temperature. h At 0 ℃. i At 5 ℃. |
To further improve the yields, the dosage of azidotrimethylsilane (TMSN3) (3) was then studied. Increasing the dosage of TMSN3 (3) did not significantly influence the azidoheteroarylation process (Table 1, Entry 12). Conversely, decreasing the dosage of azidotrimethylsilane (TMSN3) (3) markedly diminished the efficiency of the azidoheteroarylation reaction (Table 1, Entry 13). Of note, excluding azidotrimethylsilane (TMSN3) (3) from the reaction mixture led to the absence of any product (Table 1, Entry 14). Further evaluation of reaction atmosphere and temperature did not obviously improve the reactivity of the reaction.
Armed with the optimal reaction conditions, the range of substrates suitable for the azidoheteroarylation of [1.1.1]- propellane was then explored. As shown in Table 2, cyclic aldimine derivatives with a variety of substituents were found to be suitable for the reaction, yielding the corresponding products 4a~4t in moderate to excellent yields. Substrates featuring electron-donating groups, such as Me, tBu, MeO, and CF3O at the C(6)-position exhibited good reactivity, resulting in products (4a~4e) with satisfactory yields. Additionally, cyclic aldimines with C(6)-halogen substituents, which are valuable for subsequent synthetic modifications, were well compatible under optimal conditions, producing target products (4f~4h) in 55%~62% yields. Moreover, the azidoheteroarylation of [1.1.1]pro- pellane using cyclic aldimines with Me, MeO, EtO, BnO, F, Cl, Br, and MeO2C groups at the C(7)- or C(8)-positions led to the formation of the corresponding products (4i~4r) with 50%~77% yields. Of note, substrate with electron- withdrawing group provided corresponding product in lower yield probably due to the low reactivity. Furthermore, bifunctionalized cyclic aldimines also proved to be effective substrates, delivering the desired products (4s and 4t) in acceptable yields. Other substrates with iodo- (1u), alkenyl- (1v), and alkyl aldehyde moiey (1w) were not compatible under standard condition.
Table 2 Scope azidoheteroarylationa,b |

 |
a Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), 3 (0.3 mmol), PIDA (0.3 mmol), DMF (1 mL), -10 ℃, N2, 6 h. b Isolated yield. |
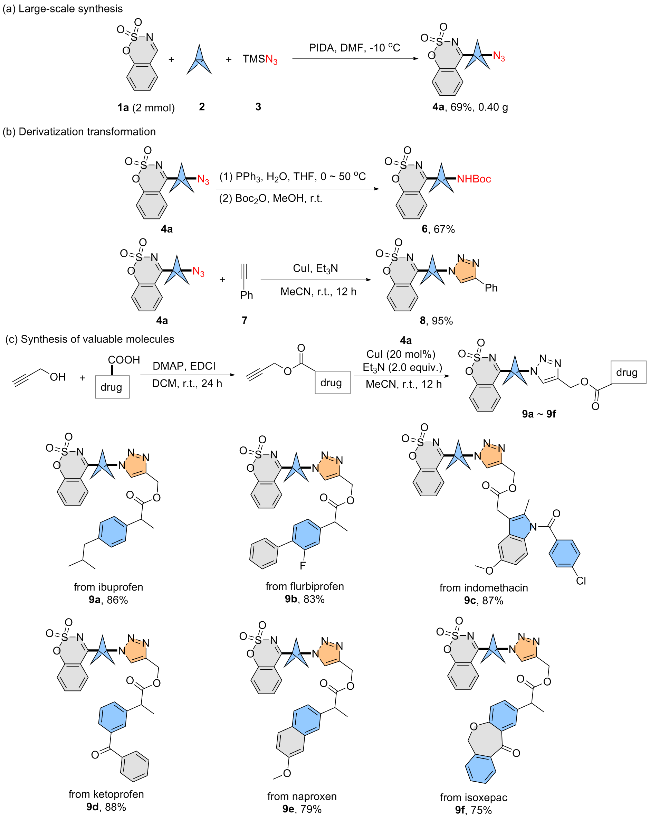
To evaluate the synthetic capabilities of this approach, gram-scale reactions, product transformations, and late- stage modifications of pharmaceuticals were conducted. The product (4a) was synthesized in 69% yield, during large-scale reaction under optimized conditions (Scheme 2, a). Furthermore, a reduction-protection sequence led to the formation of 1-amino-3-heteroaryl bicyclo[1.1.1]pentane (BCPs) (6) with 70% yield (Scheme 2, b. The 1-amino- 3-heteroaryl bicyclo[1.1.1]pentane (BCP) (4a) could also undergo click chemistry with phenylacetylene (7) to yield the corresponding bicyclo[1.1.1]pentane (BCP)-containing triazole (8). Additionally, the developed azidoheteroarylation reaction-click chemistry sequence was successfully applied for the late-stage functionalization of valuable molecules. The ibuprofen, flurbiprofen, indomethacin, ke- toprofen, naproxen, and isoxepac were effectively modified, resulting in the desired products (9a~9f) with 75%~90% yields (Scheme 2, c.
To elucidate the mechanisms underlying the solvent- controlled difunctionalization of [1.1.1]propellane, a series of control experiments were executed. Initially, the introduction of 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) into the catalytic systems resulted in the complete inhibition of azidoheteroarylation reaction of [1.1.1]propellane (Scheme 3). Additionally, the formation of the corresponding radical adduct (10) was detected by high-resolution mass spectrometry (HRMS). Moreover, the formation of azido radical (•N3) was confirmed by electron spin resonance (ESR) spectroscopy. These observations robustly indicate that a radical-mediated process is pivotal to the reaction.
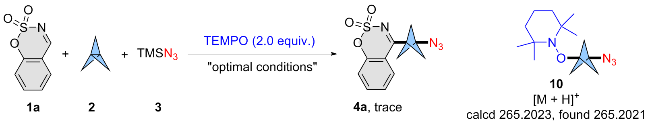
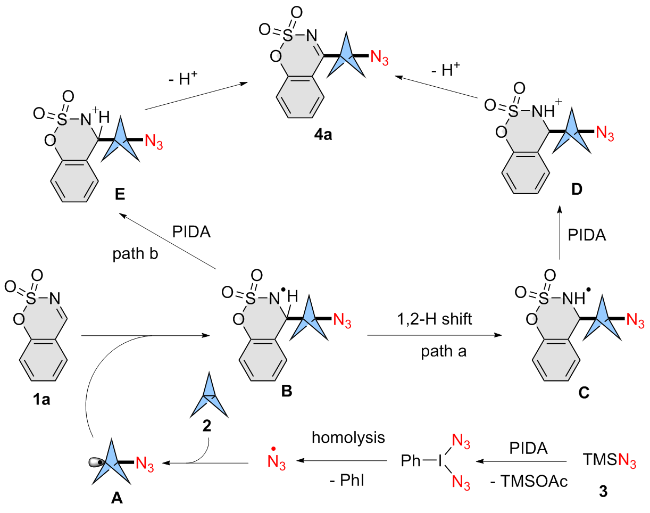
Based on the aforementioned results and existing literature,[17] we have proposed single-electron transfer (SET) mechanisms for these two reactions as depicted in Scheme 4. Initially, the reaction between (diacetoxyiodo)benzene (PIDA) and azidotrimethylsilane (TMSN3) (3) gave an unstable acyclic azido hypoiodite intermediate, which promptly decomposed to produce an azido radical (•N3). In the azidoheteroarylation reaction of [1.1.1]propellane, the azido radical (•N3) directly inserted into [1.1.1]propellane, resulting in the formation of the metastable N3-BCP-radical A, which then reacted with cyclic aldimine 1a to deliver a nitrogen radical intermediate B. In path a, the nitrogen radical intermediate B underwent a 1,2-hydrogen shift to generate a carbon radical intermediate C, which was further oxidized by (diacetoxyiodo)benzene (PIDA) to produce a carbon cation intermediate D.[6a,17b] In path b, the nitrogen radical intermediate B was directly oxidized by (diacetoxyiodo)benzene (PIDA) to produce a nitrogen cation intermediate E. Finally, the desired 1-azido-3-heteroaryl bicyclo[1.1.1]pentane (BCP) (4a) was obtained via deprotonation of the carbon cation intermediate D or nitrogen cation intermediate E.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In conclusion, an efficient protocol for direct 1,3-azido- heteroarylation of [1.1.1]propellane with cyclic aldimines and TMSN3 was developed. This approach is distinguished by its mild reaction conditions and independence from transition-metal catalysts, offering an environmentally benign and efficient route for the synthesis of a range of cyclic aldimine derivatives that incorporate the structurally advantageous bicyclo[1.1.1]pentane (BCP) motif. Our control experiments have elucidated the participation of a radical- relay mechanism in the reaction cascade.
4 Experimental section
4.1 General information
All reagents and deuterated solvents were commercially available and used without further purification. 1H NMR, 13C NMR and 19F NMR spectra were recorded on a Bruker Advance 500 spectrometer at ambient temperature with CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. Melting points were determined on an X-5 Data microscopic melting point apparatus. Analytical thin-layer chromatography (TLC) was performed on a Merk precoated TLC (silica gel 60 F254) plates. Compounds for HRMS were analyzed by positive mode electrospray ionization (ESI) using an Agilent 6530 QTOF mass spectrometer. All the cyclic aldimines was prepared according to the previous report.[6] The [1.1.1]propellane was prepared ac- cording to the previous report.[14] Caution! This procedure controlled risk by the use of millimolar concentration of highly flammable chemical n-butyllithium.
4.2 General procedure for azidoheteroarylation of [1.1.1]propellane
An oven-dried 25 mL Schlenk tube fitted with a magnetic stirring bar was charged with cyclic aldimines 1 (0.2 mmol, 1.0 equiv.), [1.1.1]propellane (2, 0.3 mmol, 1.5 equiv.) and PIDA (0.3 mmol, 1.5 equiv.) under N2 atmosphere. Then, the mixture was cooled down to 10 ℃ and TMSN3 (3) (0.3 mmol, 1.5 equiv.) in DMF (1 mL) was added dropwise under stirring. After stirring at 10 ℃ for 6 h, the mixture was poured into NaHCO3 aqueous solution, and extracted with EtOAc (5 mL×3). The collected organic layer was washed with brine, and dried with MgSO4. The solvent was removed in vacuo, and the obtained residue was further purified by silica gel column chromatography [200~300 mesh silica gel, V(petroleum ether, PE)∶V(ethyl acetate, EA)∶V(DCM)=5∶1∶1] to afford product 4.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)benzo[e][1,2,3]-oxathiazine 2,2-dioxide (4a): Yellow solid (42 mg, 72% yield). m.p. 105~106 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.92 (dd, J=8.0, 1.6 Hz, 1H), 7.73 (ddd, J=8.8, 7.5, 1.6 Hz, 1H), 7.39 (td, J=7.7, 1.2 Hz, 1H), 7.33 (dd, J=8.4, 1.1 Hz, 1H), 2.60 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 174.8, 153.7, 137.1, 128.4, 125.8, 119.5, 116.1, 56.2, 52.1, 39.1; HRMS (ESI-TOF) calcd for C12H10N4O3SNa [M+Na]+ 313.0366, found 313.0367.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-methylbenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4b): Yellow solid (42 mg, 70% yield). m.p. 146~147 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.63~7.57 (m, 1H), 7.45 (dd, J=8.5, 2.1 Hz, 1H), 7.14 (d, J=8.5 Hz, 1H), 2.52 (s, 6H), 2.37 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 174.8, 151.7, 137.9, 135.9, 128.2, 119.2, 115.9, 56.2, 52.1, 39.1, 21.1; HRMS (ESI- TOF) calcd for C13H13N4O3S [M+H]+ 305.0703, found 305.0700.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-(tert-butyl)-benzo[e][1,2,3]oxathiazine 2,2-dioxide (4c): Yellow solid (46 mg, 67% yield). m.p. 132~133 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.89 (d, J=2.4 Hz, 1H), 7.75 (dd, J=8.7, 2.4 Hz, 1H), 7.26~7.24 (m, 1H), 2.60 (s, 6H), 1.37 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 175.0, 151.5, 149.1, 134.8, 124.8, 118.9, 115.5, 56.3, 53.4, 52.1, 51.7, 39.1, 34.8, 31.2; HRMS (ESI-TOF) calcd for C16H18N4O3SNa [M+Na]+ 369.0992, found 369.0997.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-methoxybenzo-[e][1,2,3]oxathiazine 2,2-dioxide (4d): Yellow solid (47 mg, 73% yield). m.p. 137~138 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.33 (dd, J=2.1, 1.2 Hz, 1H), 7.27~7.24 (m, 2H), 3.87 (s, 3H), 2.59 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 174.5, 156.6, 147.3, 122.7, 120.3, 116.4, 112.5, 56.2, 56.1, 52.0, 39.1; HRMS (ESI-TOF) calcd for C13H12- N4O4SNa [M+Na]+ 343.0471, found 343.0471.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-(trifluorometh-oxy)benzo[e][1,2,3]oxathiazine 2,2-dioxide (4e): Yellow solid (52 mg, 70% yield). m.p. 135~136 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.79~7.72 (m, 1H), 7.61~7.57 (m, 1H), 7.39 (d, J=9.1 Hz, 1H), 2.60 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 173.8, 151.7, 145.5, 129.7, 121.3, 120.6, 120.3 (q, J=259.6 Hz), 116.5, 56.3, 52.0, 39.0; 19F NMR (471 MHz, CDCl3) δ: 58.31; HRMS (ESI-TOF) calcd for C13H9F3N4O4SNa [M+Na]+ 397.0189, found 397.0196.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-fluorobenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4f): Yellow solid (35 mg, 57% yield). m.p. 106~107 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.58 (dd, J=7.9, 3.0 Hz, 1H), 7.45 (ddd, J=9.1, 7.4, 3.0 Hz, 1H), 7.33 (dd, J=9.1, 4.4 Hz, 1H), 2.60 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 172.9 (d, J=2.5 Hz), 158.7 (d, J=254.1 Hz), 148.7 (d, J=2.5 Hz), 123.3 (d, J=23.4 Hz), 120.3 (d, J=8.8 Hz), 115.5 (d, J=8.8 Hz), 113.5 (d, J=23.4 Hz), 55.2, 50.1, 38.0; 19F NMR (471 MHz, CDCl3) δ: 112.59; HRMS (ESI-TOF) calcd for C12H9FN4O3- SNa [M+Na]+ 331.0272, found 331.0266.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-chlorobenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4g): Yellow solid (39 mg, 60% yield). m.p. 117~118 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.85 (d, J=2.4 Hz, 1H), 7.67 (dd, J=8.8, 2.5 Hz, 1H), 7.29 (d, J=8.9 Hz, 1H), 2.61 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 173.8, 152.1, 136.9, 131.3, 127.9, 121.0, 116.9, 56.3, 52.1, 39.0; HRMS (ESI-TOF) calcd for C12H9- BrN4O3SNa [M+Na]+ 346.9976, found 346.9970.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6-bromobenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4h): White solid (46 mg, 62% yield). m.p. 118~119 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.00 (d, J=2.3 Hz, 1H), 7.81 (dd, J=8.8, 2.3 Hz, 1H), 7.23 (d, J=8.8 Hz, 1H), 2.61 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 173.7, 152.6, 139.8, 130.9, 121.2, 118.4, 117.3, 56.3, 52.1, 39.0; HRMS (ESI-TOF) calcd for C12H9- BrN4O3Na [M+Na]+ 390.9471, found 390.9475.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-7-methylbenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4i): Yellow solid (44 mg, 73% yield). m.p. 165~166 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.79 (d, J=8.1 Hz, 1H), 7.20~7.16 (m, 1H), 7.12 (s, 1H), 2.58 (s, 6H), 2.49 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 174.6, 153.8, 149.7, 128.2, 126.8, 119.6, 113.7, 56.2, 52.0, 39.0, 22.2; HRMS (ESI-TOF) calcd for C13H12- N4O3Na [M+Na]+ 327.0522, found 327.0527.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-7-methoxybenzo-[e][1,2,3]oxathiazine 2,2-dioxide (4j): Yellow solid (42 mg, 65% yield). m.p. 133~134 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.82 (d, J=8.9 Hz, 1H), 6.87 (dd, J=9.0, 2.5 Hz, 1H), 6.75 (d, J=2.5 Hz, 1H), 3.93 (s, 3H), 2.56 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 172.9, 165.5, 155.3, 129.0, 112.4, 108.5, 102.3, 55.4, 55.1, 51.0, 38.0; HRMS (ESI- TOF) calcd for C13H12N4O4SNa [M+Na]+ 343.0471, found 343.0480.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-7-(benzyloxy)ben- zo[e][1,2,3]oxathiazine 2,2-dioxide (4k): Yellow solid (52 mg, 66% yield). m.p. 155~156 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.81 (d, J=9.0 Hz, 1H), 7.41 (d, J=1.4 Hz, 6H), 6.92 (dd, J=9.0, 2.5 Hz, 1H), 6.82 (d, J=2.5 Hz, 1H), 5.17 (s, 2H), 2.56 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 173.9, 165.6, 156.2, 134.7, 123.0, 129.0, 128.9, 127.6, 113.9, 109.7, 104.3, 71.2, 56.1, 52.0, 39.0; HRMS (ESI-TOF) calcd for C14H12N4O5SNa [M+Na]+ 419.1471, found 419.1475.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-7-fluorobenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4l): Yellow solid (36 mg, 59% yield). m.p. 148~149 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.96 (dd, J=8.9, 5.7 Hz, 1H), 7.11 (ddd, J=8.9, 7.7, 2.5 Hz, 1H), 7.05 (dd, J=8.3, 2.5 Hz, 1H), 2.59 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 173.9, 166.9 (d, J=263.3 Hz), 155.7 (d, J=13.8 Hz), 130.8 (d, J=11.3 Hz), 113.9 (d, J=25.2 Hz), 112.9 (d, J=3.8 Hz), 107.5 (d, J=25.2 Hz), 56.2, 52.1, 39.1; 19F NMR (471 MHz, CDCl3) δ: 94.16; HRMS (ESI-TOF) calcd for C12H9FN4O3SNa [M+Na]+ 331.0272, found 331.0267.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-7-chlorobenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4m): Yellow solid (42 mg, 65% yield). m.p. 137~138 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.85 (d, J=8.5 Hz, 1H), 7.38~7.32 (m, 2H), 2.59 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 174.1, 154.2, 143.3, 129.3, 126.4, 119.9, 114.5, 56.2, 52.1, 39.1; HRMS (ESI-TOF) calcd for C12H9ClN4O3SNa [M+Na]+ 346.9976, found 346.9976.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-8-methylbenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4n): Yellow solid (46 mg, 75% yield). m.p. 125~126 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.75 (dd, J=8.0, 1.6 Hz, 1H), 7.60~7.53 (m, 1H), 7.30~7.26 (m, 1H), 2.59 (s, 6H), 2.40 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 175.2, 152.1, 138.5, 129.3, 125.9, 125.1, 115.9, 56.2, 52.0, 39.2, 15.1; HRMS (ESI- TOF) calcd for C13H12N4O3SNa [M+Na]+ 327.0522, found 327.0527.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-8-methoxybenzo-[e][1,2,3]oxathiazine 2,2-dioxide (4o): Yellow solid (49 mg, 77% yield). m.p. 133~134 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.46 (dd, J=7.6, 1.8 Hz, 1H), 7.32~7.26 (m, 2H), 3.95 (s, 3H), 2.58 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 175.1, 149.2, 143.4, 125.5, 119.1, 119.0, 119.0, 116.8, 56.6, 56.2, 52.0, 39.3; HRMS (ESI-TOF) calcd for C13H12N4O4SNa [M+Na]+ 343.0471, found 343.0467.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-8-ethoxybenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4p): Yellow solid (48 mg, 72% yield). m.p. 169~170 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.48~7.41 (m, 1H), 7.27 (d, J=6.9 Hz, 2H), 4.15 (q, J=7.0 Hz, 2H), 2.58 (s, 6H), 1.48 (t, J=7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ: 175.2, 148.6, 143.5, 125.4, 120.2, 119.0, 116.9, 65.6, 56.2, 52.0, 39.3, 14.6; HRMS (ESI-TOF) calcd for C14H14N4O4SNa [M+Na]+ 357.0628, found 357.0629.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-8-bromobenzo[e]-[1,2,3]oxathiazine 2,2-dioxide (4q): Yellow solid (49 mg, 67% yield). m.p. 105~106 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.93 (dd, J=8.0, 1.4 Hz, 1H), 7.88 (dd, J=7.9, 1.5 Hz, 1H), 2.60 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 174.7, 150.8, 140.5, 127.3, 126.3, 117.5, 113.5, 56.3, 52.1, 39.1; HRMS (ESI-TOF) calcd for C12H9BrN4O3SNa [M+Na]+ 390.9471, found 390.9481.
Methyl 4-(3-azidobicyclo[1.1.1]pentan-1-yl)benzo[e]-[1,2,3]oxathiazine-8-carboxylate 2,2-dioxide (4r): Yellow solid (39 mg, 55% yield). m.p. 125~126 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.62 (s, 1H), 8.36 (dd, J=8.6, 2.0 Hz, 1H), 7.38 (d, J=8.7 Hz, 1H), 4.00 (s, 3H), 2.64 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 174.4, 164.5, 156.5, 137.7, 130.2, 127.9, 119.8, 115.6, 56.3, 53.1, 52.1, 39.1; HRMS (ESI-TOF) calcd for C14H12N4O5SNa [M+Na]+ 371.0421, found 371.0431.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-6,8-di-tert-butyl-benzo[e][1,2,3]oxathiazine 2,2-dioxide (4s): Yellow solid (39 mg, 49% yield). m.p. 154~155 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.75 (d, J=2.4 Hz, 1H), 7.73 (d, J=2.3 Hz, 1H), 2.58 (s, 6H), 1.46 (s, 9H), 1.36 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 175.9, 150.5, 148.1, 140.4, 132.2, 122.8, 116.4, 56.3, 52.0, 39.4, 35.3, 35.0, 31.3, 29.8; HRMS (ESI-TOF) calcd for C20H27N4O3S [M+H]+ 403.1798, found 403.1782.
4-(3-Azidobicyclo[1.1.1]pentan-1-yl)-5,7-dimethoxy-benzo[e][1,2,3]oxathiazine 2,2-dioxide (4t): Yellow solid (45 mg, 64% yield). m.p. 171~172 ℃; 1H NMR (500 MHz, CDCl3) δ: 6.40 (d, J=2.4 Hz, 1H), 6.32 (d, J=2.4 Hz, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 2.42 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 175.4, 167.1, 160.1, 157.0, 103.1, 96.4, 95.8, 56.4, 55.8, 55.0, 51.7, 41.3; HRMS (ESI-TOF) calcd for C14H14N4O5SNa [M+Na]+ 373.0577, found 373.0572.
4.3 General procedure for the synthesis of product 4a on a 2 mmol scale
An oven-dried 50 mL Schlenk tube fitted with a magnetic stirring bar was charged with cyclic aldimines 1 (2.0 mmol, 1.0 equiv.), [1.1.1]propellane (2) (3.0 mmol, 1.5 equiv.) and PIDA (3.0 mmol, 1.5 equiv.) under nitrogen atmosphere. Then, the mixture was cooled down to 10 ℃ and TMSN3 (3) (3.0 mmol, 1.5 equiv.) in DMF (10 mL) was added dropwise under stirring. After stirring at 10 ℃ for 12 h, the mixture was poured into NaHCO3 aqueous solution, and extracted with EtOAc (20 mL×3). The collected organic layer was washed with brine, and dried with MgSO4. The solvent was removed in vacuo, and the obtained residue was further purified by silica gel column chromatography [200~300 mesh silica gel, V(PE)∶V(EA)∶V(DCM)=5∶1∶1].
4.4 General procedure for the synthesis of compound 6
A mixture of compound 4a (0.2 mmol, 1.0 equiv.), tetrahydrofuran (THF) (2 mL) and H2O (0.5 mmol, 2.5 equiv.) in a 25-mL Schlenk tube was stirred at 0 ℃ under a nitrogen atmosphere for 10 min. Then, a solution of PPh3 (0.25 mmol, 1.25 equiv.) in THF (1.5 mL) was added dropwise. The mixture was then warmed up to 50 ℃ and stirred for 5 h. Upon completion, the solvent was removed under reduced pressure, and MeOH (3 mL) was added, followed by addition of Boc2O (0.4 mmol, 2.0 equiv.). After stirring at room temperature for another 12 h, the solvent was removed in vacuo, and the obtained residue was further purified by silica gel column chromatography (200~300 mesh silica gel, V(PE)∶ V(EA)=3∶1) to give N-(3-(2,2- dioxidobenzo[e][1,2,3]oxathiazin-4-yl)bicyclo[1.1.1]pen-tan-1-yl)pivalamide (6) (49 mg, 67% yield). Yellow solid, m.p. 112~113 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.01 (d, J=7.2 Hz, 1H), 7.74~7.65 (m, 1H), 7.35 (t, J=7.6 Hz, 1H), 7.30 (d, J=8.3 Hz, 1H), 5.16 (s, 1H), 2.63 (s, 6H), 1.47 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 175.9, 154.8, 153.6, 136.9, 128.8, 125.7, 119.3, 116.2, 56.4, 46.8, 41.0, 28.4; HRMS (ESI-TOF) calcd for C12H11FNO3S [M+H]+ 365.4216, found 365.4220.
4.5 General procedure for the click chemistry
A mixture of compound 4a (0.2 mmol), phenylacetylene (7) or alkyne-containing drugs (0.3 mmol), CuI (20 mol%), Et3N (0.4 mmol, 2.0 equiv.), and MeCN (5 mL) in 25-mL Schlenk tube was stirred at room temperature for 12 h. After completion of the reaction, the reaction mixture was filtered and the solution was concentrated in vacuo. The obtained residue was further purified by silica gel column chromatography (200~300 mesh silica gel, V(PE)∶V(EA)=3∶1 for compound 8, V(PE)∶V(EA)=1∶1 for compounds 9).
4-(3-(4-Phenyl-1H-1,2,3-triazol-1-yl)bicyclo[1.1.1]-pentan-1-yl)benzo[e][1,2,3]oxathiazine 2,2-dioxide (8): Yellow solid (74 mg, 95% yield). m.p. 125~126 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.02 (dd, J=8.0, 1.6 Hz, 1H), 7.89~7.82 (m, 3H), 7.77 (ddd, J=8.7, 7.4, 1.5 Hz, 1H), 7.45 (t, J=7.6 Hz, 3H), 7.40~7.34 (m, 2H), 3.05 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 173.2, 152.8, 147.1, 136.4, 129.1, 127.9, 127.5, 127.3, 125.0, 124.9, 118.6, 117.1, 114.9, 56.0, 49.2, 39.0; HRMS (ESI-TOF) calcd for C12H10- FNO3SNa [M+Na]+ 415.0835, found 415.0843.
(1-(3-(2,2-Dioxidobenzo[e][1,2,3]oxathiazin-4-yl)bi-cyclo[1.1.1]pentan-1-yl)-1H-1,2,3-triazol-4-yl)methyl 2-(4- isobutylphenyl)propanoate (9a): Yellow solid (91 mg, 86% yield). m.p. 165~166 ℃; 1H NMR (500 MHz, Chloroform-d) δ: 7.98 (dd, J=8.0, 1.6 Hz, 1H), 7.76 (ddd, J=8.7, 7.5, 1.5 Hz, 1H), 7.48 (s, 1H), 7.43 (td, J=7.7, 1.2 Hz, 1H), 7.35 (dd, J=8.4, 1.1 Hz, 1H), 7.19 (d, J=8.2 Hz, 2H), 7.09 (d, J=8.1 Hz, 2H), 5.27~5.20 (m, 2H), 3.73 (q, J=7.1 Hz, 1H), 2.95 (s, 6H), 2.44 (d, J=7.2 Hz, 2H), 1.84 (dt, J=13.5, 6.8 Hz, 1H), 1.49 (d, J=7.2 Hz, 3H), 0.89 (d, J=6.6 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ: 174.6, 174.1, 153.8, 143.4, 140.8, 137.4, 137.3, 129.4, 128.3, 127.2, 126.0, 122.1, 119.6, 115.9, 57.8, 56.9, 50.1, 45.1, 45.0, 39.9, 30.2, 22.4, 18.4; HRMS (ESI-TOF) calcd for C28H30N4- O5SNa [M+Na]+ 557.1829, found 557.1829.
(1-(3-(2,2-Dioxidobenzo[e][1,2,3]oxathiazin-4-yl)bi-cyclo[1.1.1]pentan-1-yl)-1H-1,2,3-triazol-4-yl)methyl 2-(2- fluoro-[1'-biphenyl]-4-yl)propanoate (9b): Yellow solid (95 mg, 83% yield). m.p. 175~176 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.94 (dd, J=8.0, 1.6 Hz, 1H), 7.75 (ddd, J=8.6, 7.4, 1.5 Hz, 1H), 7.56 (s, 1H), 7.52 (dt, J=8.0, 1.5 Hz, 2H), 7.44~7.40 (m, 3H), 7.39~7.32 (m, 3H), 7.14 (dd, J=7.9, 1.8 Hz, 1H), 7.08 (dd, J=11.6, 1.8 Hz, 1H), 5.27 (s, 2H), 3.79 (q, J=7.2 Hz, 1H), 2.94 (s, 6H), 1.54 (d, J=7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ: 174.1, 173.9, 159.6 (d, J=249.5 Hz), 153.7, 143.1, 141.4 (d, J=8.8 Hz), 137.4, 135.3, 130.8 (d, J=3.8 Hz), 128.9 (d, J=3.8 Hz), 128.6, 128.3, 127.9 (d, J=13.8 Hz), 127.8, 126.1, 123.7 (d, J=2.5 Hz), 122.2, 119.6, 115.9, 115.2 (d, J=23.4 Hz), 58.0, 56.9, 50.2, 44.8, 39.9, 18.2; 19F NMR (471 MHz, CDCl3) δ: 117.45; HRMS (ESI-TOF) calcd for C30H25F- N4O5SNa [M+Na]+ 595.1422, found 595.1415
(1-(3-(2,2-Dioxidobenzo[e][1,2,3]oxathiazin-4-yl)bi-cyclo[1.1.1]pentan-1-yl)-1H-1,2,3-triazol-4-yl)methyl 2-(1- (4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)ace-tate (9c): Yellow solid (119 mg, 87% yield). m.p. 154~155 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.98 (dd, J=7.9, 1.5 Hz, 1H), 7.76 (ddd, J=8.6, 7.4, 1.5 Hz, 1H), 7.65 (d, J=8.5 Hz, 2H), 7.51~7.41 (m, 4H), 7.35 (dd, J=8.4, 1.0 Hz, 1H), 6.94~6.87 (m, 2H), 6.68 (dd, J=8.9, 2.5 Hz, 1H), 5.28 (s, 2H), 3.79 (s, 3H), 3.71 (s, 2H), 2.93 (s, 6H), 2.36 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 174.1, 170.7, 168.3, 156.0, 153.8, 139.4, 137.4, 136.1, 133.8, 131.2, 130.8, 130.5, 129.2, 128.3, 126.0, 119.6, 115.9, 115.0, 112.2, 111.7, 101.4, 58.1, 56.9, 55.8, 50.2, 39.9, 30.3, 13.4; HRMS (ESI-TOF) calcd for C34H28ClN5O7SNa [M+Na]+ 708.1296, found 708.1292.
(1-(3-(2,2-Dioxidobenzo[e][1,2,3]oxathiazin-4-yl)bi-cyclo[1.1.1]pentan-1-yl)-1H-1,2,3-triazol-4-yl)methyl 2-(3- benzoylphenyl)propanoate (9d): Yellow solid (102 mg, 88% yield). m.p. 166~167 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.95 (dd, J=8.0, 1.5 Hz, 1H), 7.68 (qd, J=6.7, 3.5 Hz, 4H), 7.58~7.49 (m, 3H), 7.39 (ddt, J=22.1, 15.3, 7.8 Hz, 5H), 7.26 (d, J=8.3 Hz, 1H), 5.22~5.12 (m, 2H), 3.77 (q, J=7.1 Hz, 1H), 2.88 (s, 6H), 1.47 (d, J=7.2 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ: 196.6, 174.4, 173.8, 153.7, 143.1, 140.6, 137.9, 137.3, 132.8, 131.7, 130.1, 129.2, 129.0, 128.6, 128.5, 128.5, 126.1, 122.3, 119.5, 115.9, 58.1, 56.9, 50.2, 45.2, 39.9, 18.3; HRMS (ESI-TOF) calcd for C31H26N4O6SNa [M+Na]+ 605.1465, found 605.1428.
(1-(3-(2,2-Dioxidobenzo[e][1,2,3]oxathiazin-4-yl)bi-cyclo[1.1.1]pentan-1-yl)-1H-1,2,3-triazol-4-yl)methyl 2-(7- methoxynaphthalen-2-yl)propanoate (9e): Yellow solid (88 mg, 79% yield). m.p. 171~172 ℃; 1H NMR (500 MHz, Chloroform-d) δ: 7.91 (dd, J=8.0, 1.5 Hz, 1H), 7.74 (s, 1H), 7.71~7.65 (m, 3H), 7.44~7.38 (m, 2H), 7.34 (dd, J=8.3, 1.0 Hz, 1H), 7.16~7.10 (m, 3H), 5.34~5.20 (m, 2H), 3.90 (d, J=7.2 Hz, 1H), 3.88 (s, 3H), 2.77 (s, 6H), 1.60 (d, J=7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ: 174.4, 174.2, 157.9, 153.7, 143.5, 137.5, 135.2, 133.8, 129.3, 128.8, 128.4, 127.2, 126.4, 126.1, 126.1, 121.6, 119.6, 119.3, 115.9, 105.5, 57.9, 56.8, 55.4, 50.0, 45.3, 39.8, 18.0; HRMS (ESI-TOF) calcd for C29H27N4O6S [M+H]+ 559.1646, found 559.1629.
1-Ethyl-2-(3-(3,5-dioxo-2,4-di(prop-2-yn-1-yl)-2,3,4,5-tetrahydro-1,2,4-triazin-6-yl)bicyclo[1.1.1]pentan-1-yl)-acetate (9f): Yellow solid (92 mg, 75% yield). m.p. 162~163 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.09 (d, J=2.3 Hz, 1H), 7.97 (dd, J=7.9, 1.5 Hz, 1H), 7.86 (dd, J=7.6, 1.4 Hz, 1H), 7.77~7.69 (m, 2H), 7.54 (dd, J=7.5, 1.4 Hz, 1H), 7.47 (dd, J=7.5, 1.3 Hz, 1H), 7.41~7.33 (m, 4H), 7.02 (d, J=8.4 Hz, 1H), 5.28 (s, 2H), 5.18 (s, 2H), 3.67 (s, 2H), 2.97 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 189.9, 173.2, 170.3, 159.5, 152.7, 139.3, 136.4, 135.4, 134.5, 131.9, 131.4, 128.4, 128.3, 127.3, 126.9, 126.3, 125.0, 124.1, 120.2, 118.6, 114.9, 72.6, 57.0, 55.9, 49.2, 38.9, 38.9, 29.9; HRMS (ESI-TOF) calcd for C12H11FNO3S [M+H]+ 597.1438, found 597.1420.
Supporting Information Ineffective substrates, ESR experiments using 5,5-dimethyl-1-pyrroline N-oxide (DM- PO), and the NMR spectra of compounds 4a~4t, 6, 8, 9a~9f. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Zhao, C.)