


光催化作为一种条件温和的合成策略, 在有机合成方法学领域发挥了极为重要的推动作用[1-2]. 在过去十余年中, 一系列光催化有机合成新方法被开发和完善, 涵盖了杂环的直接官能团化、烯烃的双官能团化、金属协同偶联反应、螺环化合物的构建、骨架编辑和双环[1,1,1]戊烷的衍生化等多个方面[3-12]. 同时, 丰富易得的羧酸类化合物结构多样、性质稳定、环境友好, 被广泛应用于有机合成领域[13-15]. 近年来, 光诱导的羧酸脱羧产生自由基进而转化为高附加值分子的研究逐渐成为有机化学研究的热点之一[16-19].
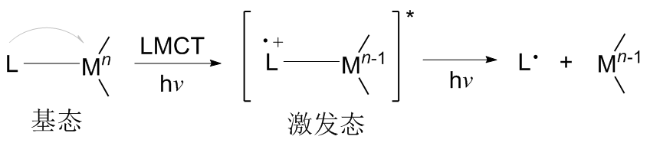
光诱导的配体-金属电荷转移(LMCT)[20-22]过程涉及从配体的填充轨道到金属中心的空轨道的电子跃迁[23], 形成激发的LMCT状态(Scheme 1). 该激发态可导致M—L键均裂, 产生高反应活性的自由基(L•). 这类自由基能够作为高效的自由基引发剂参与多种化学反应过程[24]. 在金属和配体选择方面, 由于LMCT涉及电子向金属中心的转移, 因此该过程通常发生在具有高氧化态的亲电金属上, 例如Cu2+、Fe3+、Ni3+、Ag2+、Ce4+、Co3+、Bi3+和Ti4+. 同时, 配体作为电子跃迁的内源性电子供体, 其富电子特性能够降低LMCT的跃迁能垒[21]. 因此, 诸如卤化物、羧酸盐、醇和叠氮化物等配体在与金属配位后, 可通过光诱导均裂生成相应的卤素自由基、羧基自由基、烷氧自由基和叠氮自由基. 这些自由基在多种反应中作为关键中间体或引发剂, 从而驱动反应进程. 基于过渡金属的LMCT有机光催化过程, 克服了对典型贵金属光催化剂的依赖, 提供了自由基生成的新范式, 并被认为是一种更绿色环保的替代方案. 其中, 以羧酸盐为配体, 在LMCT的作用下产生羧基自由基进而脱羧得到烷基自由基的反应机制, 已成为有机合成方法学研究中的重要工具.
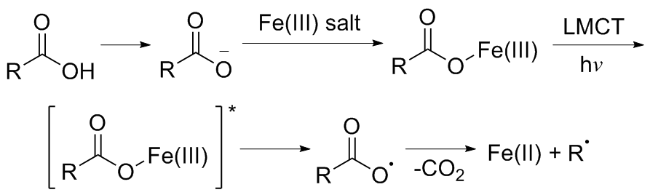
近年来, 研究者将光催化策略与廉价过渡金属(铁、铜、镍等)相结合, 在羧酸脱羧官能团化领域取得了显著进展[25-34]. 其中, 铁作为地壳中丰度最高的过渡金属, 因其低毒性及优异的生物相容性, 使得铁配合物在光催化脱羧官能团化反应中的应用备受关注[35-39]. 铁参与的LMCT诱导脱羧过程主要包括以下步骤: (1)羧酸去质子化; (2)铁盐与羧酸阴离子螯合; (3)光照下铁-羧酸配合物形成激发态, 发生羧酸盐到金属中心的电荷转移; (4)金属-氧键均裂形成羧基自由基, 随后脱羧产生相应的烷基自由基(Scheme 2). 该策略具有高度的化学选择性: 反应位点局限于金属-氧键, 避免其他易氧化的官能团的副反应[28]. 随着自由基化学的蓬勃发展, 金属/光催化羧酸脱羧反应的研究受到了广泛关注. 尽管已有研究者对LMCT参与的有机反应进行了总结[40-42], 并对光或电催化条件下芳基酸[43]及脂肪酸[44]的脱羧官能团化反应进行了概述, 但目前尚未有聚焦于LMCT介导的铁/光催化羧酸脱羧官能团化反应的综述性研究. 鉴于此, 本文系统综述了近两年基于LMCT机制的光诱导铁催化脱羧官能团化反应的研究进展, 旨在为开发羧酸脱羧的新方法奠定基础.
1 脱羧官能团化
1.1 脱羧氧化
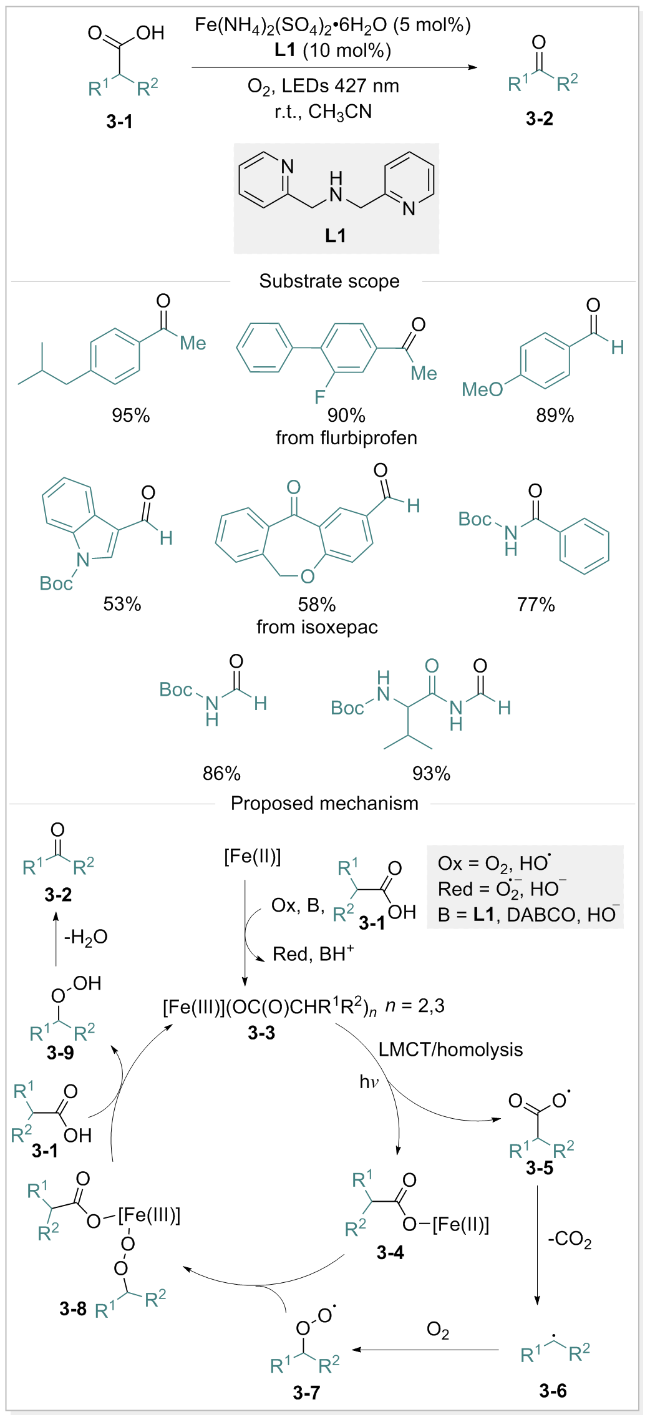
羧酸的脱羧氧化反应与人体诸多关键代谢转化过程密切相关, 其特征在于旧的C—CO2H键的选择性断裂及新的C—O键的生成[45]. 由于羧酸类化合物的丰富易得, 脱羧氧化反应逐渐成为有机化学研究的热点领域[46-47]. 特别地, 利用分子氧作为清洁氧化剂的光驱动脱羧氧化过程, 因其具有绿色化学特点而备受关注[48-49]. 2023年, Guérinot团队[50]报道了一种铁催化、可见光驱动、分子氧介导的羧酸脱羧氧化反应. 该反应以布洛芬为模板底物, 二(2-吡啶甲基)胺为配体, 在氧气氛围下通过LMCT过程实现目标产物的合成. 通过一系列酮、醛和酰胺类化合物(包括药物衍生物)的高效、选择性合成, 验证了该方法的普适性, 为酮类化合物的合成提供了一条新途径. 作者采用电子顺磁共振(EPR)、红外光谱(IR)、紫外-可见光谱(UV/vis)等多种手段深入探究了反应机制, 提出了合理的反应机制. 如Scheme 3所示: 铁催化剂与底物3-1配位形成Fe(III)羧酸根配合物3-3. 在蓝光照射下, 电子从羧酸根转移至Fe(III), 产生Fe(II)配合物3-4和羧基自由基3-5. 随后, 羧基自由基3-5脱除CO2, 转化为碳中心自由基3-6, 其被分子氧捕获形成过氧自由基3-7. 自由基3-7与Fe(II)羧酸盐3-4重组生成Fe(III)配合物3-8. 该配合物进一步与羧酸配位, 再生Fe(III)羧酸盐3-3并释放烷基氢过氧化物3-9, 后者经脱水反应生成羰基衍生物3-2.
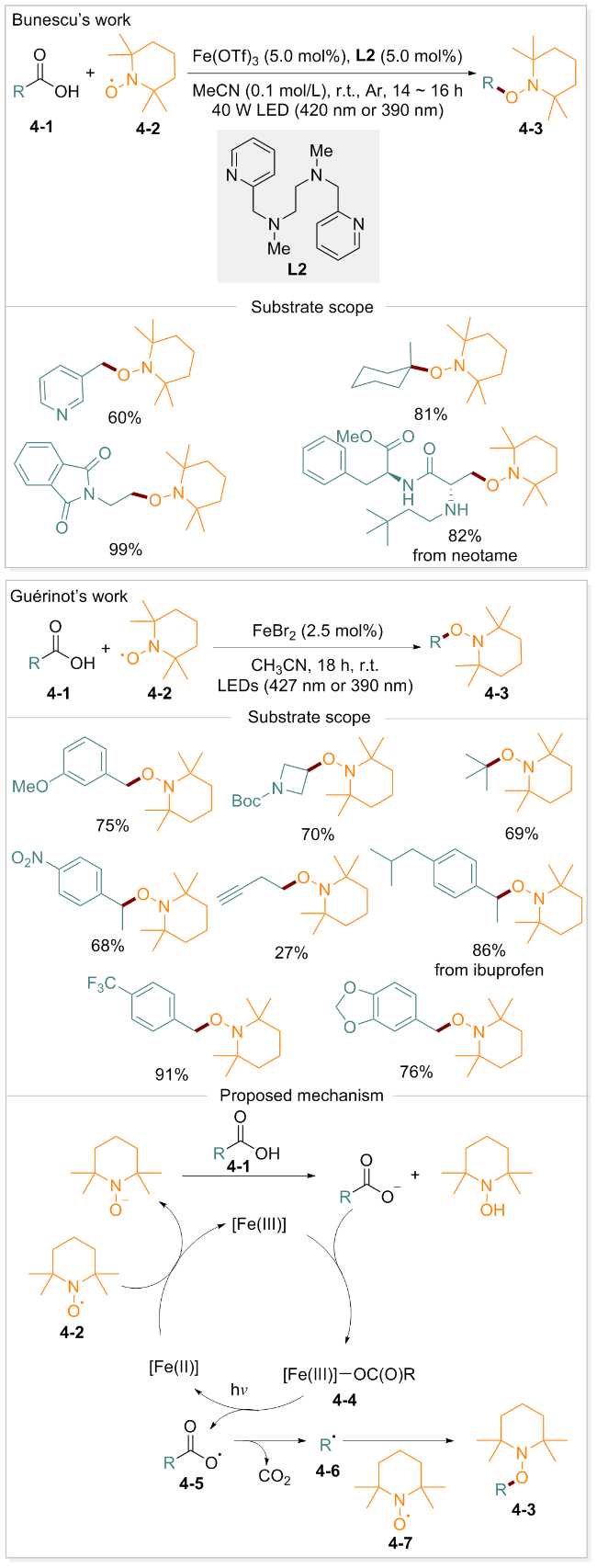
2024年, Bunescu团队[51]报道了另一种铁催化的脂肪酸脱羧氧化反应. 该反应以Fe(OTf)3为催化剂, 2,2,6,6-四甲基哌啶-1-氧基(TEMPO)为氧化剂, 在光照(390 nm或420 nm)及配体的存在下, 实现了羧酸选择性脱羧并形成C(sp3)—O键. 同年, Guérinot小组[52]报道了一种铁催化、光驱动的脱羧氧化反应. 该反应以TEMPO衍生物为氧化剂, FeBr2为催化剂, 在390 nm或427 nm的光照条件下反应18 h, 可将多种羧酸(包括苄型、非苄型、伯、仲、叔等)高效转化为相应的烷氧基衍生物. 该方法对不同类型的羧酸表现出良好的选择性, 并以中等至较高的产率获得相应的产物, 可直接应用于一系列生物活性分子的后期氧化修饰. 所得TEMPO加合物作为通用的合成中间体, 可进一步参与C—C和C—杂原子键的构建. 基于实验研究, 作者提出了如Scheme 4所示的反应机理: 羧酸4-1去质子化并与Fe(III)配位, 形成配合物4-4. 在光照条件下, 配合物4-4发生羧酸根到铁的电荷转移, 导致Fe—O键均裂, 生成Fe(II)中间体和羧基自由基4-5. 羧基自由基脱去一分子CO2得到烷基自由基R•(4-6). 烷基自由基R•被TEMPO捕获得到目标产物4-3, 同时Fe(II)中间体通过TMEPO氧化再生为Fe(III). 研究表明, TEMPO在反应中兼具自由基捕获剂、氧化剂及内部碱三重作用.
1.2 脱羧氢化
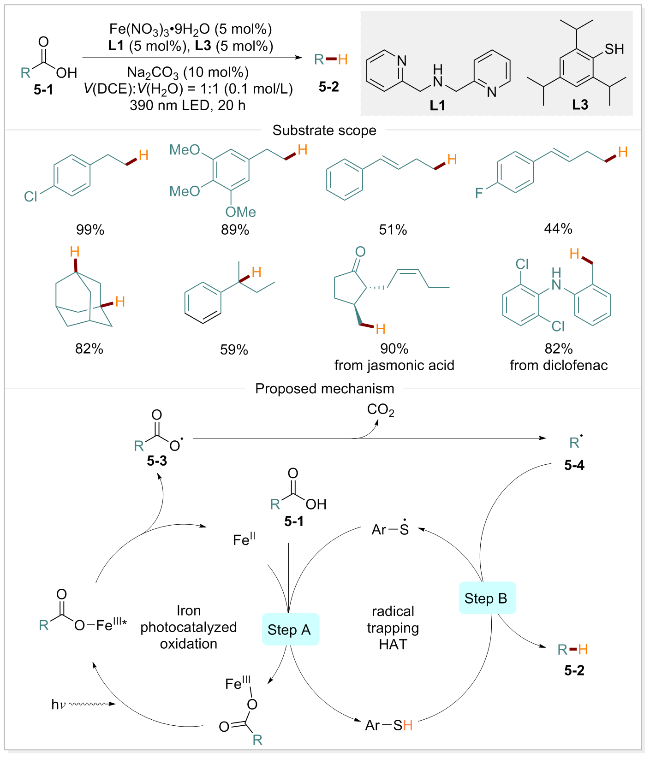
脱羧氢化是一种通过氢或其同位素取代极性羧酸基团的通用策略. 从合成的角度看, 该策略使羧酸成为一种无痕的导向基团, 常用于天然产物合成的后期阶段[53-54]. 此外, 利用合适的氘源, 可合成在材料和药物化学领域中具有重要价值的同位素标记化合物[55-57]. West课题组[58]采用铁催化剂和氢原子转移催化剂的组合, 开发了一种新型的脱羧氢化方法. 该方法摒弃了传统方法对昂贵光氧化还原催化剂、有毒氢源、高温、强氧化剂以及预活化酸性底物的需求. 基于同位素标记实验, 自由基捕获及自由基钟等实验手段, 作者提出了可能的反应机理(Scheme 5). 羧酸5-1与铁盐螯合形成Fe(III)配合物. 经光照射形成激发态, 随后在分子内部发生电子转移, 使O—Fe键均裂产生羧基自由基5-3和Fe(II). 羧基自由基5-3快速脱羧产生碳中心自由基5-4, 在硫醇配体作用下, 烷基自由基5-4攫取氢原子生成目标产物5-2和硫自由基(步骤B). 最后, 硫自由基在羧酸存在下进一步氧化Fe(II), 释放高价Fe(III)并形成新的Fe(III)配合物(步骤A), 从而实现光催化氢化循环.
1.3 脱羧氟烷基化
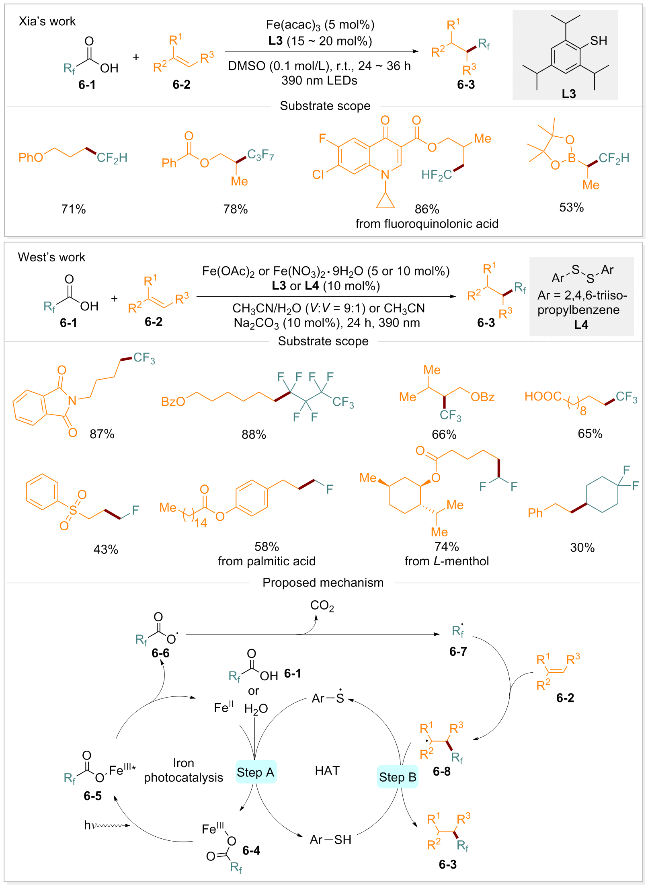
烯烃的氟烷基化反应是合成含氟化合物的高效策略, 在药物中引入氟烷基能够显著增强母体分子的治疗特性[59]. 然而, 由于氟烷基羧酸具有较高的氧化电位, 直接从氟烷基羧酸产生氟烷基自由基面临较大挑战, 通常需要借助复杂且昂贵的前体试剂. 氟烷基(CFn)与羟基(OH)和巯基(SH)具有等排性和等极性, 可作为氢键的亲脂供体[60], 因此, 在分子中选择性地引入氟烷基对于药物设计和有机合成具有极其重要的意义[61-63]. 夏吾炯团队[64]构建了一种基于光氧化还原和铁催化的LMCT的多功能脱羧策略, 成功实现了烯烃的二氟甲基化. 该方法以来源广泛且成本低廉的脂肪族羧酸和烯烃为原料, 采用Fe(acac)3为催化剂, 在二甲基亚砜中, 经390 nm光照24至36 h, 可获得二氟甲基化产物. 此外, 该策略同样适用于未活化烯烃的二氟甲基氘化、二氟甲基炔基化以及二氟甲基氯化等多种反应, 显著扩展了烯烃官能团化的范畴. 同期, West研究团队[65]报道了一种光诱导的未活化烯烃的氟烷基化反应, 以廉价易得的氟烷基羧酸作为氟烷基自由基来源, 实现了包括单氟甲基、二氟甲基、三氟甲基和全氟烷基化在内的多种氢-氟烷基化反应. 该反应以Fe(OAc)2或Fe(NO3)3•9H2O为催化剂, 乙腈为溶剂, 在碱性条件下通过390 nm光照24 h, 即可得到氟烷基化产物(Scheme 6). 该反应条件温和,无需贵金属参与, 并在一些药物分子、天然产物及生物活性分子衍生物的氟烷基化反应中表现出优异的性能, 展示了其广泛的适用性和合成潜力. 作者通过自由基捕获实验、自由基时钟实验、同位素标记实验和电子顺磁共振光谱等系统研究, 提出了合理的反应路径: 首先, 氟烷基羧酸6-1与Fe(II)螯合生成Fe(III)配合物6-4, 在光照下激发形成激发态6-5, 其分子内部发生配体到金属的电荷转移过程, 导致O—Fe键均裂, 产生羧基自由基6-6和Fe(II). 羧基自由基6-6脱去一分子CO2得到氟烷基自由基6-7. 该自由基被烯烃捕获得到烷基自由基中间体6-8. 随后, 在硫醇配体与烷基自由基中间体之间发生氢原子转移(HAT), 生成所需的氢氟烷基化产物6-3和硫代自由基(步骤B). 最后, 硫代自由基在羧酸存在下氧化Fe(II)生成高价Fe(III)并重新形成配合物6-4(步骤A), 从而实现光催化氢-氟烷基化的循环过程.
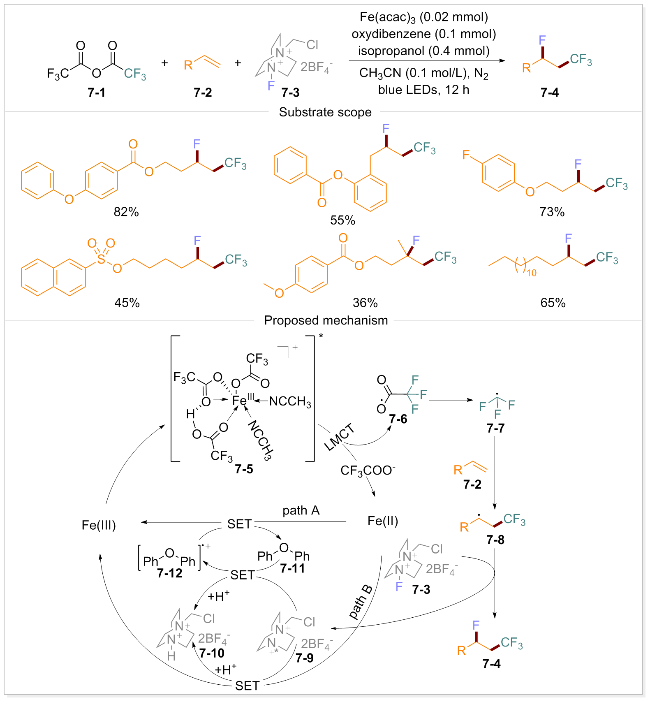
尽管上述方法已成功实现了高氧化电位羧酸的脱羧, 但其底物适用范围仍存在一定局限性. 因此, 利用含量丰富的原料作为自由基前体以增强LMCT的反应性显得尤为重要. 同年, 蓝宇团队开发了一类高效且实用的铁催化LMCT反应体系, 该体系通过使用Brønsted酸[66]或双配体[67]有效活化多种惰性卤代烷基羧酸盐(CnXmCOO−, X=F或Cl), 实现非活化烯烃的氯/氟-多卤代烷基化(Scheme 7). 分子内氢键的形成导致铁配合物的最低未占据分子轨道(LUMO)能级降低0.86 eV, 从而促进LMCT过程的进行. 在蓝光照射下, 铁催化剂与三氟乙酸酐形成的配合物被激发至激发态7-5. 随后, 该激发态经历LMCT过程形成三氟乙酸自由基7-6, 并快速脱羧形成三氟甲基自由基7-7. 该自由基进一步与烯烃7-2反应形成自由基中间体7-8. 中间体7-8与化合物7-3发生氟原子转移(FAT)过程生成目标产物7-4. 该策略具有广泛的底物适用性, 各种含有苯甲酰基的脂肪族末端烯烃在强Brønsted酸存在下均表现出良好的耐受性.
1.4 脱羧卤化
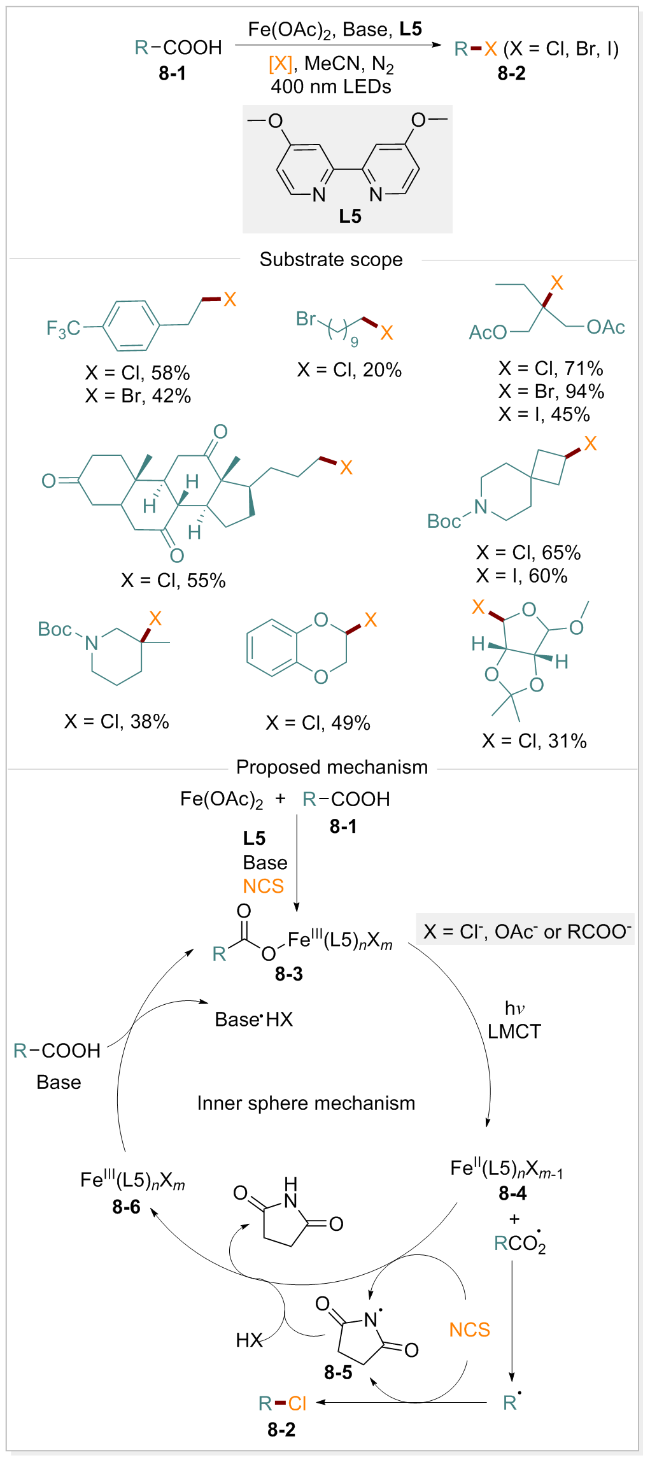
卤代烷作为一类重要的结构单元, 可通过亲核取代和交叉偶联反应实现分子结构的多样化与功能化[68-69]. 尽管脱羧卤化反应的研究可追溯至上世纪[70], 但发展符合绿色化学原则的可持续脱羧卤化策略仍面临诸多挑战. 2024年, 胡鹏课题组[71]报道了一种基于铁催化的脂肪族羧酸直接脱羧卤化的通用方案. 该方法具有广泛的底物适用性, 良好的官能团耐受性, 能够选择性地保留苄基及烯丙基C(sp3)—H键, 为复杂天然产物的后期修饰提供了高效途径. 该方法反应条件温和, 符合绿色化学可持续发展要求. 通过系统的机理研究, 作者提出了脱羧卤化反应的合理机制(Scheme 8): 羧酸8-1去质子化并与Fe(OAc)2配位, 形成具有光活性的铁羧酸盐配合物8-3. 在400 nm光照下, 配合物8-3经历LMCT过程产生Fe(II)配合物8-4和羧基自由基. 羧基自由基快速脱羧并释放CO2, 形成烷基自由基, 随后与N-卤代琥珀酰亚胺结合生成目标产物8-2, 同时产生琥珀酰亚胺自由基8-5. Fe(II)配合物8-4被N-氯代丁二酰亚胺(NCS)或酰亚胺自由基8-5氧化为Fe(III)配合物8-6, 后者进一步与羧酸配位形成铁羧酸盐配合物8-3, 从而完成光催化循环, 实现卤化产物的高效合成.
1.5 脱羧叠氮化
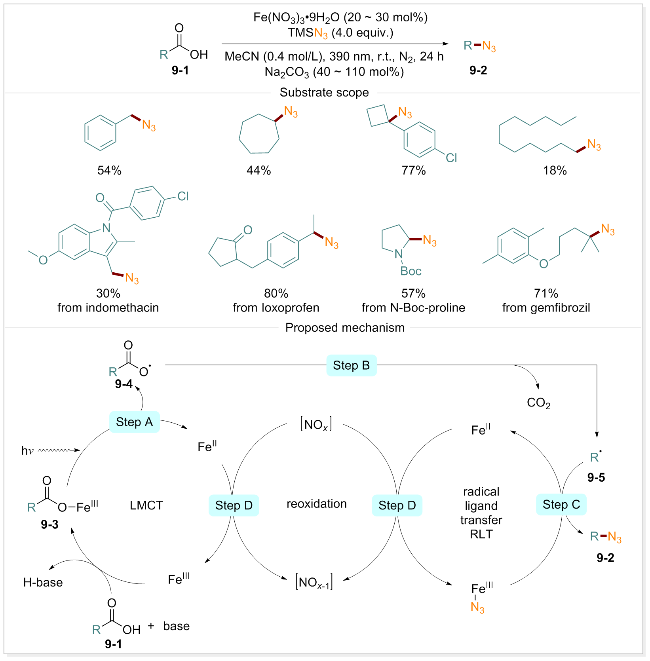
C—N键是药物化学中普遍存在的化学键. 统计数据显示, 平均每个小分子药物中含有2.3个氮原子, 且 C—N键偶联反应在已知的药物化学途径中约占8%[72]. 脱羧叠氮化是构建C—N键的一种有效策略, 具有重要的研究和应用价值. 然而, 现有方法通常依赖强氧化剂、贵金属催化剂、复杂试剂或对羧酸进行预活化, 这些限制因素阻碍了该方法的广泛应用[73-74]. 基于LMCT和自由基配体转移(RLT)的概念, West团队[75]报道了一种光化学诱导、铁催化直接脱羧叠氮化反应. 在叠氮三甲基硅烷的存在下, 仅需对廉价的硝酸铁催化剂进行紫光照射, 即可将多种羧酸直接转化为相应的有机叠氮化物. 该体系具有良好的官能团耐受性. 硝酸铁在此反应中兼具铁源和氧化剂源的双重功能, 无需额外添加外部氧化剂, 显著简化了反应流程. 通过动力学研究、自由基淬灭实验、自由基钟实验及紫外-可见光谱(UV/Vis)等实验手段, 研究者们初步揭示了光催化脱羧叠氮反应的机理(Scheme 9): 首先, 底物羧酸9-1去质子化并与铁盐配位, 形成铁羧酸盐配合物9-3. 在紫光照射下, 配合物9-3经历LMCT过程, 电子从羧酸根转移至Fe(III), 生成羧基自由基中间体9-4(步骤A)和还原态Fe(II). 随后, 羧基自由基中间体9-4迅速脱羧释放CO2(步骤B), 形成碳中心自由基9-5. 该自由基与铁-叠氮化物发生配体转移, 生成叠氮化产物9-2(步骤C)和还原性Fe(II).最后, 上述步骤中产生的Fe(II)被源自硝酸盐的氮氧化物氧化, 重新转化为Fe(III)(步骤D), 从而完成催化循环.
1.6 脱羧胺化
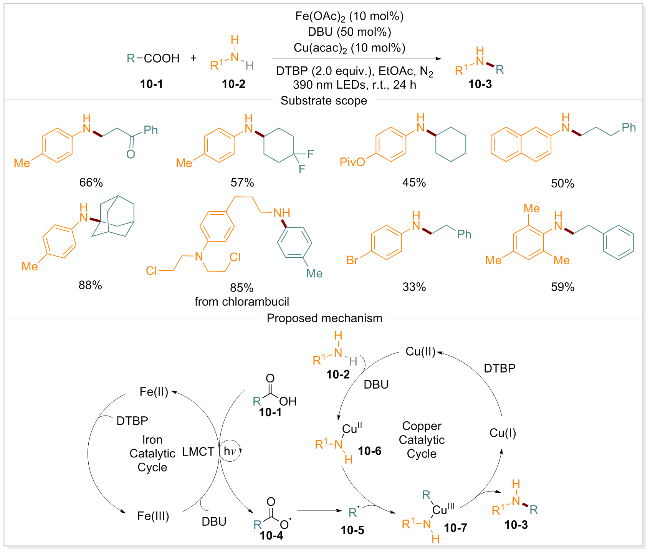
胺类化合物作为重要的有机合成中间体, 在药物研发、农药科学及功能材料等领域具有广泛的应用[76-77].近年来, 通过羧酸脱羰基化反应直接构建C(sp3)—N键的研究受到广泛关注[33]. 曾荣课题组[78]报道了一种光诱导的铁/铜双催化策略, 用于实现脂肪族羧酸的自由基脱羧胺化反应. 该策略创新性地整合了铁催化剂的单电子转移特性与铜催化剂的双电子过程优势, 在温和条件下(室温)即可高效构建脂肪族羧酸与芳香胺之间的 C—N键. 该方法具有操作简便、可扩展性强等优点, 为从多种羧酸前体制备C—N键提供了强有力的工具. 此外, 该反应体系对多种天然产物及药物分子骨架均表现出良好的兼容性, 显示出其在药物研发领域的巨大潜力. 基于实验结果, 该团队提出了脱羧胺化反应的合理途径(Scheme 10): 在二叔丁基过氧化物(DTBP)、1,8-二氮杂二环十一碳-7-烯(DBU)存在下, Fe(II)首先被氧化成Fe(III), 随后在390 nm光照下, Fe(III)与羧酸10-1配位并发生配体-铁的电荷转移, 生成羧基自由基中间体10-4, 该中间体脱去一分子CO2后形成关键的烷基自由基10-5. 与此同时, 在DBU的存在下, 芳香胺10-2与铜催化剂配位形成Cu(II)配合物10-6, 该配合物被烷基自由基10-5捕获生成烷基铜配合物10-7. 随后经还原消除反应得到目标产物10-3和铜(I). Cu(I)被DTBP氧化重新生成Cu(II), 完成催化循环. 机理研究表明, 铁催化剂通过LMCT过程实现光诱导的烷基自由基生成, 而铜催化剂则负责自由基捕获与偶联反应的进行.
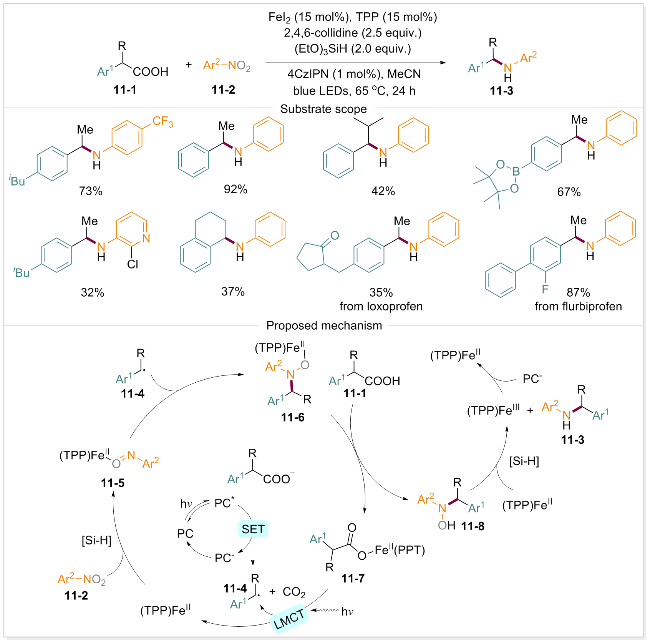
为防止过度烷基化的发生, 谢劲课题组[79]于2024年开发了一种光氧化还原催化与铁共催化的自由基C—N偶联策略, 旨在实现羧酸与硝基芳烃之间的脱羧胺化反应. 该反应以羧酸和硝基芳烃为起始原料, 避免了贵金属催化剂的使用, 且在温和反应条件下进行, 有效防止了硝基化合物的预还原, 实现了一锅法构建C—N键. 该方法展现出良好的官能团耐受性和高度的选择性, 能够以较高的产率获得多种结构各异的仲胺类化合物, 包括含有羧酸基团的药物分子前体, 均可有效转化为目标产物. 通过系统的机理研究, 作者提出了脱羧胺化反应的合理途径(Scheme 11): 首先, 羧酸11-1在碱性条件下形成羧酸阴离子, 蓝光照射下光催化剂4CzIPN形成激发态, 与羧酸阴离子发生单电子转移(SET)生成烷基自由基11-4. 同时, 铁(II)和(EtO)3SiH还原硝基芳烃, 产生亚硝基芳烃中间体11-5, 其被烷基自由基捕获形成Fe(III)配合物11-6. 随后再与羧酸进行配体交换, 得到配合物11-7和中间体11-8. 11-7在光照下发生分子内配体到金属的电荷转移, 并快速脱羧, 再次形成烷基自由基11-4, 参与后续反应链. 中间体11-8则通过铁(II)和(EtO)3SiH进一步还原, 最终转化为目标产物11-3. 烷基自由基的生成涉及SET和LMCT两个过程, 其中LMCT过程是生成关键烷基自由基中间体11-4的主要途径.
上文所述的脱羧叠氮化反应(Scheme 9)与脱羧胺化反应(Schemes 10, 11)均通过铁催化的光诱导自由基反应路径实现C—N键构建. 尽管二者均涉及羧酸脱羧生成烷基自由基的关键步骤, 但其反应机理存在显著差异. 脱羧胺化反应经历LMCT及RLT过程, 其中硝酸铁兼具催化剂与氧化剂双重功能; 而脱羧叠氮化反应则经历LMCT和Cu介导的催化循环过程, 且需额外添加氧化剂. 尽管两种胺化反应最终产物结构类似(Scheme 10, 11), 但反应路径大不相同. Scheme 10采用铜铁双金属催化体系, 需要在铜催化循环中加入DTBP氧化剂以完成自由基捕获与偶联反应; 而Scheme 11则需引入[Si-H]还原剂将硝基芳烃还原为亚硝基中间体, 进而完成后续转化过程. 这些差异化的催化路径为C—N键的构建提供了多样化的合成策略.
1.7 脱羧烯基化
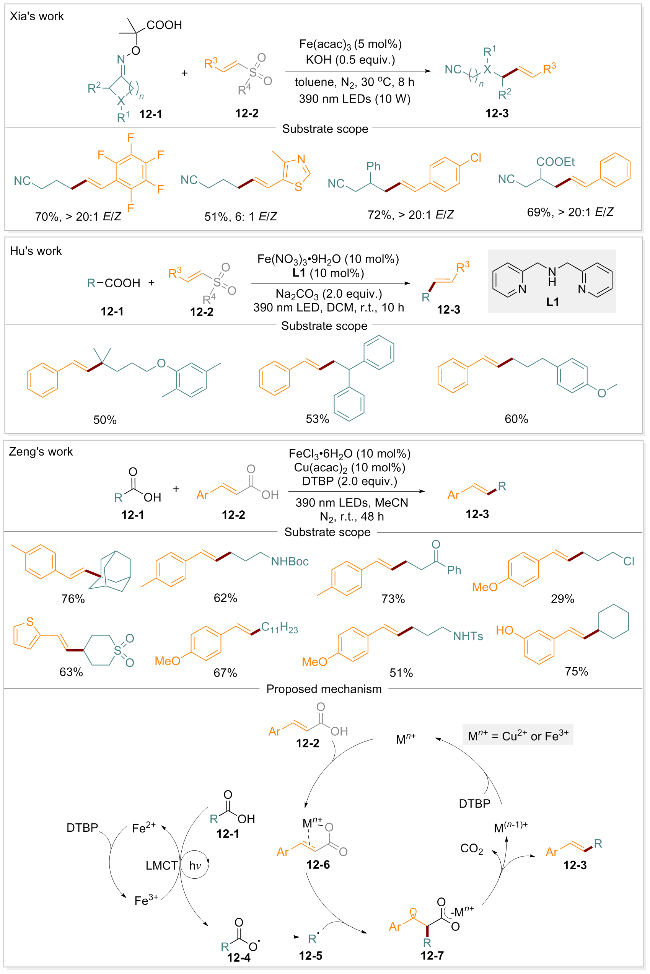
不饱和双键作为一类重要的结构单元, 广泛存在于各类功能化合物中, 是构建多种先进材料的基础骨架和多功能合成前体[80-82], 在药物化学、高分子材料、光电器件以及介晶显示技术等领域具有重要应用价值[83]. 因此, 开发用于合成不同取代的烯烃C(sp3)—C(sp2)键的合成方法尤为重要. 夏吾炯团队[84]报道了一种光诱导自由基介导的C—C键断裂-烯基化反应. 该反应以肟酸和磺酰基前体为底物, 在Fe(acac)3和碱的作用下, 以中等至良好的收率获得烯基化产物. 该体系表现出良好的底物普适性, 各类取代芳基、杂芳基(含噻唑、吡啶等)底物及开环肟酸等均能有效参与反应. 同年, 胡小强团队[85]开发了一种脱羧烯基化新方法. 该方法以反式苯基肉桂砜为自由基受体, 在九水合硝酸铁和二(2-吡啶甲基)胺催化剂作用下, 各种羧酸(包括伯、仲、叔脂肪酸)发生脱羧反应, 以良好的收率获得相应的烯基化产物. 2023年, 曾荣课题组[78]报道了光诱导的铁/铜双催化C—N和C—C偶联反应. 基于此研究基础, 该团队[86]于2024年报道了一种高效的光诱导铁催化双脱羧烯基化反应, 采用Fe/Cu双催化体系, 在DTBP作用下, 成功实现两种不同的羧酸之间的直接交叉偶联, 特别是通过C(sp2)—C(sp3)偶联构建含远程亲核基团的内烯烃结构, 为合成各类含极性官能团的烯烃化合物提供了有力工具. 同时, 为阐明烯基化反应机理, 作者开展了系统的机理实验. UV/Vis实验证实铁催化剂是一种光激发物质; 自由基捕获实验表明该反应涉及自由基过程; 自由基时钟实验验证了铁催化羧酸脱羧是生成烷基自由基的引发步骤. 基于上述实验结果, 该团队提出了一种合理的机制(Scheme 12). 在390 nm光照下, 羧酸12-1与铁催化剂配位后发生电子从羧酸根向Fe(III)的转移, 使Fe—O键断裂生成Fe(II)和羧基自由基中间体12-4, 该中间体脱去一分子CO2得到烷基自由基12-5. 同时, 烯酸12-2与过度金属催化剂(如Fe(III)或Cu(II))配位形成配合物12-6, 该配合物因其C=C双键的亲电性更易与烷基自由基12-5反应, 生成热力学稳定的苄基自由基12-7. 随后发生LMCT以及第二次脱羧反应, 得到目标产物12-3并再生低价金属催化剂(如Fe(II)或Cu(I)), 后者被DTBP氧化完成催化循环.
1.8 脱羧炔基化
当前, 通过形成C(sp3)—C(sp)键合成烷基化炔的方法存在诸多局限[87-88], 包括起始物料范围有限、反应条件苛刻(如高温)、依赖贵金属催化剂以及产生有毒副产物等. 因此, 开发一种普适性更强的炔基羧酸脱羧偶联的烷基化方法尤为重要. 2024年, 夏吾炯团队[84]进一步报道了一种光诱导自由基介导的C—C键断裂-炔基化反应. 该反应以肟酸和甲磺酰基乙炔类化合物为底物, 在Fe(acac)3和碱的共同作用下, 以中等至良好的产率获得炔基化产物. 胡小强团队[85]同期也报道了一种脱羧炔基化策略. Lee等[89]报道了一种高效的铁-光催化炔酸和烷基羧酸的双脱羧偶联方法, 为烷基化炔的合成提供了新思路. 该脱羧偶联体系展现出良好的官能团耐受性, 并对伯、仲和叔烷基酸均表现出良好的反应活性. 在蓝光照射下, 以FeCl2/TPA或Fe(NO3)3•9H2O为催化剂, 可高效构建烷基炔骨架, 获得中等收率的目标产物. 实验数据充分证实了反应机理涉及烷基羧酸脱羧生成的烷基自由基对炔C≡C键的亲核加成, 随后通过连续的单电子转移和脱羧过程形成烷基炔产物. 该体系以苯磺酰基为自由基受体, 在九水合硝酸铁和二(2-吡啶甲基)胺催化剂作用下, 使羧酸(包括伯、仲脂肪酸)发生脱羧反应, 以中等产率生成相应的炔基化产物. 基于系统的机理研究实验, 该团队提出了合理的假设(Scheme 13): 羧酸13-1首先与PhI(OAc)2反应生成关键中间体3-4. 在蓝光照射下, 铁催化剂与中间体13-4配位后发生单电子转移和脱羧反应, 形成离子自由基13-5, 进而转化为烷基自由基13-6. 该自由基与炔酸13-2的C≡C键进行亲核加成, 形成乙烯基自由基中间体13-7. 随后经历去质子化和单电子转移过程形成中间体13-8. 最终通过脱羧反应得到所需的烷基炔产物13-3.
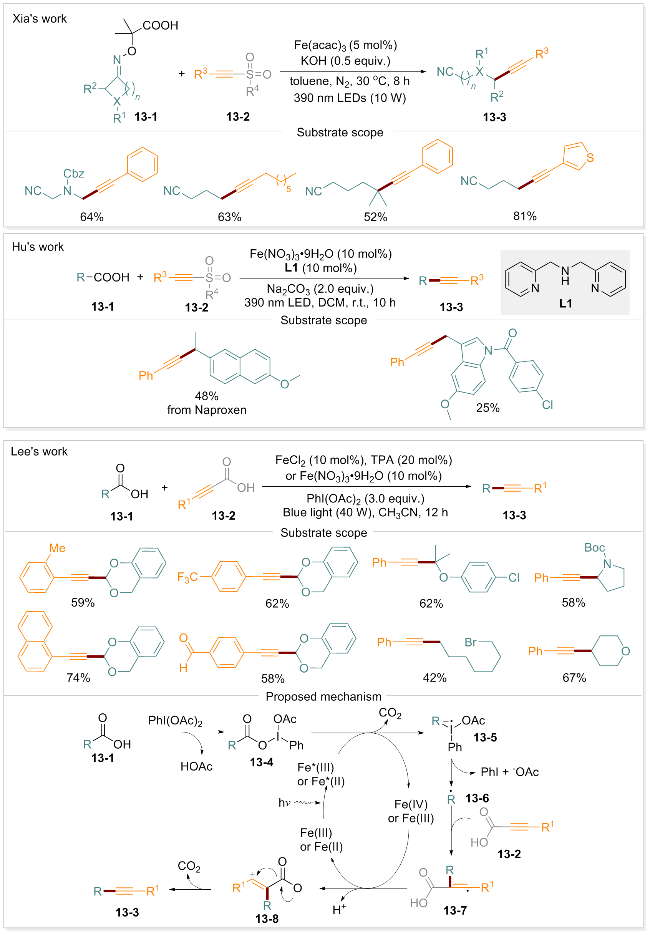
1.9 脱羧苯甲酰化
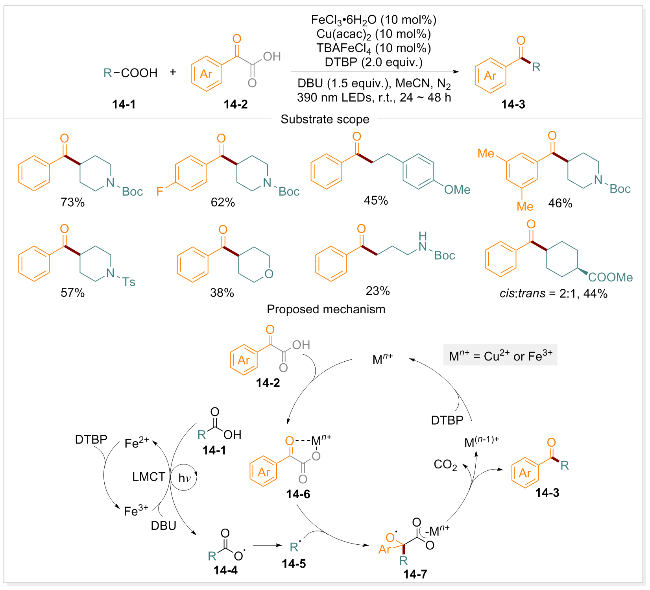
羰基结构作为一类重要的官能团, 是众多天然产物的核心结构单元, 也是有机合成中关键的构建模块[90-91]. 2024年, 曾荣课题组[86]报道了一种高效的光诱导铁催化方法, 使用Fe/Cu双催化策略, 在DTBP作用下实现了两种不同羧酸的双脱羧烯基化反应. 进一步研究发现, 烷基酸与α-酮酸发生脱羧反应时会生成酮类化合物. 这些酮类化合物能够进一步发生Wittig反应制备末端烯烃, 从而简化复杂药物类似物的合成过程. 在TBAFeCl4、Fe(OAc)2、Cu(acac)2、DTBP和DBU组成的催化体系中, α-酮酸与烷基酸通过Fe-LMCT过程生成相应的苯甲酰化产物. 由于α-酮酸在氧化条件下稳定性较低, 含有给电子基团或吸电子基团的α-酮酸, 其苯甲酰化产物的收率均处于中等水平. 为深入探究α-酮酸脱羧苯甲酰化的反应机制, 该课题组开展了系统的机理研究, 包括自由基淬灭, 自由基时钟和UV/vis分析等, 并据此提出了合理的反应机制(Scheme 14). 在DBU存在下, 配体在390 nm光照下引发铁催化的电荷转移过程, 生成Fe(II)和RCOO•自由基中间体14-4, 该自由基中间体释放CO2形成烷基自由基14-5. 与此同时, α-酮酸(14-2)与金属催化剂(如Fe(III)或Cu(II))螯合形成配合物14-6, 烷基自由基R•被配合物14-6捕获生成热力学稳定的苄基自由基14-7. 随后发生电荷转移和二次脱羧, 最终形成苯甲酰化产物14-3. 机理研究表明, 该反应由光诱导的铁催化剂通过首次CO2释放引发, 铜催化剂则参与单电子转移过程, 并介导第二次脱羧及后续的苯甲酰化反应.
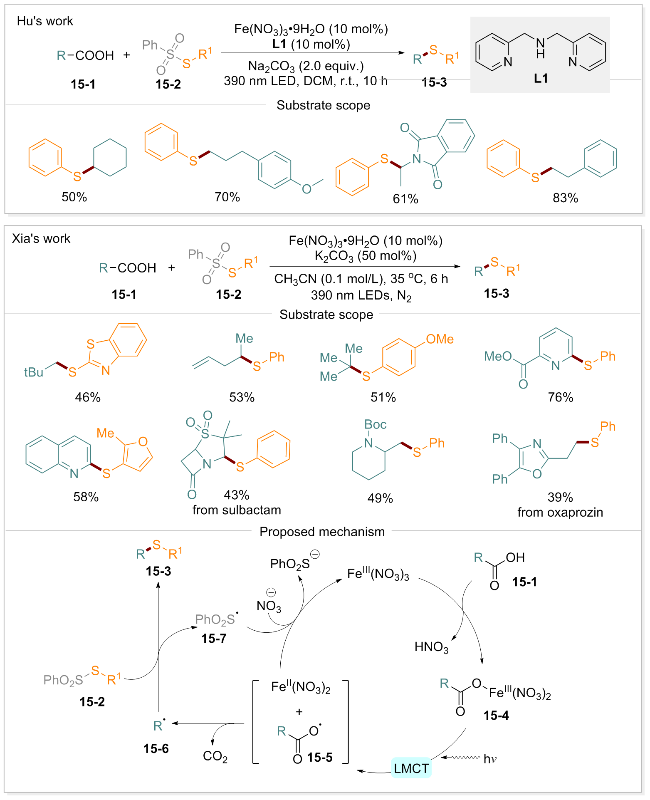
1.10 脱羧硫醇化
含硫化合物在自然界中广泛分布, 尤其是在天然和合成的生物活性分子中普遍存在. 与此同时, C—S键的构建与转化在有机合成、材料化学、农业等领域也受到广泛关注[92-94]. 然而, 在温和条件下使用廉价且易得的原料实现C—S键的构建仍是一个亟待解决的关键科学问题. 针对这一挑战, 胡小强团队[85]开发了一种基于Fe-LMCT的羧酸脱羧硫醇化方法. 该方法以S-苯基硫代苯基砜(PhSO2SPh)为自由基受体, 在二(2-吡啶甲基)胺(配体L1)存在下, 羧酸在常温下即可发生脱羧硫醇化反应, 以良好至优异的产率生成相应的硫醚产物. 夏吾炯团队[95]则通过光诱导的Fe-LMCT策略, 建立了另一种C—S键构建方法, 高效地将烷基和(杂)芳基羧酸转化成相对应的硫醚产物. 该方法以九水合硝酸铁为催化剂, 在390 nm光照及碱的作用下, 羧酸与苯基硫代磺酸盐在35 ℃氮气条件下反应6 h, 即可获得硫醇化产物,且无需使用额外的配体. 这两种方法均具有操作简单、易于扩展、官能团耐受性强的特点, 适用于复杂分子的后期修饰, 为实际应用提供了重要价值. 为深入理解光诱导铁催化脱羧硫醇化的机理, 作者通过自由基时钟实验、同位素标记等手段, 提出了合理的反应途径(Scheme 15): 羧酸底物15-1在碱的作用下去质子化并与铁催化剂螯合, 形成配合物15-4, 在紫光照射下, 电子从羧酸根转移至Fe(III), 使配合物15-4中O—Fe均裂, 形成羧基自由基15-5和Fe(II)盐. 羧基自由基15-5中CO2离去, 释放出关键的烷基自由基15-6. 该烷基自由基被硫代磺酸盐15-2捕获形成目标产物硫醚15-3, 同时生成磺酰基自由基15-7. 磺酰基自由基将Fe(II)重新氧化为Fe(III), 从而实现光催化脱羧硫醇化的循环过程.
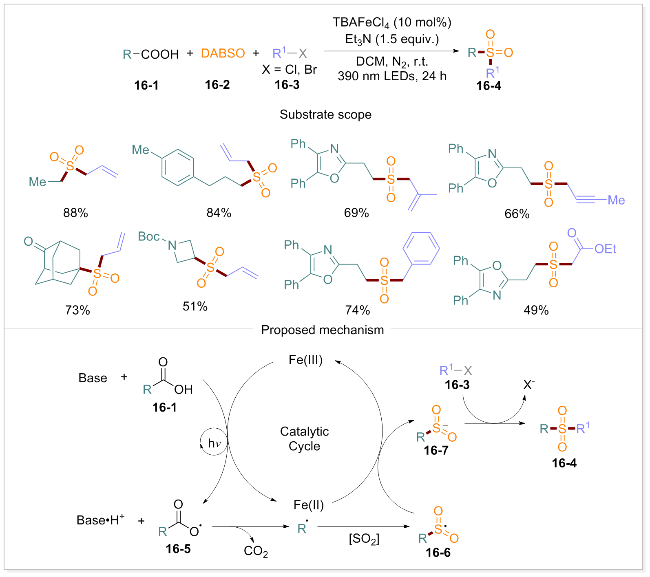
1.11 脱羧磺酰化
有机砜类化合物在有机合成、医药、功能材料及生物学等领域具有重要的应用价值[96-97]. 然而, 羧酸直接脱羧磺酰化, 尤其是以铁作为金属催化剂时, 仍面临重大挑战. 2024年, 曾荣课题组[98]报道了一种光诱导的铁催化体系, 通过羧酸与1,4-二氮杂双环[2.2.2]辛烷-1,4-二鎓-1,4-二亚磺酸(DABSO)及亲电试剂的反应实现了磺酰化产物的高效合成(Scheme 16). 该方法以DABSO为自由基受体, 在TBAFeCl4和三乙胺存在下, 经390 nm光照24 h即可获得磺酰化产物. 该反应具有广泛的底物适用性, 烷基、烯基、苯基酸等均可转化为相应的磺酰化产物. 此外, 反应过程中醚键、酯基、酰胺、羰基和氨基甲酸酯等官能团均能保持稳定, 显示出该方法在合成应用中的巨大潜力, 为有机砜类化合物的合成提供了一种简洁而高效的方案. 通过系统的机理研究实验, 该团队提出一个磺酰化反应的合理途径: 首先, 羧酸16-1去质子化后, 在390 nm光照下与Fe(III)催化剂配位并发生单电子转移, 形成Fe(II)和RCO2•自由基16-5. 随后, 该自由基脱羧形成烷基自由基R•, 其在DABSO作用下转化为砜自由基中间体RSO2• (16-6). 最终, Fe(II)和RSO2•之间发生单电子转移, 形成硫中心阴离子16-7并再生Fe(III)催化剂. 阴离子16-7被亲电试剂捕获生成所需的磺酰化产物16-4.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2 结论与展望
(Cheng, F.)