


1 Introduction
Natural products are extensively used as chiral scaffolds for the synthesis of significant compounds with diverse biological activities and functional organic molecules.[1-2] One of the most successful natural chiral building blocks is undoubtedly rosin.[3] Isosteviol, the hydrolyzed product of stevioside, has also received widespread attention owing to its special tetracyclic triterpenoid structure.
Great efforts have been made to the structural modification of isosteviol for the pharmacological activity and microbial transformation.[4] At the same time, organocatalysts with isosteviol frameworks have also received widespread attention in the field of organocatalysis.
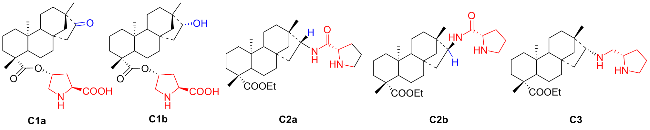
Inspired by the groundbreaking research of List and Barbas on the asymmetric Aldol condensation catalyzed by proline, much efforts have been devoted to the design of L-proline based organocatalysts and their application in asymmetric carbon-carbon bond-forming reactions.[5-9] The isosteviol-type organocatalysts derived from proline can be roughly divided into three categories: the isosteviol-proline conjugates C1, isosteviol-based prolinamides C2 and isosteviol-pyrrolidinyl C3 (Figure 1).
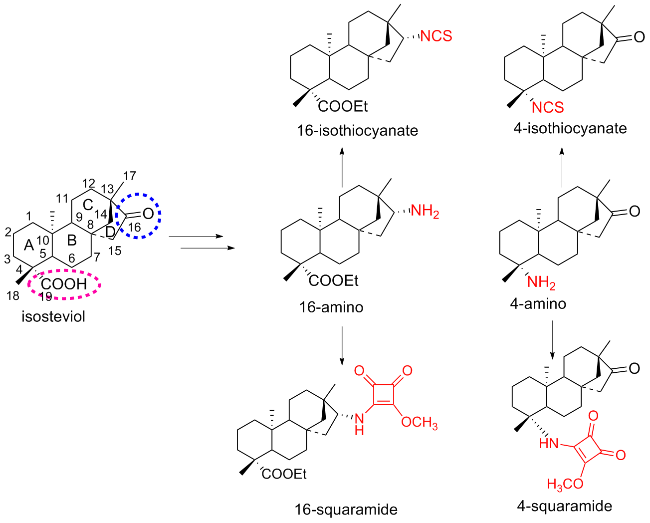
As shown in Figure 2, the main distinctive features of the isosteviol are the carbonyl group at C-16 and the carboxyl group at C-4. Both of the carbonyl and the carboxyl groups can be chemically converted to NH2. After further modification of the NH2, several isosteviol-type isothiocyanates and squaramides can be fabricated.
Chiral thiourea and squaramide, as representative dual hydrogen bond donor structural units, have been effectively utilized in enantioselective reactions owing to their unique and potent hydrogen-bonding abilities.[10-20] In the past decade, organocatalysts with natural origin of the chiral frameworks have received widespread attention in the field of organocatalysis.[21] The important accomplish-ments of isosteviol-derived bifunctional hydrogen bond donor organocatalysts have been extensively studied in various enantioselective reactions. As shown in Figure 3, the isosteviol-based H-bond donor organocatalysts can be generally classified into two groups on the basis of the catalytic active center: amine thioureas (C4~C8) and amine squaramides (C9~C12).
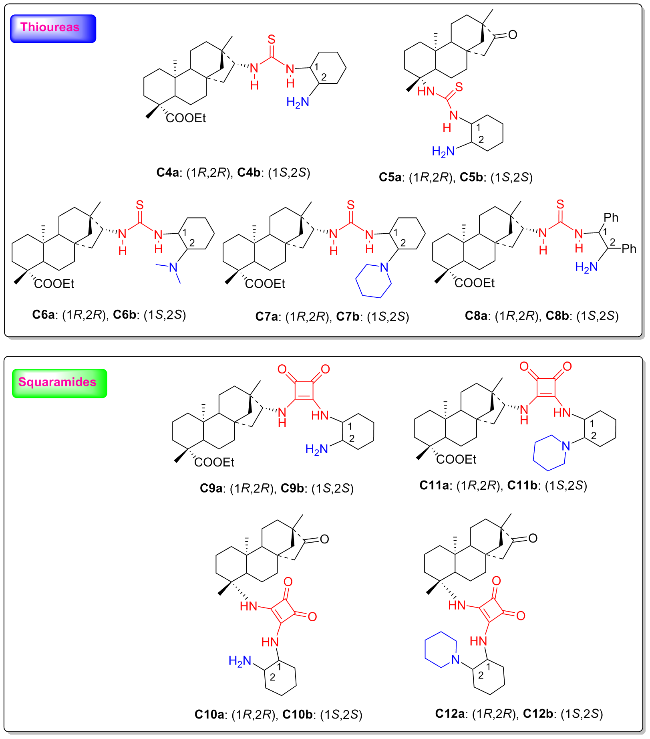
As shown in Figure 3, these isosteviol-type bifunctional thiourea and squaramide organocatalysts usually composed of two chiral parts: the isosteviol skeleton and chiral trans-1,2-diamine which reinforce high catalytic reactivity and stereoselectivity. Considering our ongoing interest in organocatalysts derived from isosteviol and the progress made in the past ten years, it is necessary to conduct a latest review to promote the sustainable utilization of natural isosteviol. The purpose of this brief overview is to afford a thorough summary of isosteviol-type organocatalysts on different enantioselective reactions (2010~2025). Isosteviol-type organocatalysts were divided into three categories: proline-based catalysts, bifunctional thiourea and bifunctional squaramide. And based on this classification, their applications in asymmetric catalysis were discussed.
2 Iosteviol-type organocatalysts derived from proline
2.1 Asymmetric Aldol reaction
In 2010, An et al.[22] described the highly enantioselective Aldol condensation between cyclohexanone and substituted benzaldehydes. The reaction was promoted by a mere 1 mol% the amphiphilic catalyst C1a in aqueous medium, resulting in β-hydroxy aldehydes with excellent diastereoselectivity (up to 99∶1 dr) and enantioselectivity (>99% ee) (Scheme 1). The influence of solvent, catalyst loading and temperature on the condensation was studied. The findings revealed that organocatalyst C1a featuring the hydrophobic substituent and a chiral concave at the 4-position of proline exhibited excellent catalytic efficiency and enantioselectivity for the Aldol condensation. The isosteviol-proline catalyst demonstrates significantly superior performance to chiral 1,2-diamines catalysis in both catalytic activity and enantioselectivity.[23]
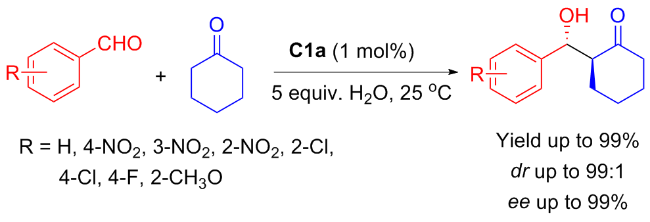
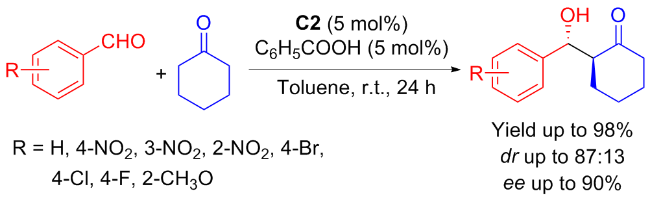
In 2018, Liuʼs group[24] developed isosteviol-type prolinamides for asymmetric Aldol condensation between cyclohexanone and aromatic aldehydes. With the cooperation of benzoic acid, excellent catalytic reactivity and good enantioselectivity were obtained (Scheme 2). And it was found that the stereoconfiguration of C-16 had almost no effect on the catalytic effect.
2.2 Asymmetric α-aminoxylation of aldehydes or ketones
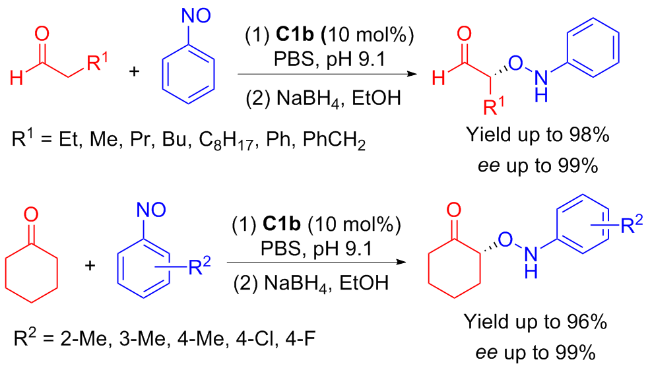
In 2011, Anʼs group[25] developed C1b, which exhibited remarkable efficiency for the enantioselective α-aminoxy- lation of aldehydes or ketones, achieving high yield of 98% and exceptional enantioselectivities (ee value up to 99%) (Scheme 3). As the results show, in phosphate buffer, the catalyst C1b exhibited pH responsive ability, which promoted excellent O-selective reaction and demonstrated a feasible method for developing asymmetric supramolecular catalysts.
2.3 Enantioselective Mannich reaction
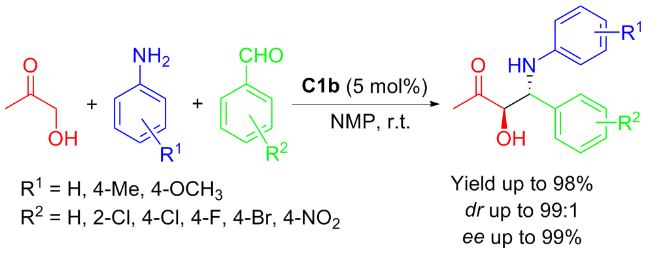
In 2011, An’s group[26] reported that the isosteviol-proline conjugate C1b was shown to effectively catalyze enantioselective three-component Mannich reaction involving hydroxyacetone, aniline and benzaldehyde (Scheme 4). The primary products obtained were the anti α-hydroxy-β-amino carbonyl compounds, achieved in up to 98% yield and good to exceptional enantioselectivities (dr up to 99∶1 and ee up to 99%) within a brief reaction period. Especially, using p-toluidine or p-nitrobenzalde- hyde, high yields along with excellent diastereo- and enantioselectivity were obtained.
In 2012, An’s group[27] reported again that C1b promoted efficiently three-component Mannich reactions involving cyclohexanone, aniline and aromatic aldehyde at the presence of water (Scheme 5). Surprisingly, syn-Man- nich products were achieved with excellent diastereoselectivity (anti/syn ratio up to 99∶1) and excellent enantioselectivity (>99% ee).
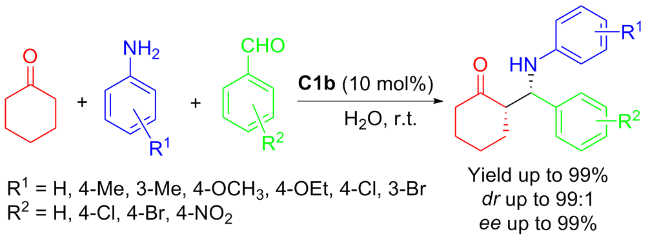
The proposed mechanism entailed the creation of a biphase system via H-bond formation, facilitated by the amphiphilic catalyst C1b. As illustrated in Figure 4, in the H2O-surrounded organic microphase, the catalytically generated enamine attacked the Si-face of imine resulting in the syn-configured Mannich products.
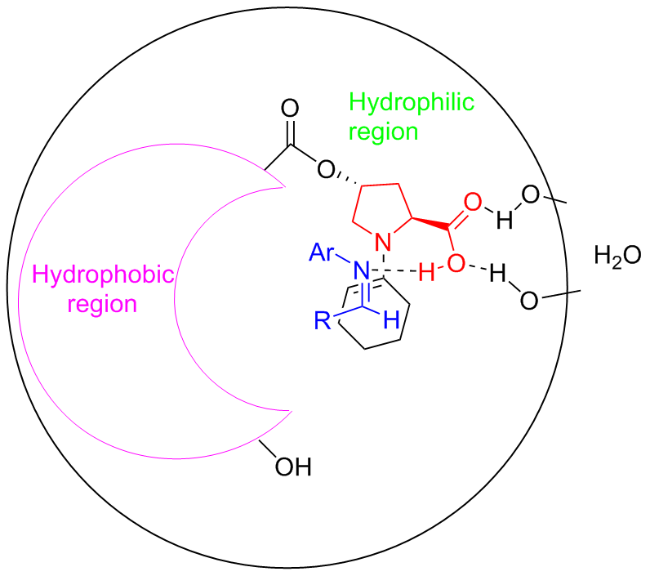
2.4 Asymmetric Michael reaction
In 2024, Liuʼs group[28] disclosed the enantioselective Michael addition reaction between cyclohexanone and nitroolefins catalyzed by C3 (Scheme 6). The catalyst C3 was readily synthesized through the reduction of C2 which demonstrated remarkable efficiency in catalyzing the asymmetric Aldol reaction. Although only good stereoselectivities (up to 90% ee) were obtained in the aqueous medium, outstanding catalytic efficiency (up to 99% yield) was achieved with 10 mol% C3.
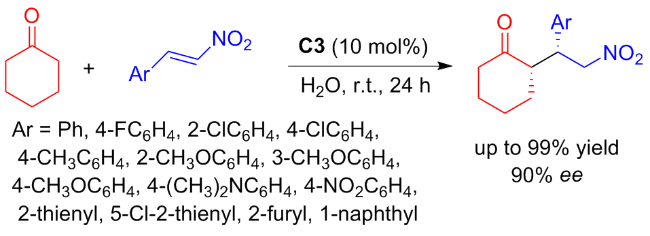
A plausible mechanism was proposed, wherein the enamine nucleophile, which generated from the carbonyl and pyrrolidine, activated the nitroolefins through the hydrogen bond with nitro groups, followed by nucleophilic attack on the Si-face of nitroolefins (Figure 5)
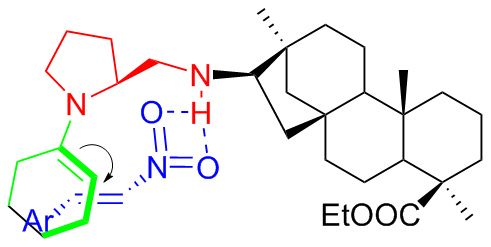
3 Isosteviol-type bifunctional thiourea organocatalysts
3.1 Bifunctional primary amine-thioureas
As early as 2011, Ma and colleagues[29] first synthesized the new type of amine-thiourea C4a and its diastereomer C4b to explore the doubly stereo controlled Michael addition between maleimide and aldehyde (yields up to 98% and ee value up to 99%) (Scheme 7). Even if the reaction scale was increased to 100 mmol, the corresponding addition products can still be achieved in high yields (reaching 98%) and with outstanding enantioselectivity (ee value up to 99%).
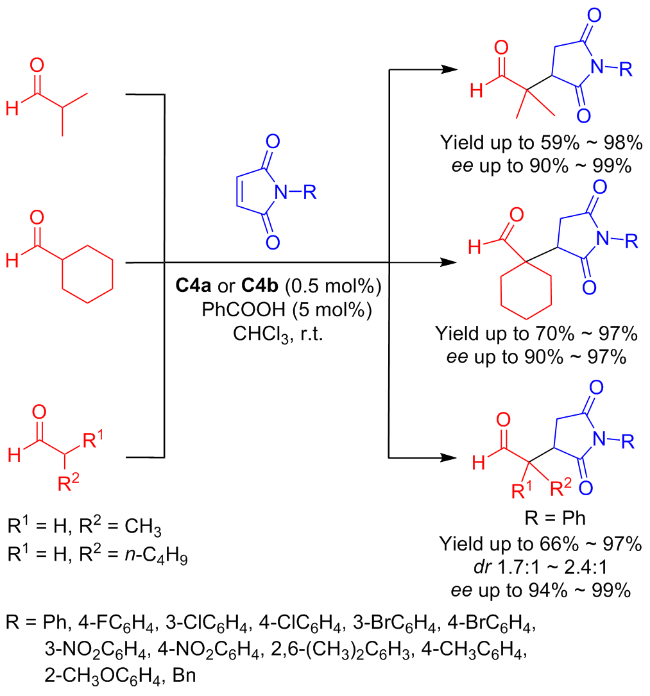
In order to broaden the adaptability of C4a and C4b in different reaction environments, nearly at the same time, Ma et al.[30] evaluated their catalytic performances in the Michael addition of aldehydes and nitroalkenes (Scheme 8). The catalytic effects in organic and aqueous phases were explored separately. The catalytic activity and enantioselectivity in CHCl3 (92% yield with 98% ee) were superior to those in aqueous phase (89% yield with 93% ee).
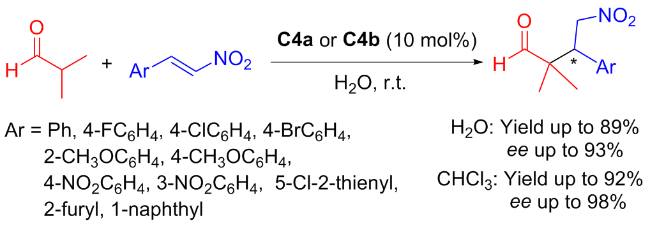
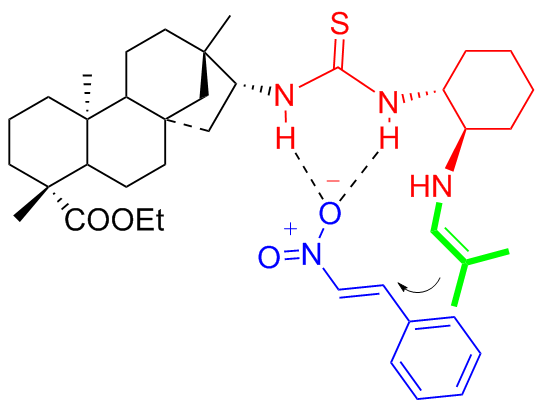
Drawing from the experimental findings, the author proposed a reasonable catalytic transition-state to account for the stereochemical outcome and verified the experimental results through the theoretical simulations. The calculation results highly matched the experimental results that the catalyst C4a catalyzed more efficiently to obtain the S configuration of the Michael adduct (Figure 6), while C4b obtained the R configuration (Figure 7).
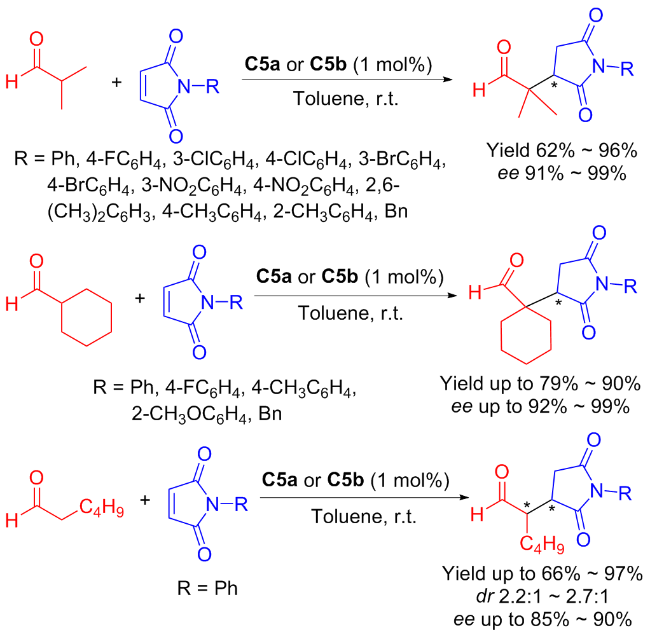
In 2014, Song et al.[31] reported another isosteviol-type of primary amine thiourea bifunctional organocatalysts C5a and C5b for catalyzing the Michael reaction of aliphatic aldehydes with maleimide (Scheme 9). The catalysts C5a and C5b showed comparable catalytic activities (96% yield) and enantioselectivities (ee value reaching 99%) utilizing a minimal amount of catalyst (1 mol%).
Both isosteviol-type of amine thiourea catalysts C4 and C5 exhibit higher catalytic activity and enantioselectivity than N-primary-amine tetrapeptide catalysts in asymmetric Michael addition of aldehydes to maleimides.[32]
3.2 Bifunctional tertiary amine-thioureas
In 2012, Maʼs group[33] presented chiral bifunctional tertiary amino-thioureas C6a and C6b for the enantioselective conjugate addition of acetylacetone to nitroolefins with dual stereocontrol (Scheme 10). The catalytic efficiency and stereoselectivity of C6a (up to 94% yield and 91% ee) were superior to that of C6b (up to 92% yield and 97% ee), whereas the reactions of aliphatic nitroolefins were not tolerated under identical reaction conditions. Furthermore, the Michael reaction of β-carbonyl ester with nitrostyrene proceeded smoothly at the presence of C6a with 92% yield and 94% ee (not depicted in the Scheme 10).
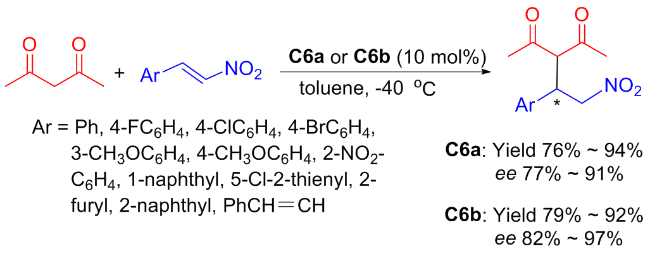
To account for the dual stereochemical outcomes observed in this addition, a possible transition-state model using C6b as catalyst was proposed based on the results of the experiment presented in Figure 8.
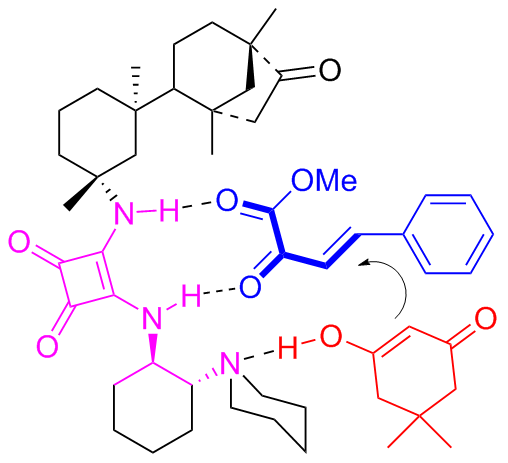
Subsequently in 2013, Maʼs group[34] discovered that the isosteviol-derived bifunctional catalyst C6a was also effective concerning the asymmetric conjugate addition between α-substituted cyanoacetate and maleimides (Scheme 11). This reaction efficiently constructed structurally diverse succinimides bearing adjacent quaternary and tertiary stereocenters with high yields (up to 97), outstanding diastereoselectivities (98∶2 dr), and good to high enantioselectivities (93% ee).
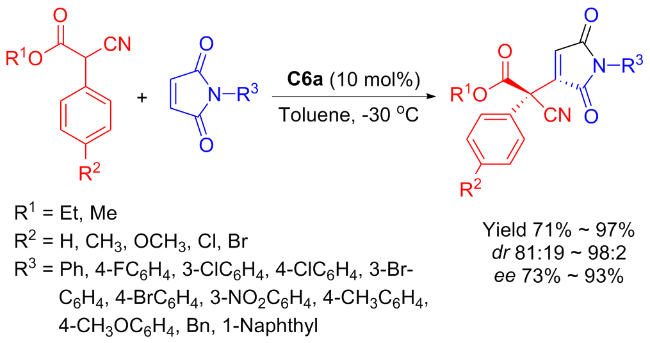
Furthermore, the author verified the application in synthesis of the catalytic method through an experiment conducted on a gram scale. In the gram-scale reaction, the catalytic performance was still maintained at the same level both in the yield and stereoselectivity.
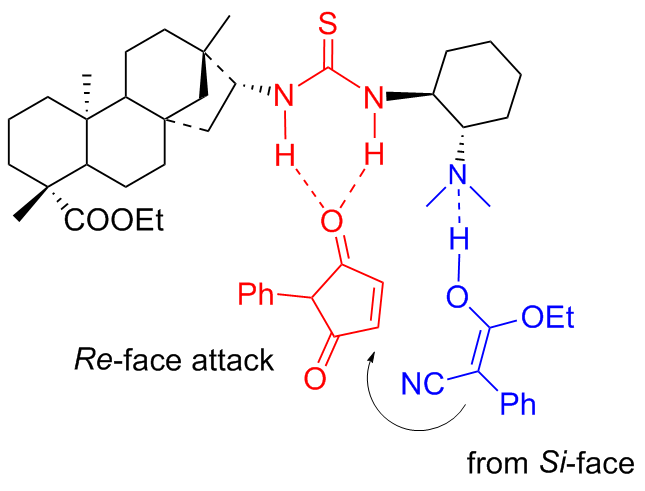
Within the catalytic cycle, the nucleophilic addition of α-substituted cyanoacetate to maleimide occurred, which was activated by the double intermolecular hydrogen bonding. The Si-face enolate attack on the Re-face maleimide was promoted by the single hydrogen bonding interaction of OH resulting the addition product (Figure 9).
In the same year, Geng and coworkers[35] discovered that the tertiary amine-thiourea C6b was also efficient in the enantioselective Michael/aromatization reaction between azlactones and α,β-unsaturated pyrazolones (Scheme 12). This reaction efficiently constructed various of heterocyclic compounds with both a protected amino acid structure and a 3-hydroxypyrazole moiety, achieving fine to excellent yields and stereoselectivities. In addition, based on the X-ray analysis of addition product, a possible catalytic transition-state was recommended as exhibited in Figure 10.
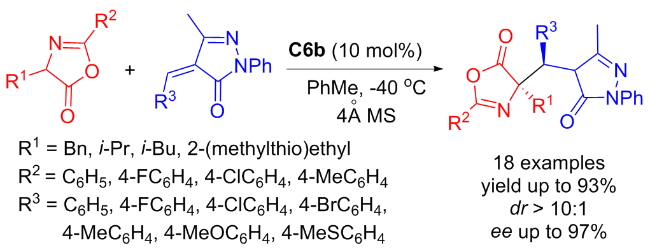
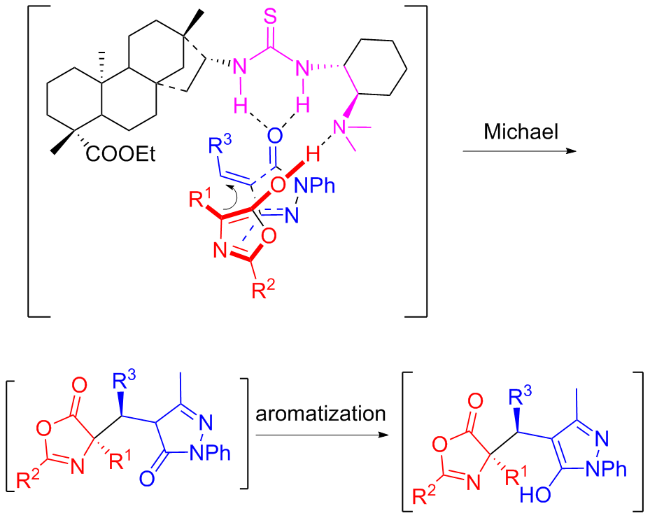
In 2022, Maʼs group[36] developed new isosteviol-type bifunctional thioureas C7a and C7b bearing tertiary amine moieties which were applied in the enantioselective Michael addition of nitroalkenes to pyrazolin-5-one (Scheme 13). High yields (up to 95%) and good enantioselectivities (up to 88% ee) of useful chiral pyrazolone derivatives were achieved with just 5 mol% catalyst. A proposed catalytic transition-state was illustrated in Figure 11.
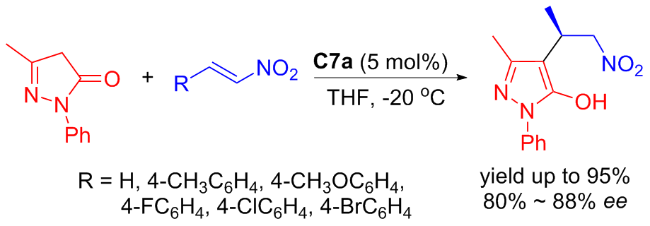
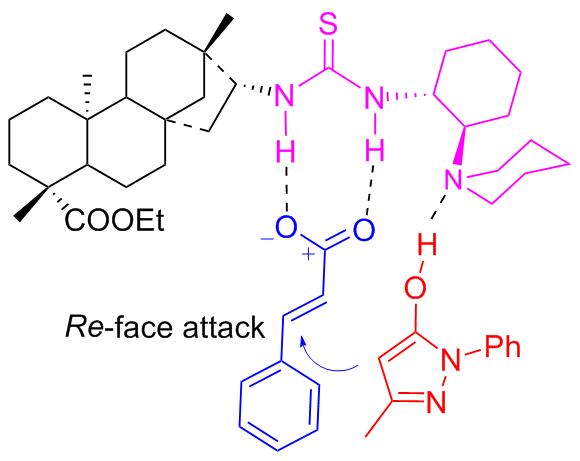
In 2025, Liuʼs group[37] developed new isosteviol-based 1,2-diphenylethylenediamine (DPEN) bifunctional thioureas C8a and C8b, which were efficient in the Michael addition of acetophenone to nitroalkenes (Scheme 14). The new isosteviol-DPEN thiourea catalysts provided high yield (up to 97%) and enantioselectivities (up to 97%) of Michael addition products with dual stereoselective control. Furthermore, the isosteviol-DPEN thiourea catalyst had sufficient recyclability and maintained the stereoselectivity of the product in the five reuse cycles of the catalyst.
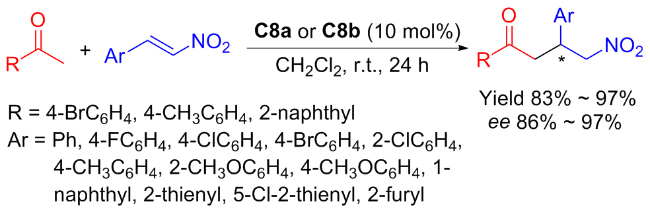
4 Isosteviol-type bifunctional squaramide organocatalysts
In addition to bifunctional thiourea organocatalysts, another efficient hydrogen bond donor is squaramide. Inspired by the application of some squaramide organocatalysts in asymmetric catalytic reactions,[38-40] some research groups have devoted to developing bifunctional squaramide organocatalysts derived from isosteviol, which have proven to be efficient for enantioselective Michael addition as well as other types of reactions.
4.1 Primary amine-squaramide bifunctional organo-catalysts
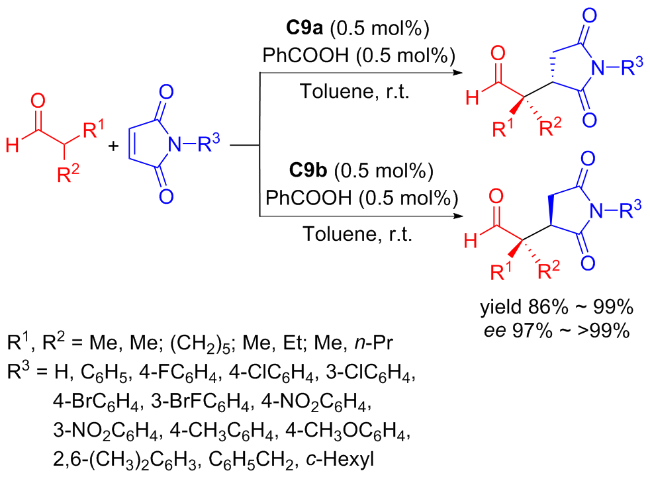
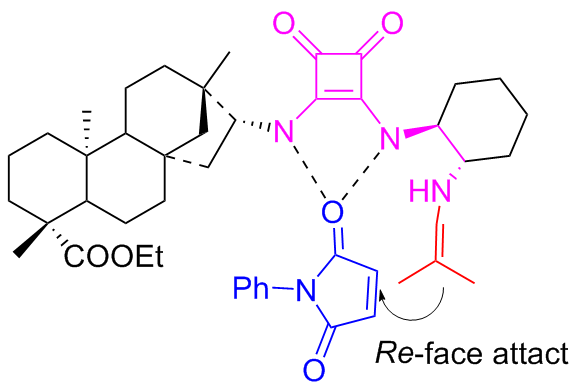
In 2017, Ma et al.[41] developed the new efficient bifunctional primary amine-squaramides C9a and C9b to catalyze the symmetric Michael addition reaction between maleimides and α,α-disubstituted aldehydes (Scheme 15). This catalyzed asymmetric reaction provided an effective pathway for obtaining substituted succinimides with a wide substrate range. The two stereoselective isomeric catalysts were used only 0.5 mol% to achieve high yields (up to 99%) of the corresponding succinimide derivatives with exceptional enantioselectivities exceeding 99% ee. The probable transition-state using C9a as an example was also provided as depicted in Figure 12.
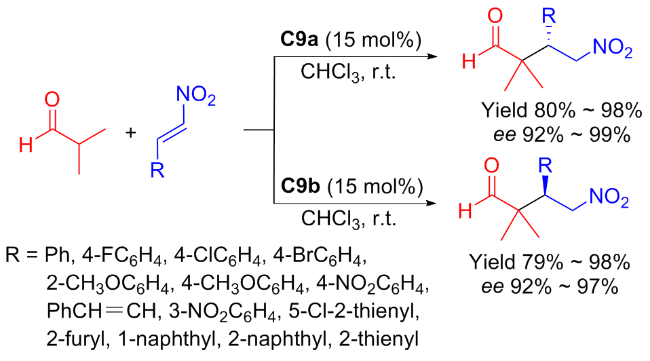
Subsequently, Maʼs group[42] revealed the efficient catalysts C9a and C9b for Michael reaction of isobutyraldehyde to nitroolefins (Scheme 16). The asymmetric addition progressed smoothly to generate a series of γ-nitroalde- hydes with both enantiomers in high yields (up to 98%) and enantioselectivities (up to 99% ee). The possible transition state as shown in Figure 13 was provided to explain the specific stereochemistry of the products in the catalytic Michael addition.
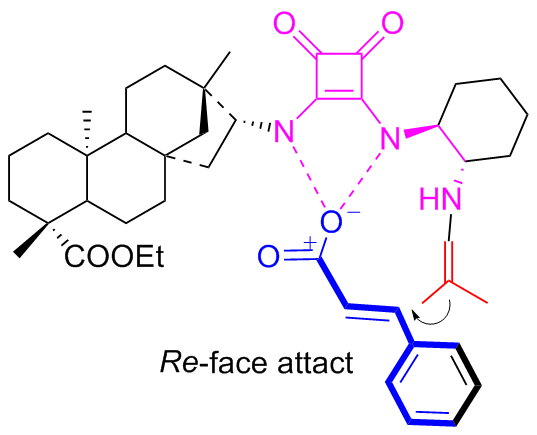
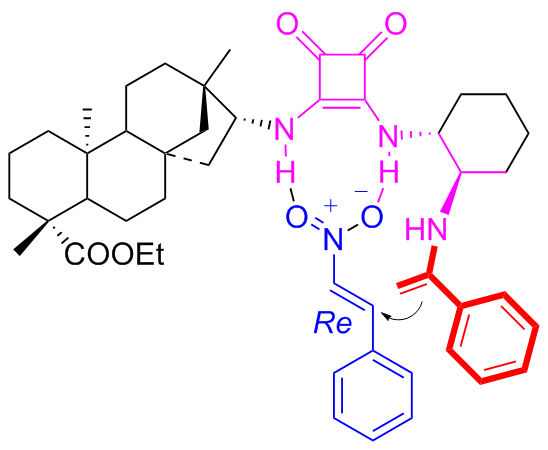
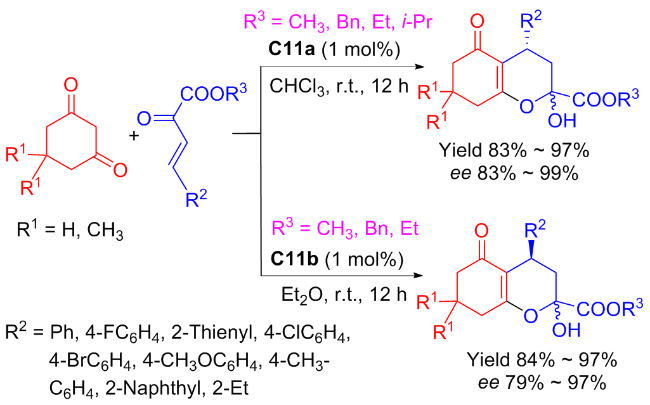
In 2018, the new isosteviol-type squaramides C10a and C10b were synthesized and utilized to catalyzed Michael addition of isobutyraldehyde to maleimide for the first time (Scheme 17).[43] Compared to the catalytic results of primary amine thiourea,[31] both the catalytic reactivity and the enantioselectivity have been improved. The addition products with opposite configurations were achieved in high yields (up to 97%) and high enantioselectivity (exceeding 99% ee). Based on the reported models of dual functional catalysts, a proposed catalytic transition-state using C10b as an example has been proposed as shown in Figure 14.
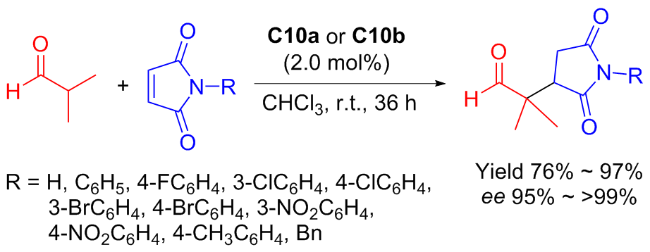
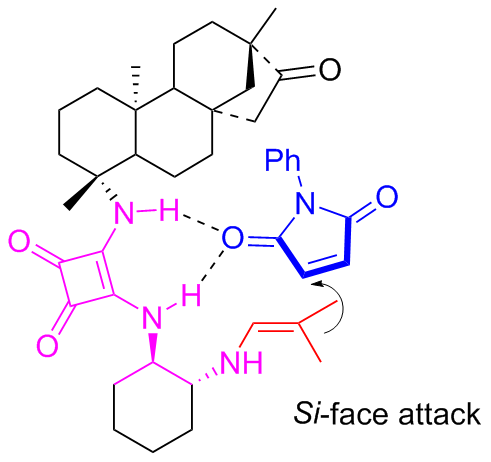
In 2022, Liuʼs group[44] found that C9a and C9b have good to excellent asymmetric catalytic performance in the Michael addition of acetophenone to nitroalkenes (Scheme 18), which enhanced the applicability of the primary amine-squaramide bifunctional organocatalysts. The possible reaction mechanism was suggested as demonstrated in Figure 15.

4.2 Tertiary amine-squaramide bifunctional organocatalysts
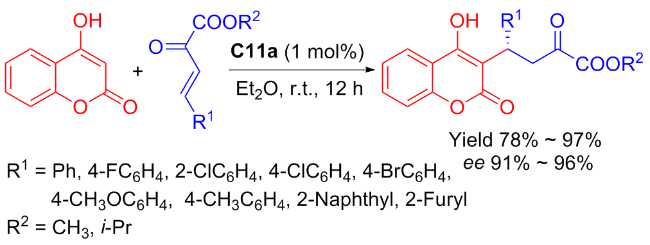
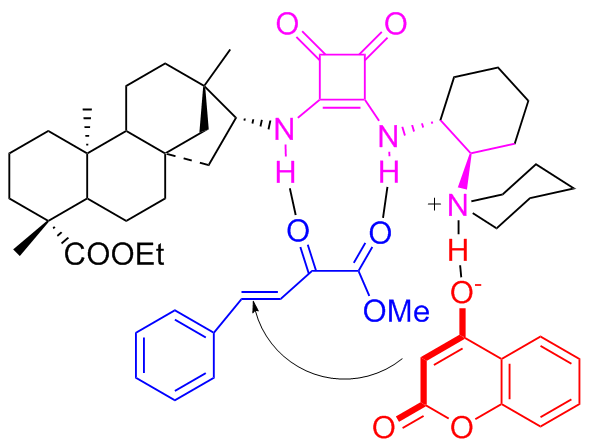
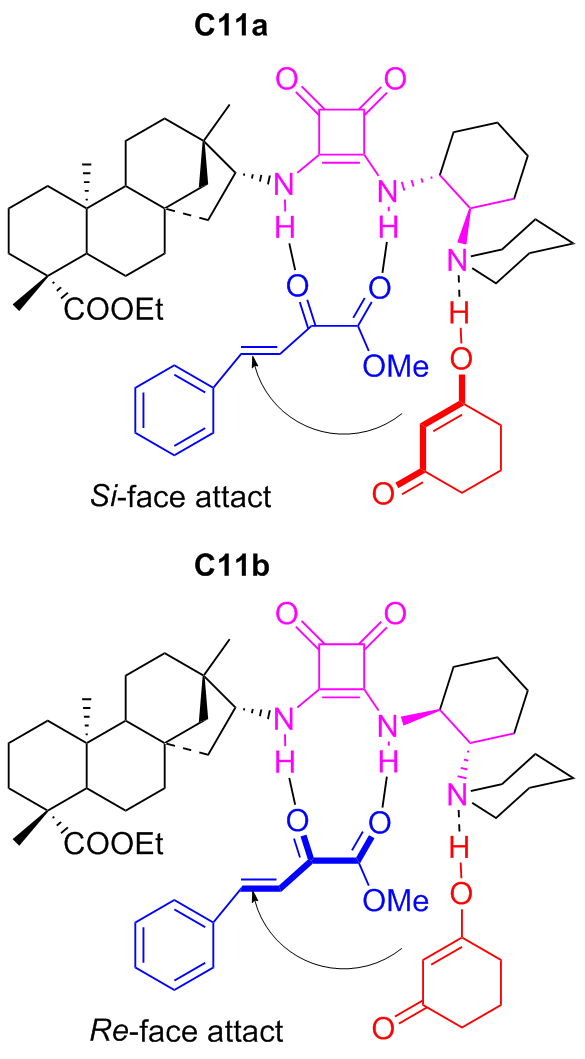
In 2022, Maʼs group[45] introduced the highly enantioselective bifunctional organocatalyst C11a featuring tertiary amine and squaramide moieties, for the synthesis of coumarins through the asymmetric Michael addition of β,γ- unsaturated α-ketoesters to 4-hydroxycoumarin (Scheme 19). Using 1 mol% catalyst, the reaction was facilitated, resulting in a series of coumarin compounds in yields ranging from moderate to high (up to 97%) and outstanding enantioselectivities (up to 96% ee). A probable transition-state was shown in Figure 16, wherein tertiary amine activated 4-hydroxycoumarin while the squaramide activated β,γ-unsaturated α-keto ester through hydrogen bonds, and subsequently, the nucleophilic attack of the 4-hydroxy- coumarin occurred on the Si-face of β,γ-unsaturated α-keto ester.
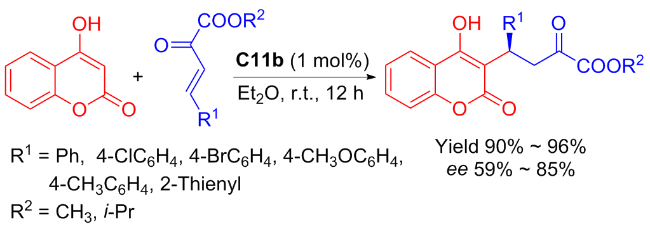
Subsequently, Liuʼs group[46] devised an enantioselective method for the synthesis of coumarins through the Michael addition of β,γ-unsaturated α-ketoesters to 4-hydroxycoumarin catalyzed by C11b (Scheme 20). The results showed that the catalyst had good tolerance to various reaction substrates and addition products were obtained with high yields (up to 96%), although the enantioselectivity was not excellent (not exceeding 85% ee).
Moreover, Maʼs group[47] discovered that the diastereomers C11b of C11a could efficiently and stereoselectively catalyzed the Michael addition of nitroolefins with acetylacetone (Scheme 21). For sure, compared to C6a and C6b, the using of catalyst C11b enhanced catalytic efficiency (yield reaching to 96%) and stereoselectivity (ee value reaching to 99%), although only 1 mol% catalyst was used. Furthermore, other Michael donors, such as 1-benzoylacetone and 1,3-diphenylacetone, were also attempted. However, only good catalytic activities (90% yield) and good enantioselectivities (76%~90% ee) were obtained (not included in the Scheme 20). The probable transition state was also speculated as presented in Figure 17.
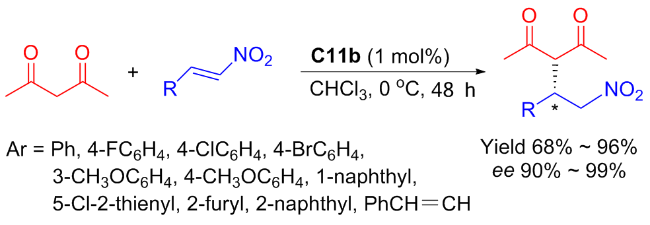
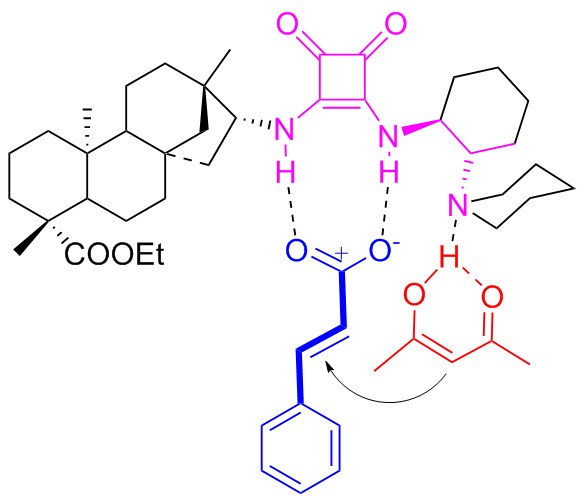
To further explore the potential of C11a and C11b, in 2022, Ma and coworkers[48-49] discovered the two catalysts were efficient (yield reaching to 97%) and highly stereoselective (ee value reaching to 99%) for the Michael addition between β,γ-unsaturated α-keto esters and cyclic 1,3-di- ketones (Scheme 22). With only 1 mol% catalyst, chiral bicyclic compounds, which are extremely important in synthesis and medicine, were dual stereoselectivitily obtained. In addition, the large-scale reaction was further performed in 100 mmol with maintained yield and enantioselectivity, which undoubtedly offered a solid foundation for its industrial application. The author also proposed a transition state speculation for the dual stereoselective control (Figure 18).
That very year, researchers from the Ma group[50] revealed Michael addition/cyclization reaction of α-bromo nitroalkene and cyclohexane 1,3-dione catalyzed by C12b (Scheme 23). High yields (90%~94%) of the desired dihydrofurans were achieved, along with stereoselectivities varying between good and excellent. Referring to the literature,[51-52] the author provided a possible catalytic mechanism as illustrated in Scheme 24.
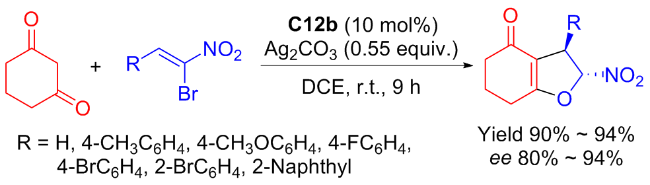
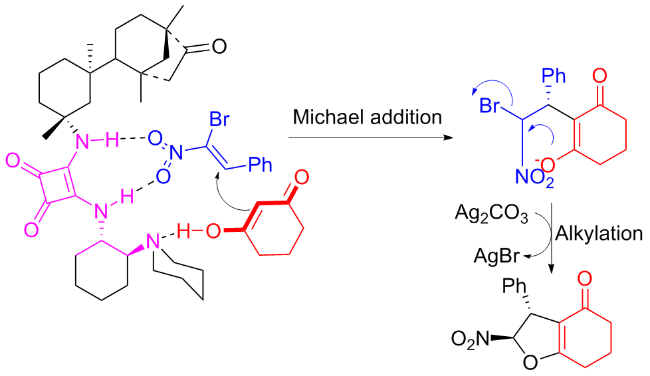
Almost simultaneously, Liu et al.[53] studied the catalytic performance of C12a on the enantioselective Michael addition between β,γ-unsaturated α-keto esters and cyclic diketones (Scheme 25). Multiple desired Michael addition compounds were generated in high yields (reaching to 97%) and excellent stereoselectivities (ee value reaching to 97%). The author speculated a possible transition state as demonstrated in Figure 19.
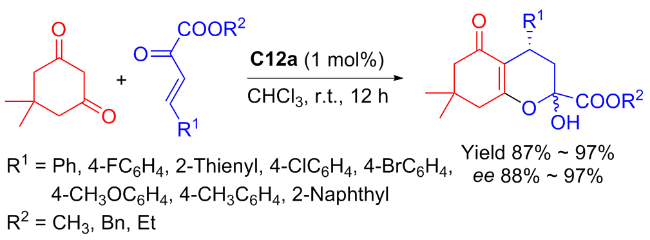
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 Conclusions
Organocatalysts derived from isoteviol, especially bifunctional thiourea and squaramide derivatives, have played an important role in promoting a wide range of enantioselective reactions as mentioned in this mini review over the past decade. Despite the remarkable contributions of the isoteviol-derived organocatalysts to the development of asymmetric organocatalysis, there are still several issues that need to be paid attention to for the development of isoteviol-tape organocatalysts. Firstly, there are currently relatively few types of organocatalysts based on isosteviol, with secondary chiral scaffold primarily being proline and chiral cyclohexane-1,2-diamine. Furthermore, the catalytic effect demonstrates that the isosteviol scaffold exhibits a synergistic effect with the catalytic active center. Therefore, other types of organocatalysts with alternative chiral skeletons derived from isosteviol are still worthy of attention. Secondly, most isosteviol-tape organocatalysts are structurally modified through the carbonyl group of the D ring or the carboxyl group of the A ring. It would be more attractive to simultaneously modify the two functional groups to achieve more desired catalytic activities and enantioselectivities. Thirdly, most of the catalytic active centers are connected to the isosteviol through squaramide or thiourea, leading to the activation site being situated at a considerable distance from the steric environment of isosteviol. Therefore, the stereoselectivity of catalytic reactions and the absolute configuration of products are mainly determined by the secondary chiral unit. Fourthly, there have been no published reports on the supporting, recycling, and reuse of isosteviol-derived organocatalysts, which represents an interesting way for the future research. Fifthly, to expand the application scope, the catalytic performance should be explored in green processes like the cycloaddition reaction between carbon dioxide and propylene oxide. These immature insights may provide direction for the further development and practical application of isosteviol-tape organocatalysts.
(Zhao, C.)