

含吸电子取代基顺式烯烃中的吸电子基团通过诱导效应可显著降低双键的电子云密度, 进而导致β-碳具有强亲电性, 这一特性使其能够发生高效亲核加成反应, 在众多领域具有广泛应用[1]. 例如, 在有机合成领域可作为关键中间体参与Michael加成、Diels-Alder环加成及过渡金属催化的交叉偶联反应(如Suzuki偶联), 高效构建碳-碳键及复杂环系结构; 在功能材料领域如导电高分子材料中作为电子受体单元提升共轭聚合物的载流子迁移率; 在生物医药领域如杀菌、降血压剂和抗肿瘤等药物中广泛存在, 通过特异性相互作用, 调节关键生物靶点的活性以实现治疗功能.
目前含吸电子取代基顺式烯烃的合成方法包括Wittig反应、Julia反应、Peterson烯基化反应、烯烃复分解反应以及炔烃选择性还原反应等(图1a)[2>>-6], 但均存在反应条件苛刻、选择性低等问题. 光催化异构化(图1b)具有反应条件温和、过程安全和产物选择性高等优点, 有望成为合成含吸电子取代基顺式烯烃的高效方法. 近年来, 多个课题组相继报道了光催化异构化反应在含醇类、酮类、醚类和叠氮结构顺式烯烃合成的应用案例[7-9], 这些研究为光催化异构化技术的发展提供了重要的理论和实验基础. 然而目前光催化异构化合成顺式烯烃研究还处于研究初期, 在难度较大的含吸电子取代基(如CF3、Cl、Br、NO2等)顺式烯烃合成方面报道较少[10-13]. 因此, 开展高效合成含吸电子取代基顺式烯烃合成新体系具有重要意义.
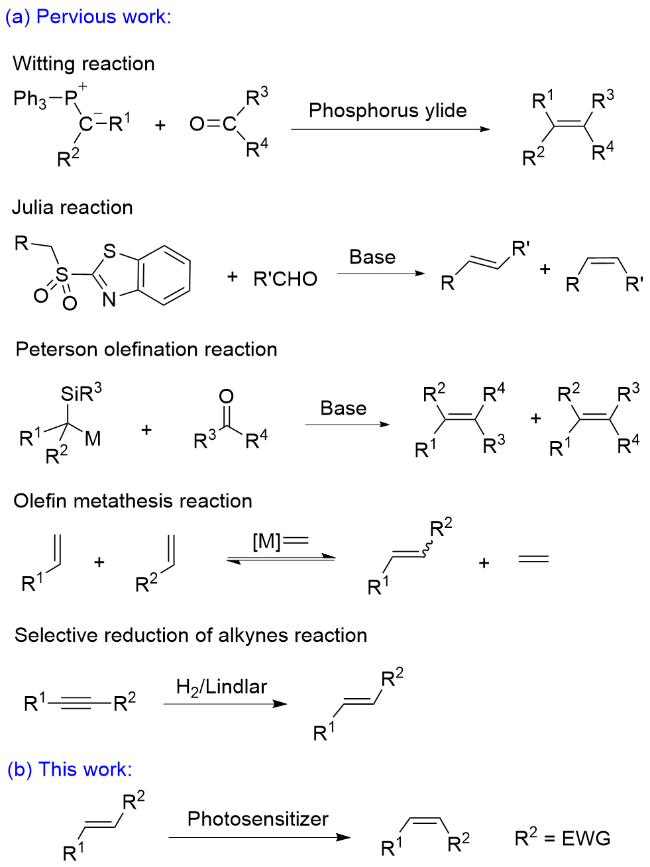
本研究以反式-β-三氟甲基苯乙烯(1a)、反式-β-硝基乙烯(5a)、反式-β-溴苯乙烯(6a)等反式烯烃为原料, 开展光催化异构化合成相应顺式烯烃反应研究; 采用密度泛函理论(DFT)计算不同取代基底物S0→T1跃迁的激发能垒, 探究光催化异构化合成顺式芳基烯烃反应机理, 为光催化异构化合成含吸电子取代基顺式烯烃提供理论指导.
1 结果与讨论
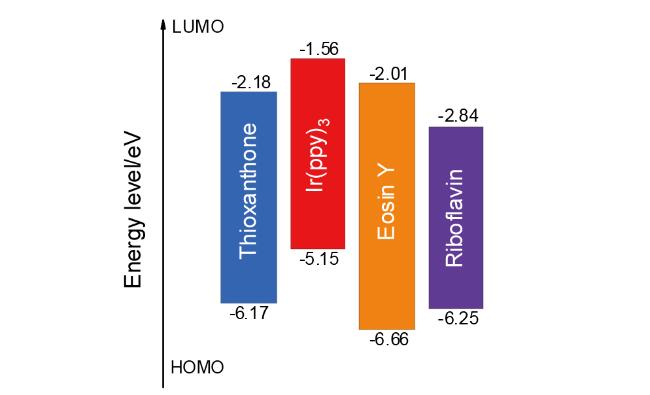
在光催化异构化反应体系中, 光敏剂是影响反应效率的关键因素. 光催化异构化反应所用的光敏剂主要包括天然色素、羰基类化合物、稠环化合物和金属配合物等. 本研究以伊红Y (Eosin Y)、核黄素(Riboflavin)、Ir(ppy)3和噻吨酮(Thioxanthone)为光敏剂, 采用紫外-可见吸收光谱测定各光敏剂的光谱吸收范围、采用氧化还原电位测试各光敏剂的氧化还原能力, 对光敏剂进行初步筛选. 实验结果如图2所示, 通过对光谱数据的分析, 发现不同光敏剂的最大吸收峰范围存在显著差异.
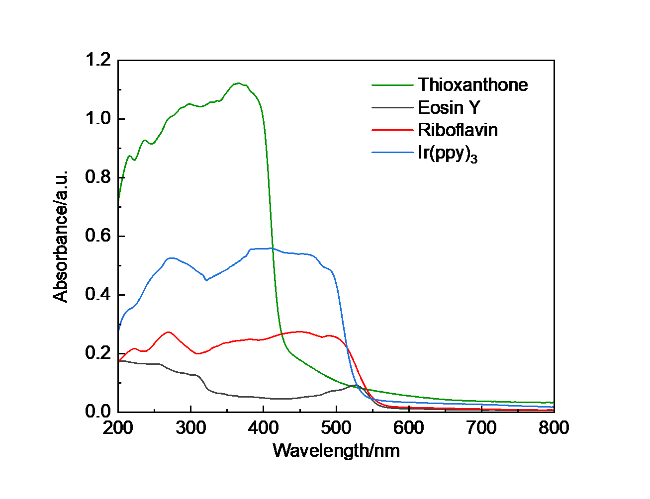
核黄素和Ir(ppy)3的最大吸收峰范围大致相同, 均在270~500 nm之间, 表明这两种光敏剂在紫外到可见光区域具有较宽的光吸收能力, 能够有效利用多种波长的光源驱动光催化反应. 其中, 核黄素在370和440 nm附近表现出较强的吸收峰, 而Ir(ppy)₃在380和450 nm附近也有明显的吸收峰. 这种宽谱吸收特性使得它们在光催化反应中具有较高的光能利用效率.
噻吨酮的最大吸收峰范围较窄, 主要集中在300~420 nm之间, 尤其在350和390 nm附近表现出较强的吸收峰. 这种吸收特性表明噻吨酮更适合在紫外到蓝光区域进行光催化反应, 但其光吸收范围相对较窄, 可能限制了其在某些反应中的应用; 伊红Y的最大吸收峰范围最宽, 覆盖了200~520 nm的波长范围, 尤其在280, 350和520 nm附近表现出显著的吸收峰. 这种宽谱吸收特性使得伊红Y能够有效利用从紫外到绿光区域的光源, 具有较高的光能利用潜力. 然而, 其吸收峰强度相对较低, 可能影响其在某些高能量需求反应中的催化效率.
进一步测定Ir(ppy)3、核黄素、噻吨酮以及伊红Y的氧化电位, 实验结果如图3所示: 伊红Y的氧化电势为EP/2=+1.743 V vs. SCE; Ir(ppy)3的氧化电势为EP1/2=+0.759 V vs. SCE, EP2/2=+1.071 V vs. SCE, EP3/2=+1.701 V vs. SCE; 噻吨酮的氧化电势为EP/2=+1.852 V vs. SCE; 核黄素的氧化电势为EP1/2=+0.959 V vs. SCE, EP2/2=+1.606 V vs. SCE.
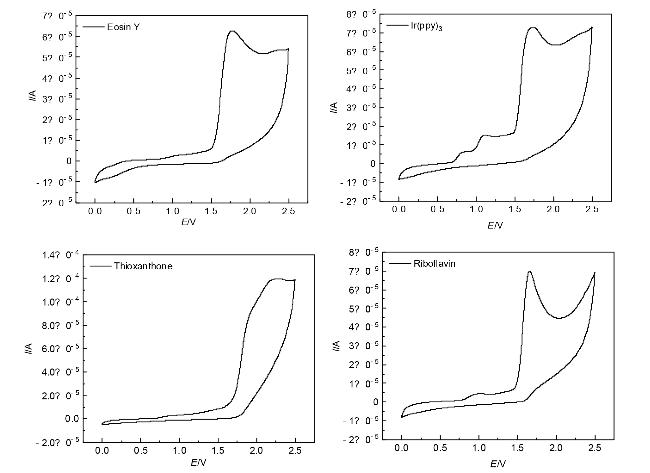
实验结果表明, Ir(ppy)3和核黄素表现出多个氧化还原电势, 而噻吨酮和伊红Y仅显示一个主要的氧化电势. 这一差异主要源于光敏剂的电子结构和激发态特性的不同: Ir(ppy)3和核黄素均属于三线态光敏剂, 其激发态具有较长的寿命和较高的能量, 因而能够发生多步电子转移过程. 当这些光敏剂被激发至三线态时, 其氧化还原能力显著增强, 能够参与更复杂的氧化还原反应, 从而在光催化异构化反应中展现出更高的催化活性和选择性; 相比之下, 噻吨酮和伊红Y的氧化还原特性较为单一, 其激发态寿命较短, 氧化还原能力相对有限, 因此在光催化反应中的效率较低. 这一发现为光敏剂的选择和光催化反应优化提供了重要的理论依据.
在上述研究基础上进行光催化异构化合成含吸电子基团顺式烯烃反应研究. 首先, 以1a及其衍生物为底物, 在40 ℃反应温度下搅拌反应24 h. 反应选择性专一且并未检测到副产物生成, 产物可通过柱层析分离, 不同光敏剂条件下光催化异构化反应结果如表1所示. 实验数据表明, 以Ir(ppy)3为光敏剂反应转化率最高, 与紫外-可见吸收光谱、氧化还原电位测试结果相符. 同时, 1a反应转化率显著低于反式-β-三氟甲基对甲氧基苯乙烯(3a)的反应转化率, 推测在烯烃中引入给电子取代基(OCH3)可以部分抵消三氟甲基的吸电子效应, 从而提高反应转化率.
表1 光催化异构化合成顺式-β-三氟甲基苯乙烯及其衍生物a,bTable 1 Photocatalytic isomerization synthesis of cis-β-trifluo- romethylstyrene and its derivatives  |
| Substrate | Photosensitizer | λ/nm | Conversion rate/%/% |
|---|---|---|---|
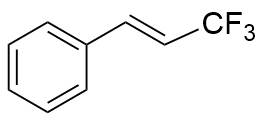 | Riboflavin | 365 | 34.9 |
| 455 | 33.2 | ||
| Ir(ppy)3 | 365 | 59.3 | |
| 455 | 51.8 | ||
| Eosin Y | 365 | 25.1 | |
| 455 | 20.3 | ||
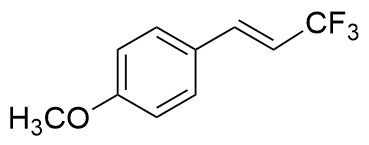 | Riboflavin | 365 | 38.7 |
| 455 | 35.1 | ||
| Ir(ppy)3 | 365 | 67.2 | |
| 455 | 60.3 | ||
| Eosin Y | 365 | 24.9 | |
| 455 | 20.1 |
a Reaction conditions (unless otherwise specified): substrate (1.0 mmol, 1.0 equiv.), Ir(ppy)3 (2 mol%), solvent (15.0 mL), 30 ℃, 25 W LEDs, b NMR yield determined by 19F NMR. |
除了光催化异构化合成1a及其衍生物外, 进一步研究了其他含不同取代基的烯烃底物的光催化异构化反应, 以探究取代基结构对反应效率的影响. 具体实验包括5a、6a及反式-[3-三氟-1-(三氟甲基)-1-丙烯基]氧基-苯(13a)等化合物的光催化异构化反应. 实验所用的底物均为1 mmol, 光敏剂用量为2 mol%.
实验结果如图4所示, 图中标注了各底物在相同反应条件下的转化率(光敏剂为2 mol% Ir(ppy)3、溶剂为丙酮、光激发波长为365 nm, 40 ℃下反应24 h). 实验结果表明, 烯烃取代基对光催化异构化反应具有显著影响. 具体而言, 取代基的吸电子能力(CF3>Br>NO2)越弱, 顺式烯烃的转化率越高. 这种现象可以归因于吸电子基团降低了C=C的电子云密度, 使得双键难以被光敏剂有效活化, 从而抑制了异构化反应的进行. 相反, 当底物中含有给电子基团(3b、4b)时, 顺式烯烃的转化率有所提高. 这是因为供电子基团增加了C=C的电子云密度, 增强了双键的反应活性, 从而促进了异构化反应的进行.
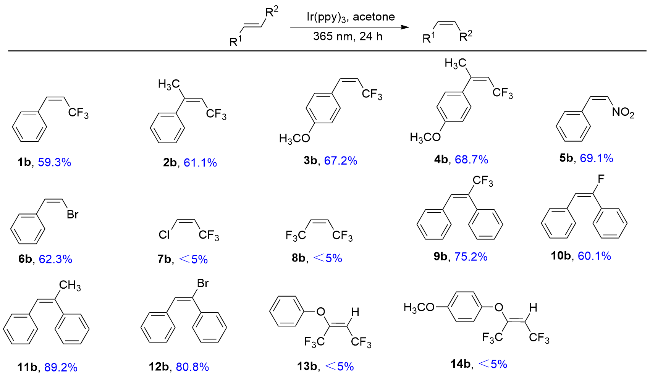
此外, 烯烃中苯环与C=C之间的π-π共轭是光催化异构化反应发生的必要条件(1b、5b、6b), 苯环与C=C之间的π-π共轭作用能够有效传递光敏剂提供的能量, 从而促进异构化反应的进行. 同时, 氧原子的引入破坏了苯环与C=C之间的π-π共轭作用(1b、13b、14b), 阻断了苯环与C=C之间的电子离域路径, 导致π-π共轭体系被中断, 光催化异构化反应难以进行.
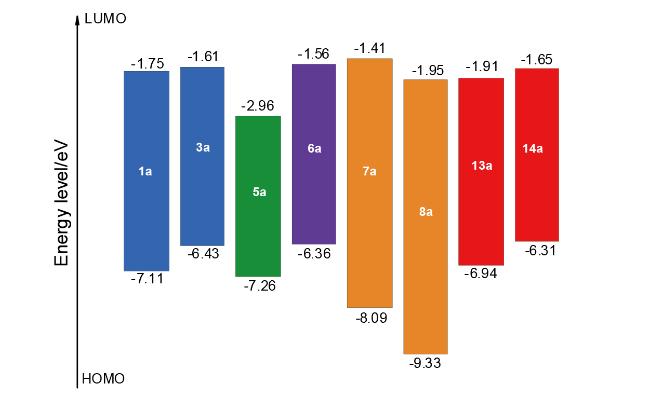
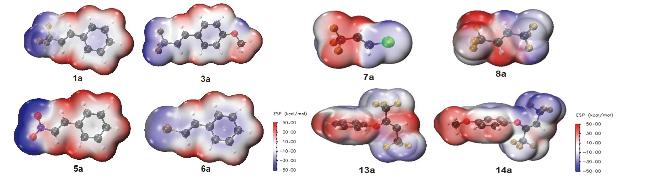
通过密度泛函理论(Density functional theory, DFT)计算光敏剂(表2)与底物(表3)的三线态激发态能垒, 计算结果如表2所示, 同时绘制能级分布图(图5、图6)和静电势图(图7). 从表2可以发现光敏剂的激发能垒大小顺序为Ir(ppy)3>核黄素>噻吨酮>伊红Y, 这与光催化异构化实验结果相符. 其原因是多方面的: 首先, 三线态能垒高的光敏剂具有较长的激发态寿命, 能够更长时间地维持激发态, 从而有更多机会与底物发生能量传递或电子转移, 促进异构化反应, 因此转化率较高. 其次, 三线态能垒高的光敏剂激发态能量更大, 能够更有效地驱动底物的激发和异构化反应, 从而显著提高反应效率. 此外, 三线态激发能垒较高的光敏剂通常具有更好的光稳定性, 能够在反应过程中保持较高的催化活性, 减少光降解或副反应的发生, 进一步提升了反应的稳定性和转化率. 综上所述, 通过合理调控光敏剂的三线态激发能垒, 可以优化其激发态寿命、能量传递效率以及光稳定性, 从而显著提升光催化异构化反应的性能. 从图5也可以发现, Ir(ppy)3的最高占据分子轨道(HOMO) (-5.15 eV)最易失去电子, 最低未占分子轨道(LUMO) (-1.56 eV)也最易接受电子, 电荷注入/传输能力强, 有利于光催化中电荷分离与传递, 因而其催化活性最高.
表2 光敏剂的激发能垒Table 2 Excited energy barrier of photosensitizers |
| Compd. | S0/a.u. | T1/a.u. | Excited energy barrier/(kJ•mol-1) |
|---|---|---|---|
| Ir(ppy)3 | -2518.7198827 | -2518.6319118 | 230.96 |
| Riboflavin | -1330.7519889 | -1330.72582644 | 208.76 |
| Thioxanthone | -973.8742644 | -973.7974316 | 201.72 |
| Eosin Y | -11440.2303443 | -11440.1591998 | 186.79 |
表3 底物的激发能垒Table 3 Excited energy barrier of substrates |
| Compd. | S0/a.u. | T1/a.u. | Excited energy barrier/(kJ•mol-1) |
|---|---|---|---|
| 1a | -646.9630444 | -646.867191653 | 251.66 |
| 3a | -761.5343183 | -761.444961102 | 234.61 |
| 5a | -514.3580475 | -514.283814466 | 194.90 |
| 6a | -2883.3464238 | -2883.25594811 | 237.54 |
| 7a | -875.4448938 | -875.307474280 | 360.79 |
| 8a | -753.0124878 | -752.867234534 | 381.36 |
| 13a | -1059.3803693 | -1059.25534541 | 328.25 |
| 14a | -1173.9495601 | -1173.82929702 | 315.75 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
从表4可以发现烯烃取代基结构对其三线态激发能垒有明显影响. 对比可以发现取代基的吸电子效应为CF3>Br>NO2, 这与光催化异构化实验结果相符. 对比1a、7a与8a的三线态激发能垒, 发现不含苯环的7a与8a激发能垒显著增大. 其主要原因是底物中的苯环被CF3或Cl取代, 吸电子能力过强导致C=C上的电子云密度降低, 且其π-π共轭效应被破坏使激发能垒大幅提高, 光催化异构化反应难以进行. 此外, 吸电子基团的引入还会影响底物的分子轨道能级分布. 强吸电子基团如CF3和Cl会降低最高占据分子轨道(HOMO)的能量, 从而增加S0→T1跃迁所需的能量(图6). 这种效应在7a、8a中尤为明显, 因为它们的分子结构中不仅含有强吸电子基团, 还缺乏有效的π-π共轭体系来稳定激发态.
然而, 在13b中, 氧原子的引入破坏了苯环与C=C之间的π-π共轭作用. 氧原子由于其孤对电子的存在, 形成了局部的电子云集中, 阻断了苯环与C=C之间的电子离域路径, 导致π-π共轭体系被中断. 这种结构上的变化使得电子云无法在分子内有效离域, 从而显著提高了激发能垒. 具体而言, 氧原子的引入不仅增加了分子轨道的局域化程度, 还使得HOMO和最低未占分子轨道(LUMO)之间的能隙增大, 导致S0→T1跃迁所需的能量显著增加. 此外, 氧原子的电负性较强, 进一步拉低了C=C上的电子云密度, 使得激发态更加不稳定. 这种双重效应(共轭作用的破坏和电子云密度的降低)共同导致了激发能垒的显著提高, 使得光催化异构化反应难以进行. 因此, 在设计光催化反应的底物时, 应尽量避免引入可能破坏π-π共轭体系的原子或基团, 以确保反应的高效进行.
2 结论
本研究开发了一种光催化异构化合成含吸电子取代基顺式烯烃方法. 反应以Ir(ppy)3为光敏剂, 以365 nm紫外光为激发光源, 反应条件温和且转化率较高. 通过紫外-可见吸收光谱、氧化还原电位测试发现Ir(ppy)3表现出最宽的光谱吸收范围、最长的激发态寿命和最优的氧化还原能力, 相较而言更适合用于光催化异构化反应. 同时, 通过对不同取代基烯烃的光催化异构化反应研究, 揭示了烯烃取代基结构对光催化异构化反应的影响规律, 为未来设计更高效的光催化反应体系提供了理论依据.
3 实验部分
3.1 仪器与试剂
仪器: Lambda1050+型紫外可见近红外分光光度计(UV-VIS-NIR), 珀金埃尔默企业管理(上海)有限公司; 气相色谱仪(GC), 日本岛津公司; 辰华CHI660E电化学工作站, 上海辰华仪器有限公司.
试剂: Ir(ppy)3, 99%, 北京伊诺凯科技有限公司; 伊红Y, 85%, 阿拉丁化学试剂有限公司; 核黄素, 98%, 北京百灵威科技有限公司; 苯乙烯, 分析纯, 成都市科龙化工试剂场; 1-(三氟甲基)-1,2-苯碘酰-3(1H)-酮, 97%, 阿拉丁化学试剂有限公司; N,N-二乙基羟胺(DEHA), 98%, 阿拉丁化学试剂有限公司; 乙腈(MeCN)、二氯甲烷(DCM)、四氢呋喃(THF)、二甲基亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)、甲醇(MeOH)、乙醇(EtOH)、无水碳酸钠(Na2CO3)、无水硫酸钠(Na2SO4)环己烷, 分析纯, 沪市化学试剂有限公司; 丙酮, 分析纯, 四川西陇科学有限公司.
3.2 实验方法
3.2.1 化合物1a的合成
向25 mL反应瓶中依次加入苯乙烯(0.104 g, 1.0 mmol)、1-(三氟甲基)-1,2-苯碘酰-3(1H)-酮(0.790 g, 2.5 mmol)、Na2CO3 (0.212 g, 2.0 mmol)、MeCN (3.3 mL)、DEHA (0.178 g, 2.0 mmol)和THF (6.6 mL). 80 ℃搅拌2 h, 加入DMF (10.0 mL), 120 ℃搅拌反应18 h, 反应瓶中溶液由无色液体变为棕色液体. 取出反应液, 在反应液中加入去离子水(10.0 mL), DCM (15.0 mL)萃取, 再使用去离子水(10.0 mL)洗涤3次后. 加入Na2SO4干燥, 旋蒸, 以正戊烷为洗脱剂, 经硅胶柱层析分离纯化得到反式-β-三氟甲基苯乙烯(1a)[14]. 1H NMR (400 MHz, CDCl3) δ: 7.42~7.48(m, 2H), 7.36~7.40 (m, 3H), 7.16 (d, J=16.0 Hz, 1H), 6.25~6.16 (m, 1H); 19F NMR (376 MHz, CDCl3) δ: -63.4 (d, J=6.4 Hz, 3F).
3.2.2 光催化异构化反应
向20 mL光反应管中依次加入一定量的反式烯烃、光敏剂和丙酮(15.0 mL). N2氛围下, 在365 nm的LED灯光照条件下40 ℃搅拌反应24 h. 取出反应液, 在反应液中加入1 g硅胶旋干, 取出固体粉末, 以正己烷为洗脱剂, 经柱层析分离纯化得到对应顺式结构产物.
顺式-β-三氟甲基苯乙烯(1b)[15]: 1H NMR (400 MHz, CDCl3) δ: 7.46~7.52 (m, 2H), 7.27~7.30 (m, 3H), 6.71 (d, J=16.0 Hz, 1H), 5.54~5.45 (m, 1H); 19F NMR (376 MHz, CDCl3) δ: -58.2 (d, J=6.4 Hz, 3F).
反式-β-对甲氧基三氟甲基苯乙烯(3a)[16]: 1H NMR (500 MHz, CDCl3) δ: 6.97 (d, J=9 Hz, 2H), 6.84 (d, J=9 Hz, 2H), 6.06 (q, J=7.5 Hz, 1H), 3.77 (s, 3H); 19F NMR (470.0 MHz, CDCl3) δ: 68.8 (s, 3F), 59.2 (d, J=6.6 Hz, 3F).
顺式-β-对甲氧基三氟甲基苯乙烯(3b)[16]: 1H NMR (500 MHz, CDCl3) δ: 6.95 (d, J=9 Hz, 2H), 6.82 (d, J=9 Hz, 2H), 6.35 (q, J=7.5 Hz, 1H), 3.76 (s, 3H); 19F NMR (470.0 MHz, CDCl3) δ: 57.3~58.0 (m, 3F), 60.0 (q, J=6.6 Hz, 3F).
反式-β-硝基乙烯(5a)[17]: 1H NMR (300 MHz, CDCl3) δ: 7.43~7.49 (m, 2 H), 7.53~7.54 (m, 1 H), 7.57 (s, 2 H), 7.61 (s, 1 H), 8.01 (d, J=13.7 Hz, 1 H) ; 13C NMR (75 MHz, CDCl3) δ: 128.5, 129.2, 129.5, 130.1, 130.2, 132.2, 137.2, 139.1.
顺式-β-硝基乙烯(5b)[16]: 1H NMR (300 MHz, CDCl3) δ: 7.58~7.26 (m, 5 H), 6.94 (d, J=9.6 Hz, 1 H), 6.75 (d, J=9.6 Hz, 1 H); 13C NMR (75 MHz, CDCl3) δ: 135.5, 133.9, 130.6, 130.4, 129.8, 128.5.
反式-β-溴苯乙烯(6a)[18]: 1H NMR (400 MHz, CDCl3) δ: 7.26~7.15 (m, 5H), 7.02 (d, J=14.4 Hz, 1H), 6.67 (d, J=14.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 137.2, 136.0, 128.8, 128.2, 126.1, 106.5.
顺式-β- 溴苯乙烯(6b)[19]: 1H NMR (400 MHz, CDCl3) δ: 7.69 (d, J=7.1 Hz, 2H), 7.41~7.31 (m, 3H), 7.08 (d, J=8.1 Hz, 1H), 6.44 (d, J=8.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 135.1, 132.5, 129.1, 128.5, 128.4, 106.5.
反式[3-三氟-1-(三氟甲基)-1-丙烯基]甲氧基-苯(14a)[14]: 1H NMR (400 MHz, CDCl3) δ: 7.39 (d, J=8.8 Hz, 2H), 7.08 (dq, J=16.0 Hz, 1H), 6.90 (d, J=8.8 Hz, 2H), 6.05 (dq, J=16.0 Hz, 1H), 3.83 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -63.3 (d, J=6.4 Hz, 3F).
顺式[3-三氟-1-(三氟甲基)-1-丙烯基]甲氧基-苯 (14b)14]: 1H NMR (400 MHz, CDCl3) δ: 7.40 (d, J=8.8 Hz, 2H), 7.09 (dq, J=16.0 Hz, 1H), 6.91 (d, J=8.8 Hz, 2H), 6.07 (dq, J=16.0 Hz, 1H), 3.84 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -62.9 (d, J=6.4 Hz, 3F).
3.2.3 光催化异构化反应转化率测定
本实验中5a、6a、7a和8a的转化率通过气相色谱积分计算, 将顺式烯烃的峰面积除以反式与顺式烯烃的总峰面积, 其中顺式与反式烯烃的保留时间通过纯品测得.
X=SZ/(SZ+SE)×100%
式中, X: 顺式烯烃的转化率; SZ: 顺式烯烃的峰面积; SE: 反式烯烃的峰面积.
3.2.4 紫外-可见吸收光谱
所有光敏剂的紫外-可见吸收光谱均采用漫反射法测定其固体粉末样品. 首先使用标准白板(BaSO₄)进行基线校正, 以消除仪器和环境的背景干扰; 取100 mg光敏剂样品, 经过充分研磨以确保粉末均匀. 将研磨后的粉末均匀填充至直径为1 cm的漫反射样品池中, 确保样品表面平整, 并轻轻压实以避免过度压缩; 将样品池放入积分球样品室中, 测定各光敏剂在200~800 nm波长范围内的吸光度, 并记录漫反射光谱数据.
使用Kubelka-Munk函数[20]将漫反射数据转换为吸收光谱:
F(R∞)=(1-R∞)2/2R∞
A=lg[1/F(R∞)]
式中F是吸收峰强度, R∞是样品的漫反射率, A是样品的吸光度.
3.2.5 氧化还原电位测试
工作站具有标准的三电极装置, 工作电极(gold plate, d=0.3 cm)、参比电极(Ag tables)和辅助电极(Pt tables). 使用二茂铁/二茂铁(Fc+/Fc0)作参照, 在脱气的MeCN溶液中以0.1 mol/L六氟磷酸四丁基铵作为电解质, 用浓度3.0 mmol/L进行所有的电化学测量. 实验以100 mV/s的扫描速率进行, 扫描范围为0~2.5 V. 对于完全可逆波, 其氧化还原电位用[E1/2(C/C-)]表示; 对于不可逆波, 其氧化还原电位用(EP/2)表示. 实验所测得的电势减去0.04 V (0.04 V的差值经过测试二茂铁的电势标定), 获得的值即为参考饱和甘汞电极(SCE)所对应的电势.
3.2.6 DFT计算
在本研究中, 采用DFT方法对底物的几何结构进行了优化和计算. 首先各光敏剂和底物的几何结构在其三线态(T1)局部极小值下进行了优化. 优化过程中使用了B3LYP杂化泛函[21], 这是一种广泛用于量子化学计算的泛函, 能够较好地平衡计算精度和效率. 同时, 采用6-31G*基组[22-24], 该基组在描述分子电子结构时具有较高的准确性. T1态的计算是在标准的Kohn-Sham框架下进行的, 自旋多重度设为3. 通过从相应的T1局部极小值能量中减去基态(S0)优化几何结构的能量, 计算了S0→T1跃迁的0-0激发能. 0-0激发能是指分子从基态到激发态的最低能量跃迁, 反映了光敏剂在光催化反应中的激发能力. 为了更真实地模拟实验条件, 溶剂效应通过可极化连续介质模型(PCM)进行考虑. PCM[25]模型将溶剂视为连续介质, 能够有效描述溶剂对分子电子结构的影响. 在本研究中, 使用Universal force field (UFF)原子半径[26]来描述乙腈溶剂, 以更准确地反映溶剂环境对分子性质的影响. 所有计算均使用Gaussian 16 Revision A.03量子化学软件包完成[27].
辅助材料(Supporting Information) 化合物13a和14a的1H NMR、13C NMR、19F NMR, 化合物5b和6b的质谱以及3a、5a、6a、7a、8a、13a、14a、核黄素、伊红Y和Ir(ppy)3的DFT计算笛卡尔坐标数据. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(Lu, Y.)