


轮烷是机械互锁分子(Mechanically interlocked molecules, MIMs)的一类, 是一个或多个环状分子大环和一个或多个链状分子轴体组成的分子集合[1]. 轴体穿过大环的空腔通过机械键相互连接, 轴体的两端由于有位阻较大的基团, 使得大环不能从轴体的两端脱离下来, 从而形成了稳定的轮烷结构, 它们不能在不破坏共价键的情况下分离. 最简单的[2]轮烷由一个大环和一个轴构成, 本综述中讨论的主要是[2]轮烷, 为简洁表示, 只表述为轮烷. 随着合成技术的发展, 轮烷在刺激响应材料、聚合物太阳能电池、发光性材料和网状框架等研究领域[2]得到了越来越多的应用.
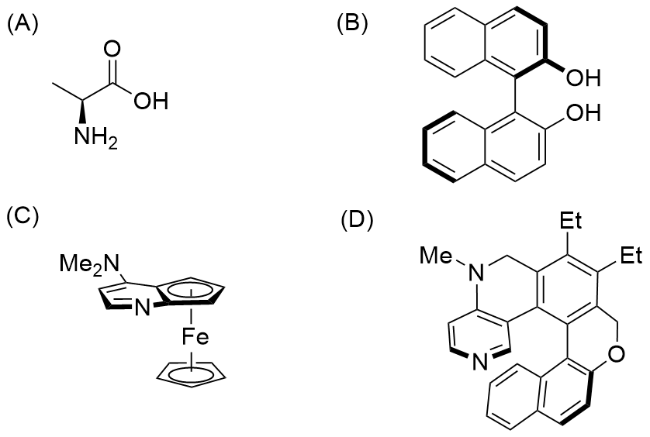
机械键的一个不寻常的性质是在没有共价立体化学的情况下生成手性立体单元的能力, 这种由机械键引起的新型手性形式有别于传统的中心手性、平面手性、轴向手性和螺旋手性等[3]共价立体化学(图1), 被称为机械立体化学[4]. 机械立体化学在空间尺度上比共价立体化学大一个数量级, 可能给手性轮烷带来一些不一样的性质和应用. 与共价立体化学高效的合成与广泛的应用相比, 机械立体化学的应用远远落后, 一个重要的原因是机械立体化学合成发展的滞后. 直到1997年, Vögtle和Okamoto等[5]利用手性固定相高效液相色谱分离一对对映异构体, 实现了光学纯机械面手性轮烷的制备.
1 机械手性轮烷的分类
纯机械手性轮烷由非手性的大环和轴体两种非手性组件组成. 根据机械键的特征和组件的对称面分析, 轮烷的非手性大环和轴体分别包括三种不同的对称类型(图3). 具有平行和垂直于大环平面的两种对称面的大环M0; 只具有平行于大环平面的对称面的大环, 即旋转不对称大环M1; 只具有垂直于大环平面的对称面的大环, 即面不对称大环M2. 具有平行于和垂直于轴体的两种对称面的轴体A0; 只具有垂直于轴体的对称面的轴体, 即旋转不对称轴体A1; 只具有平行于轴体的对称面的轴体, 即方向性轴体A2. 值得一提的是, 不具有对称面的非手性有机分子非常少见, 本文中不讨论这种分子作为轮烷组件的情况.
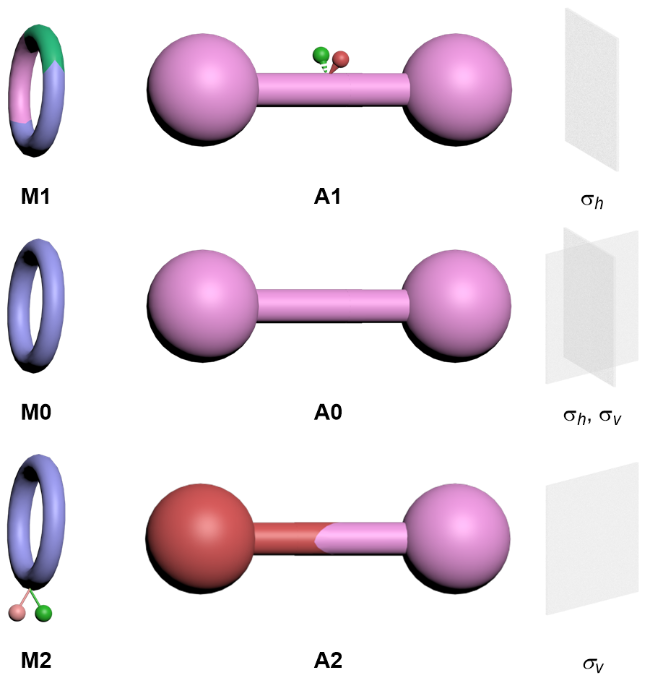
将以上各种类型大环和轴体组合, 要得到机械手性轮烷, 需要组合后的轮烷不具有对称面, 即两种组件的对称面在自由移动的情况下无法重合. 可行的组合方案只有两种情况, 大环和轴体的对称面互相垂直, 无法重叠, 分别对应于机械面手性轮烷和机械轴手性轮烷.
1.1 机械面手性轮烷
1.2 机械轴手性轮烷
以上所讨论的轮烷都是基于大环可以在轴体上自由来回穿梭的情况, 产生机械手性只能是两个组件的对称面互相垂直的情形. 当大环在轴体上的运动被限制时, 则可能出现大环和轴的对称面平行而不能重叠的情形, 导致整体轮烷分子没有对称面, 产生新的机械手性类型.
1.3 机械点手性轮烷
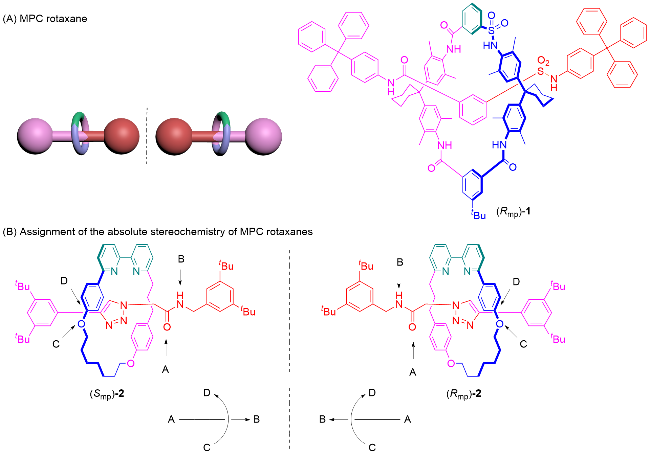
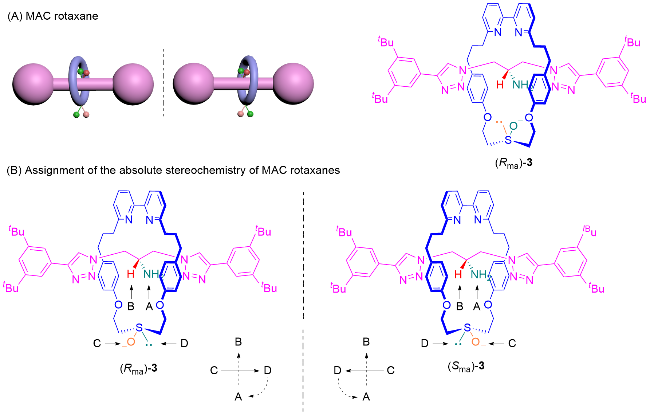
具有机械点手性(Mechanical point-chirality)的轮烷是由大环M0和旋转不对称轴体A1组成, 如图7A所示. 其组成组件中轴体的中心为前手性中心, 轴体的中心存在大位阻基团, 具有过轴体的中心垂直于轴的对称面. 由于位阻的原因, 大环只可以在半轴的范围内自由移动, 此时轴体的对称面和大环的对称面是无法重合的, 导致整个轮烷分子没有对称面. 将大环和所在半轴视为一个整体, 在轴体中心处就形成了手性中心, 该轮烷就具有机械点手性. 具有机械点手性的轮烷分子中, 手性可以用R或S来标记, 根据传统的CIP规则判定. 机械点手性轮烷最早由Leigh课题组[12a]于2008年报道, 他们通过手性催化动态动力学拆分的方法得到了轮烷4, 并使用手性HPLC分析方法测试了该轮烷的对映体过量.
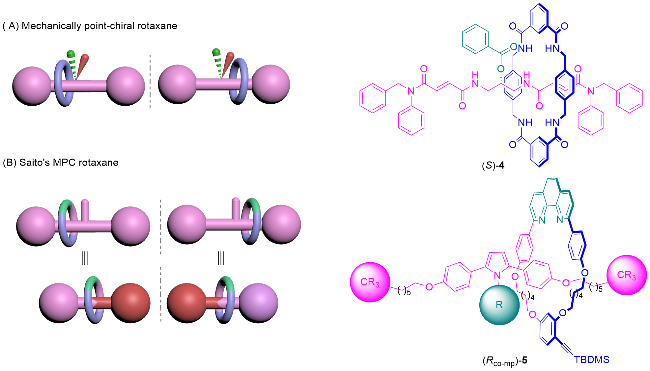
Goldup等[4a,13]将机械点手性轮烷归类为共构象机械手性轮烷. 当大环限制在半轴上移动时, 除了上述的机械点手性轮烷外, 还会出现另一种共构象机械手性的情况. 由旋转不对称大环M1和中心具有阻碍大环运动基团的轴体A0组合成的轮烷也可以表现出机械手性, 如图7B所示. Saito课题组[14]于2017年报道的轮烷5中, 旋转不对称大环M1被限制在轴体的一端, 此时大环和轴体的对称面是平行的, 无法重合, 产生机械手性. 如果将轴体中心和没有大环的一端整体视为一个封端基团, 则等同于具有方向性的轴体A2, 此时该轮烷就是由旋转不对称大环M1和方向性轴体A2组合, 可以视为机械面手性轮烷.
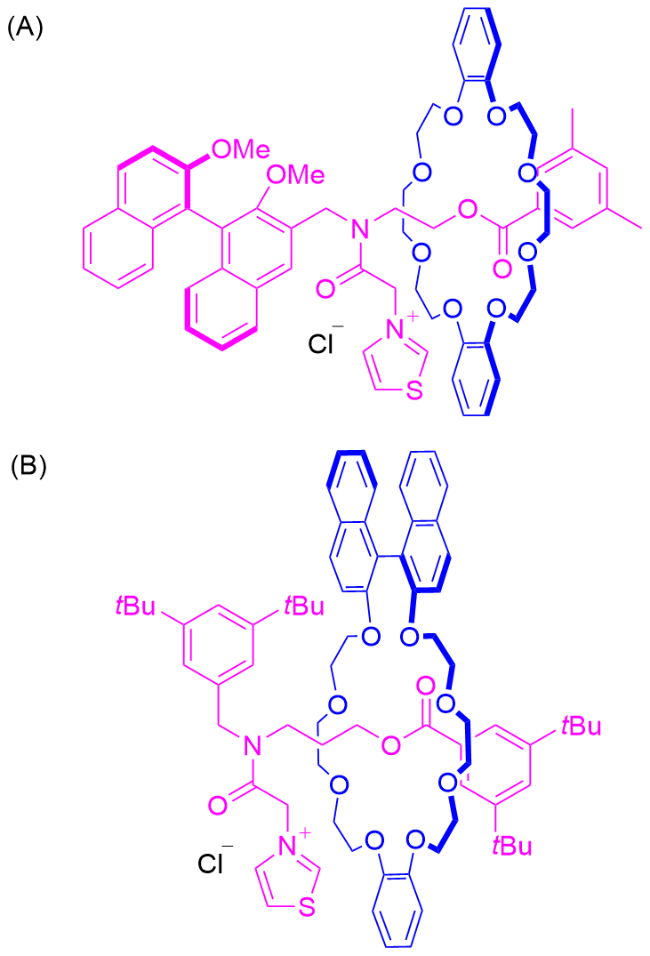
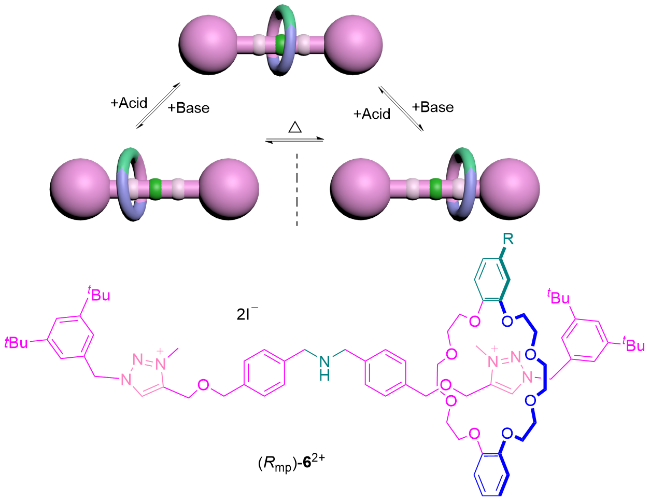
还有一类特殊的情况是, 轴体中心没有大位阻基团阻碍大环的运动, 但通过非共价相互作用, 大环可被调控固定在轴体的不同站点上. 2019年, Credi课题组[15]报道了一种可调控的机械手性轮烷6 (Scheme 1). 他们利用轮烷分子中旋转不对称冠醚大环与轴上站点之间的非共价相互作用, 在可逆的酸碱机制下实现大环分子的单向选择性穿梭. 在该研究中, 当轴体中心质子化带正电时, 大环与质子化的铵盐结合, 大环和轴体的对称面是重叠的, 轮烷分子没有手性; 被碱处理后, 质子化的轮烷发生去质子化使得大环离开轴体中心胺的位置, 与轴体一端的三唑盐站点结合, 导致大环和轴体的对称面变为平行但不重合的状态, 产生机械手性. 此时将轴体中心和其中没有大环的一端视为一个封端基团, 该类型轮烷亦可归类为机械面手性轮烷的一种表现形式.
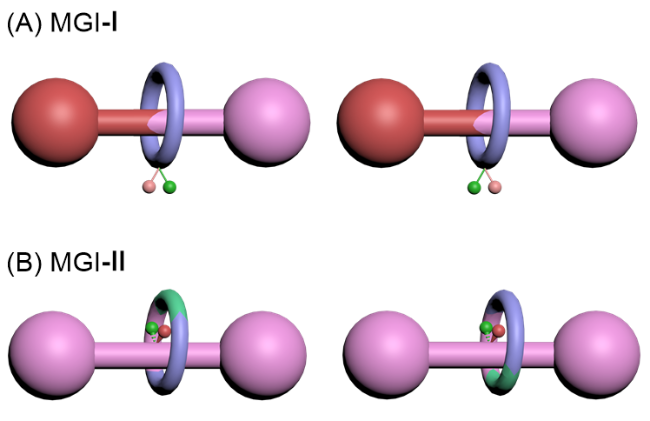
综合以上讨论结果, 非手性的大环和轴体组成的机械手性轮烷, 包括机械面手性、机械轴手性和机械点手性三种情况. 非手性的大环和轴体组合还会产生2种顺反异构, 即Ⅰ型和Ⅱ型机械几何异构体. 因位阻等原因导致的大环在轴上限制移动的情况产生的两种共构象机械手性可以分别归属为机械点手性和机械面手性轮烷.
2 轮烷的合成策略
构筑机械键、合成轮烷分子是制备机械手性轮烷的前提. 在过去的几十年中, 轮烷的合成取得了巨大的进展[16], 主要是基于模板导向合成方法. 根据轮烷的结构, 常见的两种合成策略分别是大环的缝接[17]和轴体的封端[18]. 模板导向合成方法是通过非共价相互作用, 将各组件组装在一起, 先形成假轮烷, 之后通过共价键的形成, 锁住组件形成机械键, 得到轮烷. 常用的非共价作用包括金属配位作用、氢键和π-π相互作用等. 这种模板导向合成法也被称为被动模板合成法, 模板作用不参与共价键的形成过程. 此外, 还有主动模板合成策略, 组件不需要预先通过强相互作用集合到一起, 可以在大环空腔中促进轴体的形成. 以下本文将举例来介绍这些代表性的合成策略.
2.1 被动模板合成法合成轮烷
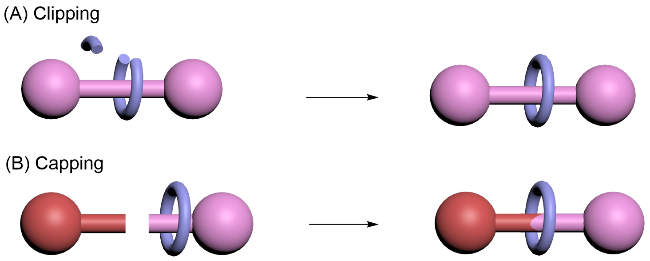
轮烷作为机械互锁分子的典型代表, 其合成策略中缝接法(Clipping)的核心在于动态共价键的闭环反应. 首先, 在轴体上设计含有反应活性位点的“圆弧”结构, 通过可逆反应使圆弧片段动态开环, 随后在模板引导下, 将圆弧片段共价连接形成大环, 最终形成机械键(Scheme 2A). 该方法的优势在于步骤简洁与动态调控能力, 但需精准设计圆弧结构的反应活性与空间匹配性. 封端法(Capping)则基于假轮烷中间体的稳定化. 首先通过非共价作用将大环预组装至部分轴体上, 形成热力学稳定的假轮烷, 随后通过快速共价反应在轴体的另一端引入大体积封端基团, 阻止大环脱出(Scheme 2B). 此策略的普适性较高, 但依赖预组装假轮烷的稳定性, 且封端反应需兼具高效性与选择性.
2.2 金属主动模板合成法合成轮烷
金属主动模板合成法是构建机械互锁分子的核心技术, 在非金属主动模板合成策略问世之前, 被称为“主动金属模板”合成法(Active metal template synthesis)[21]. 该策略的核心机理在于利用金属离子的配位作用, 精准调控大环空腔内的化学反应位点, 从而实现机械键的高效构筑(Scheme 5). 具体而言, 金属离子[如Cu(I)、Pd(II)等]首先通过配位作用预组装至大环的空腔内部, 形成稳定的金属-大环复合物. 随后, 金属离子作为动态模板, 通过其配位能力将两个半轴前体(如炔烃与叠氮化物)固定于大环空腔的两端, 并在金属催化下促使两者发生共价键形成反应, 如点击化学中的叠氮-炔烃环加成反应(Active template Cu-mediated alkyne- azide cycloaddition, 即AT-CuAAC). 这一过程不仅实现了轴体与大环的机械互锁, 还通过金属的配位-解离循环特性, 使金属催化剂得以重复利用, 从而显著降低金属试剂的用量, 大幅提升了合成效率.
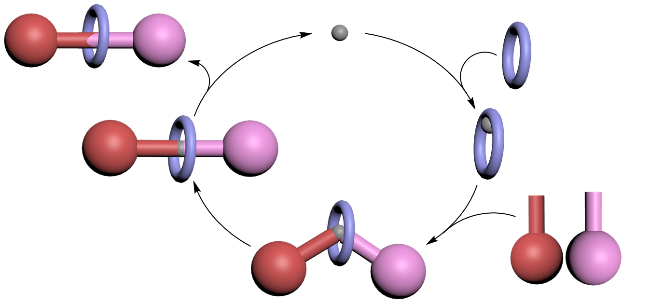
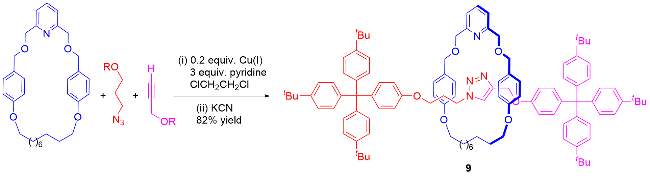
主动模板铜催化叠氮化物-炔烃环加成(AT-CuAAC)反应为机械立体化学的精准合成奠定了基础, 并推动了分子机器领域的快速发展, 是目前广泛使用的机械互锁分子合成策略, 应用于复杂体系的构建.
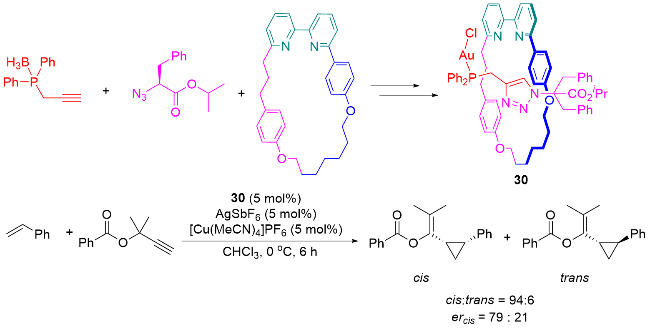
2.3 无金属主动模板合成法合成轮烷
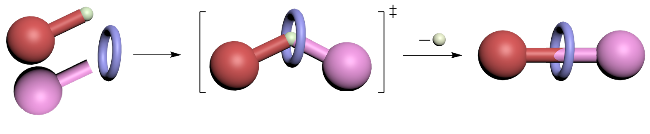
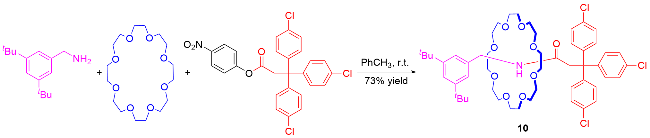
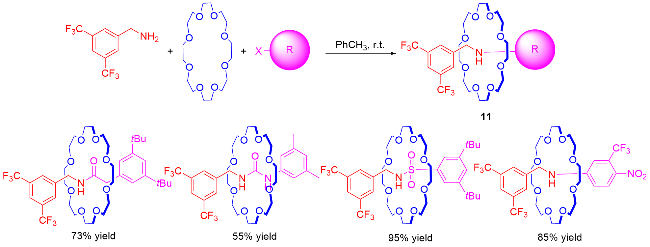
3 非对映选择性合成机械手性轮烷
3.1 手性池策略合成机械点手性轮烷
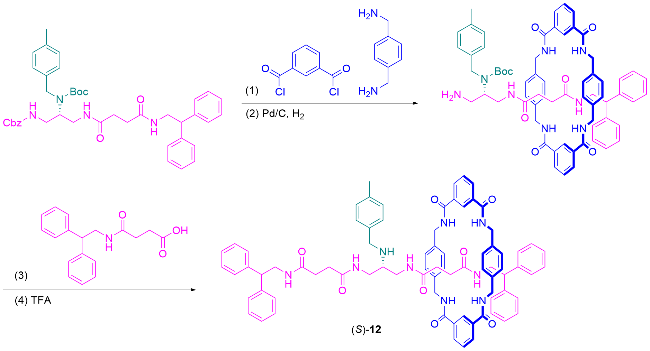
3.2 手性池和手性辅基策略合成机械面手性轮烷
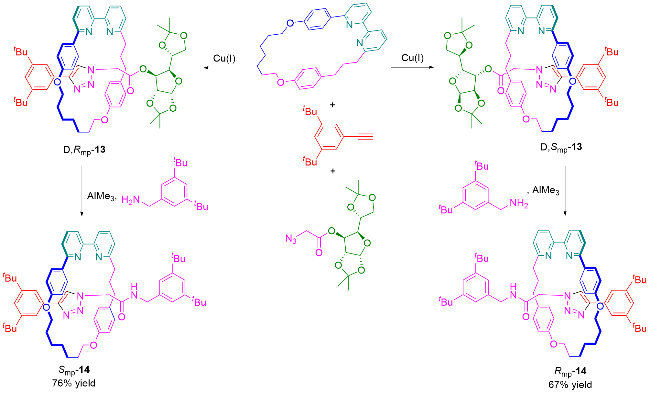
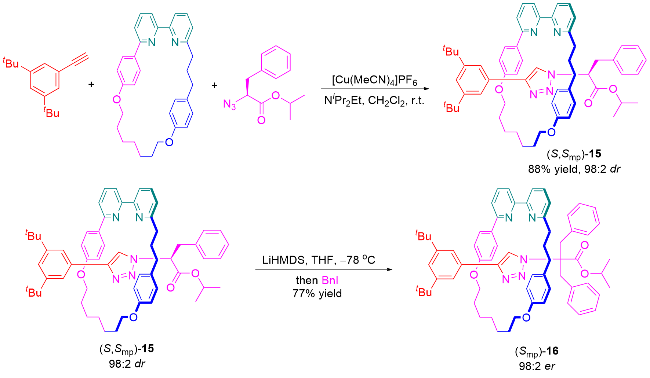
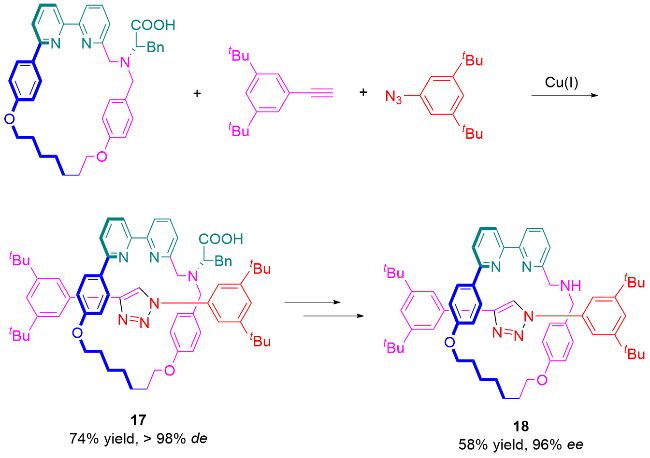
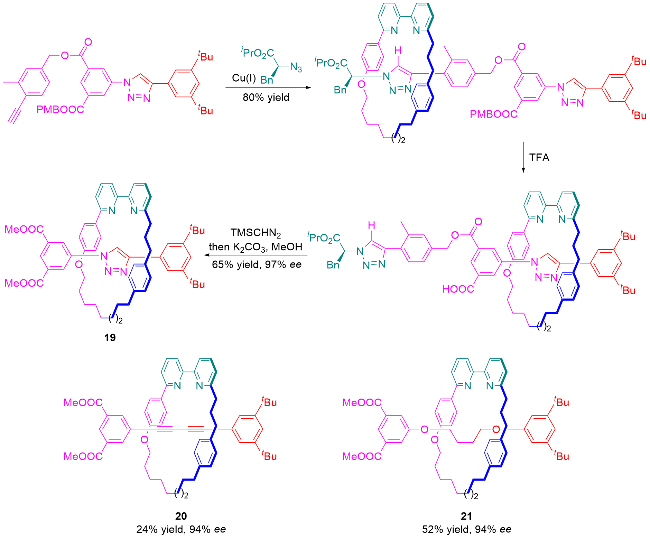
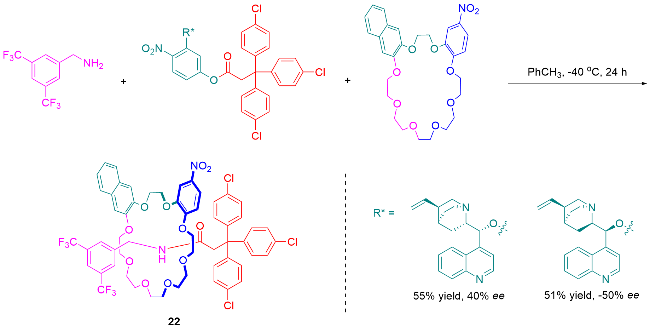
3.3 手性池策略合成机械轴手性轮烷
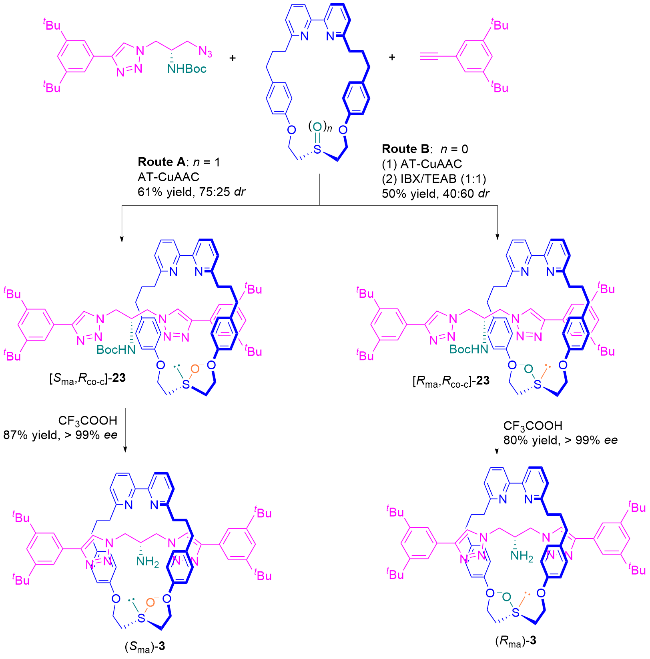
2022年, Goldup课题组[9]使用AT-CuAAC反应, 将半轴前体手性叠氮化合物与前手性亚砜大环偶联, 以75∶25 dr得到非对映异构体轮烷23, 分离主要非对映异构体并除去Boc基团, 以>99% ee得到机械轴手性轮烷3 (Scheme 16, 路线A). 之后, 他们提出利用共构立体化学控制大环面的去对称化, 硫醚大环发生立体选择性氧化, 以40∶60 dr得到非对映异构体轮烷23 (Scheme 16, 路线B). 有趣的是, 共构立体化学控制氧化得到的主要立体异构体与路线A中直接合成同一分子23得到的立体结果相反, 因此, 使用同样的半轴前体手性叠氮化合物, 可以得到机械轴手性轮烷3的一对对映异构体.
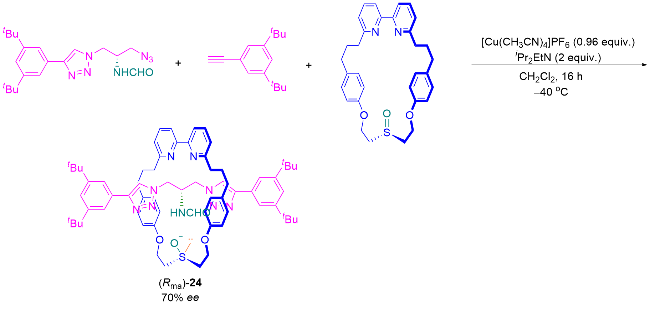
手性池合成机械手性轮烷的方法虽然可以得到目标产物, 但是需要多步转化, 效率较低, 而且需要使用昂贵的手性试剂; 手性辅基的方法同样效率较低. Goldup课题组最新发展的手性互锁辅助基团策略, 在互锁辅助合成轮烷和手性诱导上均有其优越性, 但是仍然效率较低, 不仅需要使用手性试剂, 且无法回收利用. Scheme 17中, 直接从轴前体的共价手性向轮烷的机械轴手性转化, 是简洁高效地合成机械轴手性轮烷的方法, 该方法的挑战在于实现从底物到产物高手性转化效率.
4 催化对映选择性合成机械手性轮烷
对映选择性催化作为不对称合成领域的重要策略, 通过手性催化剂精准调控反应的立体化学过程, 已成为合成手性分子最高效的途径之一. 在机械手性轮烷的构建中, 这一技术展现出独特的机遇与挑战. 目前该领域的研究仍处于探索阶段, 存在挑战包括: 现有的手性催化剂对机械键的空间识别能力不足; 轮烷组件间的弱相互作用难以稳定特定对映体等. 针对机械手性的特点, 研究者目前主要采用三种不对称催化合成策略: 动力学拆分、动态动力学拆分以及去对称化. 虽然目前机械手性轮烷的对映选择性合成的研究例子尚较少, 但是仍有较大的发展空间.
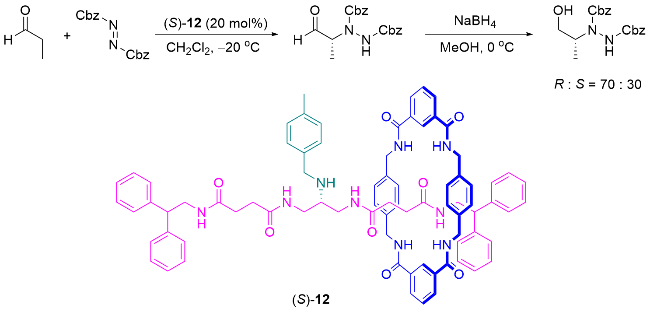
4.1 催化去对称化策略合成机械点手性轮烷
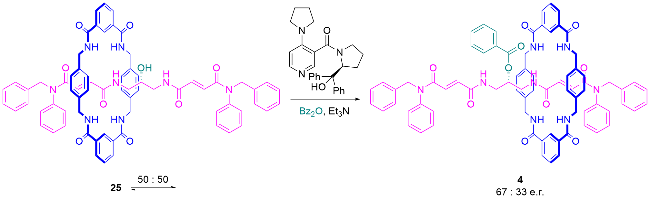
4.2 动力学拆分和催化去对称化策略合成机械面手性轮烷
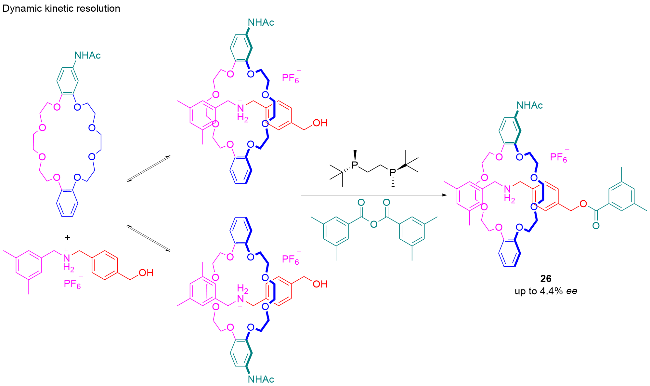
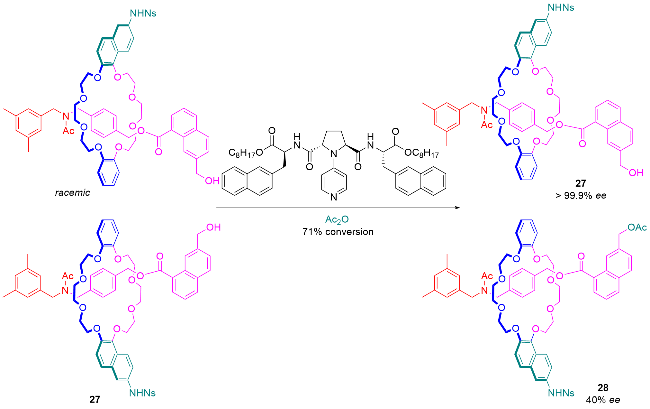
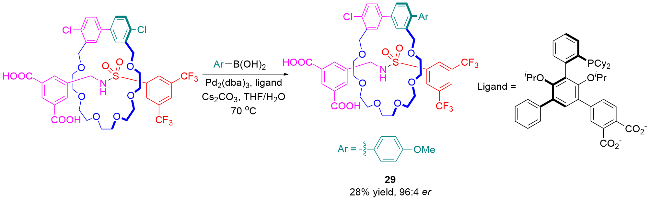
5 机械手性轮烷的应用
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 总结与展望
本综述对近年来轮烷机械手性的研究进行了归纳总结. 机械手性轮烷作为机械互锁分子的一种, 凭借其独特的机械键特性, 在分子机器、手性催化、材料科学等领域展现出巨大的潜在应用价值和需求. 基于对轮烷组件的对称面的分析, 组合各种类型非手性的大环和轴体, 本文系统地阐述了机械手性轮烷的类型. 本文中的分析方法仅考虑组件的对称面, 避免使用群论中复杂的对称性表示方法, 更加简化, 易于理解. 基于组件对称面的分析方法, 将对分析三组件或更多组件轮烷的机械手性[4a], 以及索烃的拓扑机械手性[37]等提供启发. 基于组件对称面的分析方法, 也可应用于设计相应的前手性轮烷底物, 为发展机械手性轮烷的不对称催化合成方法提供指导.
本文简要概述了轮烷的被动模板合成法, 并重点介绍了金属主动模板合成法和无金属主动模板合成法. 轮烷的主动模板合成方法更加高效, 合成的轮烷分子构象更加稳定, 为机械手性轮烷的合成研究奠定了基础. 轮烷机械键的构筑仍将是合成化学领域重要的研究内容. 机械手性轮烷的合成方法, 包括基于手性池、手性辅基的非对映选择性合成以及基于手性催化剂的对映选择性催化合成. 手性池方法将手性分子融入轮烷结构, 手性辅基在合成后可移除. 对映选择性催化包括动态动力学拆分、动力学拆分及催化去对称化等策略. 基于手性催化剂对映选择性催化是合成手性分子最高效的途径, 是未来的发展方向. 机械点手性轮烷和机械面手性轮烷的合成方法均已相对较完善, 近年来不对称催化研究也得到充分发展, 然而目前机械轴手性轮烷的合成仅有手性池一种方案的报道, 其催化不对称合成仍有待研究. 机械手性轮烷的研究应开发更高效、绿色、经济的合成方法, 以提升合成效率、选择性, 并降低成本, 深入探究其结构与性能关系, 精准调控手性与机械运动特性, 拓展其在手性催化、药物研发、传感器、分子存储等领域的应用.
(Lu, Y.)