

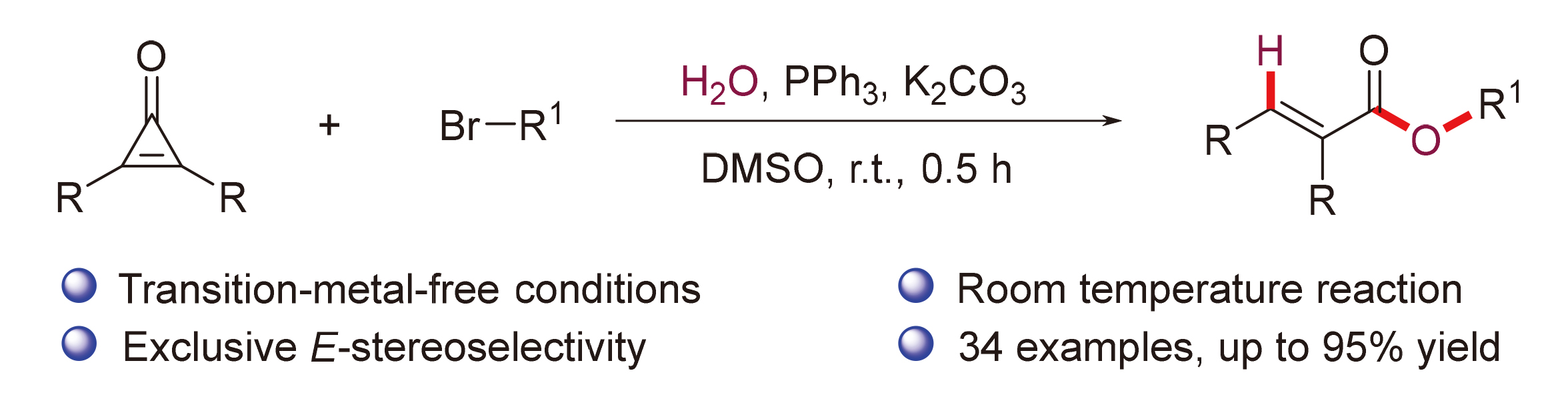
1 Introduction
α,β-Disubstituted acrylates represent a privileged structural motif that are prevalently distributed in numerous bioactive molecules[1] and natural products,[2] exhibiting a broad spectrum of remarkable pharmacological activities including antiinflammatory, anticancer and antiviral effects (Figure 1). Moreover, these molecule series can serve as precursors for Michael addition and radical acceptors, acting as versatile building blocks in organic synthesis.[3] They are also utilized for the synthesis of polymer materials through self-polymerization or co-polymerization with other monomers.[4] Consequently, the development of effective methods for their preparation continues to arouse the interest of chemists due to their significant value. The typical approaches to synthesize these prominent scaffolds encompasses the aldol condensation,[5] Knoevenagel reaction,[6] Galat reaction,[7] Wittig reaction,[8] Reppe carbonylation reaction,[9] and the hydroesterification of alkynes.[10] Nevertheless, the existing routes are generally confined to relatively rigorous operational conditions, limited substrate scopes, and poor selectivity, the exploration of innovative techniques for the efficient and stereoselective construction of α,β-disubstituted acrylates under mild conditions is still in demand.
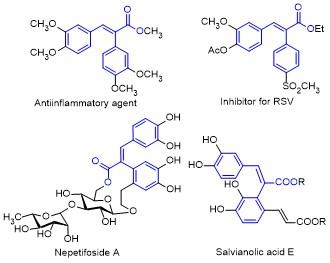
Cyclopropenones, one of the smallest Hückel aromatic carbocycles, exhibit a distinctive structure that comprises both ketone and olefin moieties within their highly strained three-membered ring.[11] The employment of cyclopropenones as flexible 3C synthons has recently garnered considerable interest from organic synthetic chemists.[12] Among the transformations exploiting their versatile reactivity, ring-opening addition processes between cyclopropenones and different nucleophiles via strain-driven C—C bond cleavage are considered as an extremely effective approach for creating structurally diverse α,β-unsaturated carbonyl derivatives.[13-17] For instance, in 2018, Sun’s research group[14] reported a pioneering AgOTf-catalyzed reaction for the direct synthesis of α,β-unsaturated acrylates from cyclopropenones and formic esters (Scheme 1a). In 2021, Ravikumar and co-workers[15] presented an appealing procedure to access trisubstituted α,β-unsaturated esters via a Pd(OAc)2-catalyzed C—C bond activation reaction of cyclopropenones with phenols (Scheme 1b). Moreover, Liu and Zhou et al.[16] developed an innovative copper(II) halide-mediated ring-opening dual halogenation reaction of cyclopropenones with saturated cyclic ethers, establishing a streamlined pathway to construct 3-halo- acrylates (Scheme 1c). Soon afterwards, Zhou and Wu et al.[17] also established a Pd-catalyzed selective ring-open- ing protocol of cyclopropenones with vinyl epoxides to afford conjugated alkadienyl carboxylates (Scheme 1d). While these reports predominantly focus on transition- metal catalysis, the activation of cyclopropenones through organocatalytic methods remains relatively underdeveloped. Phosphine has been definitively verified as an efficient organic catalyst for initiating diverse reactions.[18] Specifically, the phosphine-catalyzed C—C bond activation of cyclopropenones could generate an intriguing α- ketenyl phosphorus ylide species, which can be transformed into numerous functionalized molecules.[19] Herein, we disclosed a novel PPh3-catalyzed ring-opening addition reaction of cyclopropenones with alkyl bromides in the presence of H2O in dimethyl sulfoxide (DMSO) at room temperature, offering a concise approach to forge α,β-disu- bstituted acrylates with exclusive E-stereoselectivity under transition-metal-free conditions (Scheme 1e).
2 Results and discussion
The initial investigation was carried out by employing 2,3-diphenylcyclopropenone (1a) and 1-bromobutane (2a) as model substrates for reaction condition optimization. When reaction was performed using 30 mol% PPh3 as a catalyst and 2.0 equiv. of K2CO3 as a base in CH3CN at 100 ℃ for 0.5 h, butyl (E)-2,3-diphenylacrylate (3a) was produced in 76% yield with exclusive stereoselectivity. Notably, H2O in the environment also participated in this reaction (Table 1, Entry 1). This outcome spurred extensive research on various reaction parameters to further improve the yield. Several commonly used bases, such as Cs2CO3, CsOAc, NaOH, tBuOK, Et3N, pyridine, and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), were systematically evaluated to determine their impact on this reaction. Nevertheless, all these bases exhibited relatively lower capacity compared with K2CO3 (Table 1, Entries 2~8). Subsequent screening of solvents revealed that DMSO was more favorable (Table 1, Entries 9~15). The regulation of the temperature gradient demonstrated that conducting the reaction at room temperature gave better result (Table 1, Entries 16 and 17). Finally, the influence of the equivalent of the PPh3 catalyst was assessed. Product 3a was not detected in the absence of PPh3, while decreasing the amount of PPh3 to 10 mol% delivered 3a in the highest yield (Table 1, Entries 18~20).
Table 1 Optimization of reaction conditionsa |
| Entry | Base | Solvent | Temp./℃ | Yieldb/% |
|---|---|---|---|---|
| 1 | K2CO3 | CH3CN | 100 | 76 |
| 2 | Cs2CO3 | CH3CN | 100 | 72 |
| 3 | CsOAc | CH3CN | 100 | 64 |
| 4 | NaOH | CH3CN | 100 | 42 |
| 5 | tBuOK | CH3CN | 100 | 12 |
| 6 | Et3N | CH3CN | 100 | 11 |
| 7 | Pyridine | CH3CN | 100 | Trace |
| 8 | DBU | CH3CN | 100 | 10 |
| 9 | K2CO3 | DMSO | 100 | 82 |
| 10 | K2CO3 | DMF | 100 | 73 |
| 11 | K2CO3 | Toluene | 100 | Trace |
| 12 | K2CO3 | Dioxane | 100 | 25 |
| 13 | K2CO3 | THF | 100 | 31 |
| 14 | K2CO3 | DCM | 100 | Trace |
| 15 | K2CO3 | Acetone | 100 | 37 |
| 16 | K2CO3 | DMSO | 60 | 88 |
| 17 | K2CO3 | DMSO | 25 | 91 |
| 18c | K2CO3 | DMSO | 25 | Trace |
| 19d | K2CO3 | DMSO | 25 | 88 |
| 20e | K2CO3 | DMSO | 25 | 93 |
a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), PPh3 (0.06 mmol) and base (0.4 mmol) were reacted in 3 mL of solvent for 0.5 h under air; b Isolated yields; c Without PPh3; d PPh3 (0.04 mmol); e PPh3 (0.02 mmol). |
After determining the optimal conditions, the substrate scope of alkyl bromides for this transformation was explored (Table 2). Reaction of different primary bromoalkanes with 2,3-diphenylcyclopropenone (1a) delivered the desired products 3a~3i with high efficiency (80%~95%). Moreover, the configuration of 3h was unambiguously verified through X-ray single crystal diffraction (CCDC: 2449462). Substrates bearing vinyl, cyano and ester groups demonstrated good compatibility, affording products 3j~3l in satisfactory yields (71%~78%). However, 2-bromo-1- (4-methoxyphenyl)ethan-1-one only delivered 3m in 32% yield. 1-Bromo-2-chloroethane and 1-bromo-4-chlorobu- tane gave rise to products 3n and 3o in 81% and 79% yields, respectively, through the selective C—Br bond dissociation. Notably, it was found that the reaction furnished symmetrical methylene (2E,2'E)-bis(2,3-diphenylacrylate) (3p) in 87% yield from dibromomethane. Several secondary bromoalkanes were also proved to be compatible, and the corresponding products 3q~3v were obtained in moderate to good yields (45%~89%). In addition, tert-butyl bromide was amenable, affording 3w in 43% yield. However, the conversion of 1-bromoadamantane to 3x was unsuccessful, probably due to its high steric hindrance.
Table 2 Substrate scope of alkyl bromidesa |
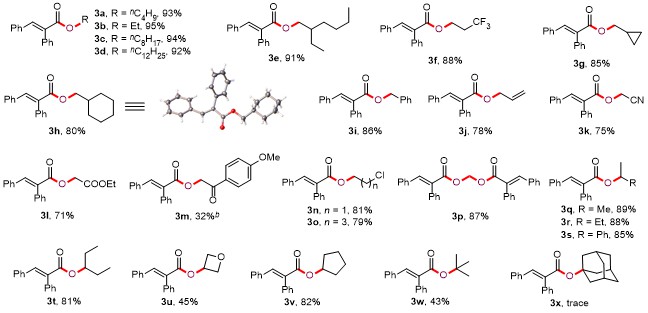 |
a Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), PPh3 (0.02 mmol), K2CO3 (0.4 mmol), in DMSO (3 mL) at room temperature for 0.5 h under air. b CsOAc (0.4 mmol) instead of K2CO3, 100 ℃ for 3 h; isolated yields. |
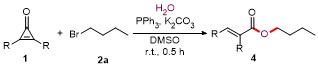
We then proceeded to evaluate the scope regarding cyclopropenones (Table 3). The results revealed that the substituents on the benzene ring exert negligible influence on the reaction, and the anticipated products were successfully achieved from substrates bearing both the electron-donating (Me, tBu) and the electron-withdrawing groups (F, Cl, Br) in good to excellent yields (4a~4i, 74%~89%). Unfortunately, when 2,3-diethylcycloprop-2-en-1-one was employed as the substrate, the desired product 4j was not isolated. Unsymmetrical 2-phenyl-3-(p-tolyl)cycloprop-2-en- 1-one led to the formation of regioisomers 4k and 4k' (1∶1 ratio) in 81% yield, whereas 2-methyl-3-phenylcycloprop- 2-en-1-one produced a single regioisomer 4l with a yield of 45%.
Table 3 Substrate scope of cyclopropenonesa |
 |
a Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol), PPh3 (0.02 mmol), K2CO3 (0.4 mmol), in DMSO (3 mL) at room temperature for 0.5 h under air; isolated yields. |
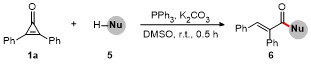
To further broaden the scope of this PPh3-catalyzed ring- opening addition reaction, the reactions of 2,3-diphenyl- cyclopropenone (1a) with other nucleophiles 5 were explored. As shown in Table 4, different types of nucleophiles, including cyclohexylamine, aniline, phenol, and p- toluenethiol, could all be transformed into the corresponding products in good yields (6a~6d, 76~82%).
Table 4 Substrate scope of cyclopropenonesa |
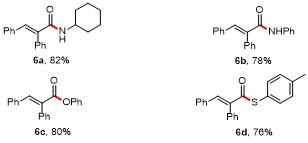 |
a Reaction conditions: 1a (0.2 mmol), 5 (0.2 mmol), PPh3 (0.02 mmol), K2CO3 (0.4 mmol), in DMSO (3 mL) at room temperature for 0.5 h; isolated yields. |
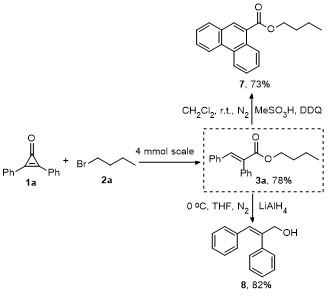
To validate the practicability and application potential of our methodology, 2,3-diphenylcyclopropenone (1a) and 1-bromobutane (2a) were subjected to the identified reaction conditions on a 4 mmol scale, and product 3a was readily achieved in 78% isolated yield. Subsequent late- stage modification of 3a was also implemented. 2,3-Di- chloro-5,6-dicyano-1,4-benzoquinone (DDQ)/MeSO3H mediated oxidative dehydrocyclization of 3a provided butyl phenanthrene-9-carboxylate (7) in 73% yield. Besides, the ester group of 3a can be selectivity reduced to deliver (E)-2,3-diphenylprop-2-en-1-ol (8) in 82% yield in the presence of LiAlH4 (Scheme 2).
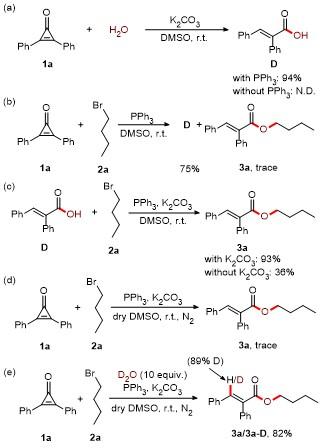
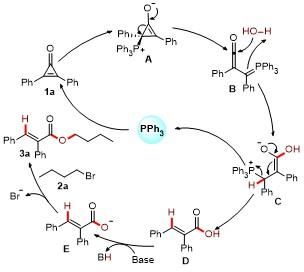
Several control experiments were conducted to comprehensively elucidate the mechanism. The reaction of 2,3- diphenylcyclopropenone (1a) with H2O in DMSO produced (E)-2,3-diphenylacrylic acid (D) in 94% yield under the standard reaction conditions, while in the absence of the PPh3 catalyst, D was not detected (Scheme 3a). The above experimental results clearly showed that PPh3 plays a crucial role in the catalytic ring-opening of cyclopropenones. In the absence of K2CO3, the reaction between 1a and 1-bromobutane (2a) did not produce butyl (E)-2,3-di- phenylacrylate (3a). Instead, compound D was isolated with a 75% yield. When D reacted with 2a under the standard conditions, product 3a was obtained in 93% yield. Without using K2CO3 as a base, the reaction of D with 2a only gave 3a in 36% yield (Schemes 3b and 3c). These findings suggested that (E)-2,3-diphenylacrylic acid (D) served as a crucial intermediate in the transformation, and the subsequent nucleophilic substitution necessitated the promotion of K2CO3. Only trace mount of product 3a was detected when 1a and 2a were reacted in dry DMSO under N2 (Scheme 3d). Furthermore, additional 10 equiv. of D2O was added to the reaction using dry DMSO as the solvent, and a mixture of 3a/3a-D was isolated in 82% yield with a ratio of 1∶8 (Scheme 3e). These observations indicated that the hydrogen atom on the vinyl group and one oxygen atom on the ester group of 3a derive from H2O in the reaction system.
Based on the experimental facts and relevant literature reports,[19-20] a plausible reaction mechanism was proposed (Scheme 4). First, PPh3 undergoes 1,4-addition reaction with 2,3-diphenylcyclopropenone (1a), giving intermediate A. Ring strain-driven C—C bond cleavage of A generates the α-ketenyl phosphorus ylide B, which is promptly intercepted by H2O via intermolecular nucleophilic addition to form intermediate C. Subsequently, the phosphine catalyst elimination from C deliveres pivotal intermediate (E)-2,3-diphenylacrylic acid (D), and regenerates PPh3 to participate in the next catalytic cycle. D is converted into carboxylate ion E under the basic conditions. Finally, a base promotes nucleophilic substitution reaction of E with 1- bromobutane (2a) will furnish product butyl (E)-2,3-di- phenylacrylate (3a) as desired.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In summary, an efficient PPh3-catalyzed ring-opening addition reaction of cyclopropenones and alkyl bromides for stereoselective construction of α,β-disubstituted acrylates at room temperature was established. Mechanistic investigation revealed that the reaction sequences involved ring strain-driven cleavage of C—C bond to generate the α-ketenyl phosphorus ylide, and the hydrogen atom on vinyl group and one oxygen atom on ester group of products were found to originate from H2O in solvent DMSO. The mild conditions, excellent compatibility with diverse functional groups, simple operation procedure, and ready availability of feedstocks render this reaction highly competitive with currently known methods. Further exploration of cyclopropenone-based ring-opening reactions to construct diverse captivating scaffolds is under investigation in our laboratory.
4 Experimental section
4.1 General experimental information
Unless specified otherwise, all chemical reagents and solvents were purchased from commercial suppliers and used directly without purification. Cyclopropenones were prepared following the reported protocols.[13] Reaction progress was monitored by thin-layer chromatographic (TLC) analysis using precoated glass plates under UV light irradiation (254 or 365 nm). Purification of the products was carried out through column chromatography using 200~300 mesh silica gel. 1H NMR spectra were recorded on a Bruker Avance II 300 MHz instrument using CDCl3 as the solvent. 13C NMR spectra were acquired in CDCl3 on a 75 MHz instrument. Melting points were measured on an XT-4 digital melting point apparatus and not corrected. High- resolution mass spectrometry (HRMS) data were acquired on an Agilent QTOF 6540 MS or a Thermo Scientific LTQ Orbitrap XL, equipped with an electrospray source. The X-ray crystallographic analysis was conducted with a Bruker SMART APEX CCD diffractometer.
4.2 Experimental procedures
4.2.1 Procedure for the synthesis of products 3 and 4
A mixture of cyclopropenones 1 (0.2 mmol), alkyl bromides 2 (0.3 mmol), PPh3 (5.2 mg, 0.02 mmol), K2CO3 (55.3 mg, 0.4 mmol), and DMSO (3.0 mL) was stirred at room temperature under air for 0.5 h. Following complete consumption of starting materials (verified by TLC analysis), the reaction underwent extraction using EtOAc (30 mL×2), followed by sequential washing with saturated NaCl solution (30 mL). The collected organic phase was dried over anhydrous Na2SO4 and evaporated under vacuum. Final purification of the residue through silica gel column chromatography (petroleum ether/ethyl acetate, V∶V 10∶1~15∶1) afforded products 3 and 4.
Butyl (E)-2,3-diphenylacrylate (3a): Yellow oil (52.1 mg, 93% yield). 1H NMR (300 MHz, CDCl3) δ: 7.83 (s, 1H), 7.36~7.33 (m, 3H), 7.24~7.11 (m, 5H), 7.04 (d, J=7.5 Hz, 2H), 4.20 (t, J=6.6 Hz, 2H), 1.68~1.59 (m, 2H), 1.41~1.32 (m, 2H), 0.91 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.8, 140.0, 135.9, 134.7, 132.8, 130.5, 129.7, 128.9, 128.5, 128.1, 127.7, 65.0, 30.6, 19.1, 13.7; HRMS (ESI) calcd for C19H21O2 [M+H]+ 281.1536, found 281.1532.
Ethyl (E)-2,3-diphenylacrylate (3b): Yellow oil (47.9 mg, 95% yield). 1H NMR (300 MHz, CDCl3) δ: 7.84 (s, 1H), 7.36~7.34 (m, 3H), 7.23~7.21 (m, 2H), 7.16 (t, J=6.6 Hz, 3H), 7.05~7.04 (m, 2H), 4.28 (q, J=6.9 Hz, 2H), 1.31 (t, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.7, 140.0, 135.8, 134.6, 132.7, 130.5, 129.7, 128.9, 128.5, 128.1, 127.7, 61.1, 14.2; HRMS (ESI) calcd for C17H17O2 [M+H]+ 253.1223, found 253.1219.
Octyl (E)-2,3-diphenylacrylate (3c): Yellow oil (63.3 mg, 94% yield). 1H NMR (300 MHz, CDCl3) δ: 7.83 (s, 1H), 7.37~7.32 (m, 3H), 7.23~7.10 (m, 5H), 7.04 (d, J=6.6 Hz, 2H), 4.19 (t, J=6.6 Hz, 2H), 1.66~1.59 (m, 2H), 1.28~1.26 (m, 10H), 0.88 (t, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.8, 140.0, 135.9, 134.6, 132.8, 130.5, 129.7, 128.9, 128.5, 128.1, 127.6, 65.3, 31.7, 29.12, 29.08, 28.5, 25.8, 22.6, 14.1; HRMS (ESI) calcd for C23H29O2 [M+H]+ 337.2162, found 337.2157.
Dodecyl (E)-2,3-diphenylacrylate (3d): White solid (72.2 mg, 92% yield). m.p. 31~32 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.83 (s, 1H), 7.37~7.33 (m, 3H), 7.24~7.11 (m, 5H), 7.04 (d, J=6.6 Hz, 2H), 4.19 (t, J=6.9 Hz, 2H), 1.66~1.61 (m, 2H), 1.26~1.25 (m, 18H), 0.88 (t, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.8, 140.0, 135.9, 134.7, 132.9, 130.5, 129.7, 128.9, 128.5, 128.1, 127.7, 65.3, 31.9, 29.63, 29.62, 29.52, 29.49, 29.3, 29.2, 28.6, 25.9, 22.7, 14.1; HRMS (ESI) calcd for C27H37O2 [M+H]+ 393.2788, found 393.2782.
2-Ethylhexyl (E)-2,3-diphenylacrylate (3e): Yellow oil (61.2 mg, 91% yield). 1H NMR (300 MHz, CDCl3) δ: 7.83 (s, 1H), 7.38~7.33 (m, 3H), 7.25~7.12 (m, 5H), 7.06 (d, J=6.9 Hz, 2H), 4.11 (d, J=5.7 Hz, 2H), 1.59~1.54 (m, 1H), 1.30~1.23 (m, 8H), 0.89~0.82 (m, 6H); 13C NMR (75 MHz, CDCl3) δ: 167.9, 139.9, 136.0, 134.7, 132.9, 130.6, 129.6, 128.9, 128.5, 128.1, 127.6, 67.5, 38.8, 30.5, 28.8, 23.9, 22.9, 14.0, 11.0; HRMS (ESI) calcd for C23H29- O2 [M+H]+ 337.2162, found 337.2161.
3,3,3-Trifluoropropyl (E)-2,3-diphenylacrylate (3f): Yellow oil (56.4 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ: 7.86 (s, 1H), 7.37~7.35 (m, 3H), 7.24~7.13 (m, 5H), 7.04 (d, J=6.9 Hz, 2H), 4.42 (t, J=6.6 Hz, 2H), 2.57~2.42 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 167.3, 141.1, 135.4, 134.4, 131.9, 130.7, 129.7, 129.2, 128.6, 128.2, 127.9, 125.8 (q, 1JC-F=274.5 Hz), 57.8 (q, 3JC-F=3.8 Hz), 33.3 (q, 2JC-F=29.3 Hz); HRMS (ESI) calcd for C18H16F3O2 [M+H]+ 321.1097, found 321.1091.
Cyclopropylmethyl (E)-2,3-diphenylacrylate (3g): Yellow solid (47.3 mg, 85% yield). m.p. 40~41 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.86 (s, 1H), 7.33~7.31 (m, 3H), 7.23~7.21 (m, 2H), 7.15~7.08 (m, 3H), 7.03 (d, J=7.2 Hz, 2H), 4.04 (d, J=6.9 Hz, 2H), 1.22~1.09 (m, 1H), 0.55~0.49 (m, 2H), 0.29~0.25 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 167.6, 139.9, 135.8, 134.5, 132.7, 130.4, 129.6, 128.8, 128.4, 128.0, 127.6, 69.5, 9.8, 3.1; HRMS (ESI) calcd for C19H19O2 [M+H]+ 279.1380, found 279.1374.
Cyclohexylmethyl (E)-2,3-diphenylacrylate (3h): Yellow solid (51.3 mg, 80% yield). m.p. 73~74 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.74 (s, 1H), 7.27~7.23 (m, 3H), 7.13~7.11 (m, 2H), 7.08~7.00 (m, 3H), 6.96 (d, J=7.2 Hz, 2H), 3.92 (d, J=6.0 Hz, 2H), 1.61~1.58 (m, 6H), 1.17~1.00 (m, 3H), 0.90~0.79 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 167.7, 139.9, 135.9, 134.6, 132.8, 130.5, 129.6, 128.8, 128.4, 128.0, 127.6, 70.2, 37.0, 29.5, 26.3, 25.6; HRMS (ESI) calcd for C22H25O2 [M+H]+ 321.1849, found 321.1849.
Benzyl (E)-2,3-diphenylacrylate (3i): Yellow solid (54.1 mg, 86% yield). m.p. 78~79 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.87 (s, 1H), 7.36~7.32 (m, 8H), 7.24~7.21 (m, 2H), 7.18~7.13 (m, 3H), 7.03 (d, J=6.9 Hz, 2H), 5.25 (s, 2H); 13C NMR (75 MHz, CDCl3) δ: 167.5, 140.6, 136.1, 135.7, 134.5, 132.4, 130.6, 129.7, 129.0, 128.6, 128.4, 128.1, 127.9, 127.8, 127.7, 66.7; HRMS (ESI) calcd for C22H19O2 [M+H]+ 315.1380, found 315.1378.
Allyl (E)-2,3-diphenylacrylate (3j): Yellow oil (41.2 mg, 78% yield). 1H NMR (300 MHz, CDCl3) δ: 7.87 (s, 1H), 7.36~7.35 (m, 3H), 7.24~7.13 (m, 5H), 7.05 (d, J=6.6 Hz, 2H), 6.00~5.88 (m, 1H), 5.26 (d, J=17.4 Hz, 1H), 5.20 (d, J=10.8 Hz, 1H), 4.71 (d, J=5.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 167.4, 140.5, 135.7, 134.5, 132.4, 132.1, 130.6, 129.7, 129.0, 128.6, 128.2, 127.8, 117.7, 65.6; HRMS (ESI) calcd for C18H17O2 [M+H]+ 265.1223, found 265.1219.
Cyanomethyl (E)-2,3-diphenylacrylate (3k): White solid (39.5 mg, 75% yield). m.p. 84~85 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.92 (s, 1H), 7.39~7.36 (m, 3H), 7.23~7.20 (m, 3H), 7.15 (t, J=7.5 Hz, 2H), 7.04 (d, J=7.5 Hz, 2H), 4.77 (s, 2H); 13C NMR (75 MHz, CDCl3) δ: 166.0, 143.0, 134.6, 133.8, 130.8, 130.1, 129.7, 129.6, 128.8, 128.22, 128.19, 114.5, 48.8; HRMS (ESI) calcd for C17H14NO2 [M+H]+ 264.1019, found 264.1020.
2-Ethoxy-2-oxoethyl (E)-2,3-diphenylacrylate (3l): White solid (44.1 mg, 71% yield). m.p. 68~69 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.93 (s, 1H), 7.40~7.36 (m, 3H), 7.30~7.25 (m, 2H), 7.22~7.13 (m, 3H), 7.06 (d, J=6.9 Hz, 2H), 4.72 (s, 2H), 4.24 (q, J=7.2 Hz, 2H), 1.30 (t, J=7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.8, 167.2, 141.7, 135.4, 134.4, 131.5, 130.7, 129.8, 129.3, 128.6, 128.2, 127.9, 61.4, 61.3, 14.1; HRMS (ESI) calcd for C19H19O4 [M+H]+ 311.1278, found 311.1274.
2-(4-Methoxyphenyl)-2-oxoethyl (E)-2,3-diphenylacryl- ate (3m): White solid (23.8 mg, 32% yield). m.p. 131~132 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.97 (s, 1H), 7.91 (d, J=8.7 Hz, 2H), 7.41~7.33 (m, 5H), 7.26~7.14 (m, 3H), 7.08 (d, J=7.5 Hz, 2H), 6.95 (d, J=9.0 Hz, 2H), 5.41 (s, 2H), 3.88 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 190.6, 167.3, 163.9, 141.5, 135.5, 134.5, 131.7, 130.7, 130.1, 129.9, 129.1, 128.6, 128.2, 127.9, 127.2, 114.0, 66.5, 55.5; HRMS (ESI) calcd for C24H21O4 [M+H]+ 373.1434, found 373.1433.
2-Chloroethyl (E)-2,3-diphenylacrylate (3n): White solid (46.5 mg, 81% yield). m.p. 49~50 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.88 (s, 1H), 7.35~7.31 (m, 3H), 7.23~7.20 (m, 2H), 7.16~7.08 (m, 3H), 7.03 (d, J=6.9 Hz, 2H), 4.40 (t, J=5.7 Hz, 2H), 3.65 (t, J=5.7 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 167.1, 140.9, 135.3, 134.3, 131.8, 130.5, 129.6, 129.0, 128.4, 128.0, 127.7, 64.4, 41.4; HRMS (ESI) calcd for C17H16ClO2 [M+H]+ 287.0833, found 287.0833.
4-Chlorobutyl (E)-2,3-diphenylacrylate (3o): Yellow oil (49.7 mg, 79% yield). 1H NMR (300 MHz, CDCl3) δ: 7.84 (s, 1H), 7.37~7.33 (m, 3H), 7.22~7.19 (m, 2H), 7.16~7.10 (m, 3H), 7.04 (d, J=6.6 Hz, 2H), 4.22 (t, J=5.7 Hz, 2H), 3.47 (t, J=6.3 Hz, 2H), 1.84~1.74 (m, 4H); 13C NMR (75 MHz, CDCl3) δ: 167.6, 140.3, 135.8, 134.4, 132.4, 130.5, 129.5, 129.0, 128.5, 128.1, 127.7, 64.2, 44.3, 29.1, 25.9; HRMS (ESI) calcd for C19H20ClO2 [M+H]+ 315.1146, found 315.1144.
Methylene (2E,2'E)-bis(2,3-diphenylacrylate) (3p): White solid (40.1 mg, 87% yield). m.p. 155~156 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.83 (s, 2H), 7.30~7.28 (m, 6H), 7.17~7.06 (m, 10H), 6.98 (d, J=6.9 Hz, 4H), 5.94 (s, 2H); 13C NMR (75 MHz, CDCl3) δ: 166.3, 141.9, 135.1, 134.3, 131.4, 130.8, 129.8, 129.4, 128.7, 128.2, 128.0, 80.8; HRMS (ESI) calcd for C31H25O4 [M+H]+ 461.1747, found 461.1743.
Isopropyl (E)-2,3-diphenylacrylate (3q): White solid (47.4 mg, 89% yield). m.p. 57~58 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.80 (s, 1H), 7.35~7.32 (m, 3H), 7.22~7.19 (m, 2H), 7.17~7.09 (m, 3H), 7.03 (d, J=6.9 Hz, 2H), 5.18~5.09 (m, 1H), 1.27 (d, J=6.3 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ: 167.2, 139.6, 135.9, 134.7, 133.2, 130.4, 129.7, 128.7, 128.4, 128.1, 127.6, 68.5, 21.8; HRMS (ESI) calcd for C18H19O2 [M+H]+ 267.1380, found 267.1378.
sec-Butyl (E)-2,3-diphenylacrylate (3r): Yellow oil (49.3 mg, 88% yield). 1H NMR (300 MHz, CDCl3) δ: 7.82 (s, 1H), 7.35~7.30 (m, 3H), 7.22~7.19 (m, 2H), 7.16~7.09 (m, 3H), 7.04 (d, J=6.9 Hz, 2H), 5.02~4.92 (m, 1H), 1.63~1.52 (m, 2H), 1.24 (d, J=6.3 Hz, 3H), 0.87 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.3, 139.6, 136.0, 134.7, 133.2, 130.4, 129.7, 128.7, 128.4, 128.0, 127.5, 73.0, 28.7, 19.3, 9.6; HRMS (ESI) calcd for C19H21O2 [M+H]+ 281.1536, found 281.1534.
1-Phenylethyl (E)-2,3-diphenylacrylate (3s): Yellow oil (55.8 mg, 85% yield). 1H NMR (300 MHz, CDCl3) δ: 7.87 (s, 1H), 7.33~7.19 (m, 10H), 7.13~7.06 (m, 3H), 7.03 (d, J=7.5 Hz, 2H), 6.01 (q, J=6.6 Hz, 1H), 1.52 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 166.7, 141.7, 140.1, 135.8, 134.5, 132.7, 130.4, 129.6, 128.8, 128.4, 128.3, 128.0, 127.6, 127.5, 125.7, 72.9, 22.5; HRMS (ESI) calcd for C23H21O2 [M+H]+ 329.1536, found 329.1532.
Pentan-3-yl (E)-2,3-diphenylacrylate (3t): Yellow oil (47.7 mg, 81% yield). 1H NMR (300 MHz, CDCl3) δ: 7.83 (s, 1H), 7.36~7.32 (m, 3H), 7.23~7.19 (m, 2H), 7.17~7.09 (m, 3H), 7.04 (d, J=7.8 Hz, 2H), 4.94~4.88 (m, 1H), 1.63~1.53 (m, 4H), 0.87 (t, J=7.5 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ: 167.6, 139.5, 136.1, 134.7, 133.2, 130.5, 129.6, 128.8, 128.4, 128.1, 127.5, 77.4, 26.3, 9.5; HRMS (ESI) calcd for C20H23O2 [M+H]+ 295.1693, found 295.1686.
Oxetan-3-yl (E)-2,3-diphenylacrylate (3u): White solid (25.2 mg, 45% yield). m.p. 86~87 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.89 (s, 1H), 7.32~7.30 (m, 3H), 7.22~7.20 (m, 2H), 7.15~7.08 (m, 3H), 7.04 (d, J=6.9 Hz, 2H), 5.51~5.44 (m, 1H), 4.84 (t, J=6.9 Hz, 2H), 4.62 (t, J=6.6 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 166.5, 141.0, 135.0, 134.0, 131.3, 130.4, 129.4, 129.0, 128.4, 127.9, 127.7, 77.1, 68.2; HRMS (ESI) calcd for C18H17O3 [M+H]+ 281.1172, found 281.1169.
Cyclopentyl (E)-2,3-diphenylacrylate (3v): White solid (48.0 mg, 82% yield). m.p. 60~61 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.78 (s, 1H), 7.35~7.31 (m, 3H), 7.21~7.10 (m, 5H), 7.04 (d, J=7.2 Hz, 2H), 5.31~5.26 (m, 1H), 1.92~1.83 (m, 2H), 1.74~1.56 (m, 6H); 13C NMR (75 MHz, CDCl3) δ: 167.4, 139.6, 135.9, 134.7, 133.2, 130.4, 129.7, 128.8, 128.3, 128.1, 127.5, 77.8, 32.6, 23.6; HRMS (ESI) calcd for C20H21O2 [M+H]+ 293.1536, found 293.1534.
tert-Butyl (E)-2,3-diphenylacrylate (3w): Yellow oil (24.1 mg, 43% yield). 1H NMR (300 MHz, CDCl3) δ: 7.73 (s, 1H), 7.38~7.32 (m, 3H), 7.21~7.12 (m, 5H), 7.03 (d, J=6.6 Hz, 2H), 1.50 (s, 9H); 13C NMR (75 MHz, CDCl3) δ: 166.9, 139.0, 136.2, 134.9, 134.3, 130.4, 129.8, 128.7, 128.4, 128.1, 127.5, 81.0, 28.1; HRMS (ESI) calcd for C19H21O2 [M+H]+ 281.1536, found 281.1532.
Butyl (E)-2,3-di-m-tolylacrylate (4a): Yellow oil (54.9 mg, 89% yield). 1H NMR (300 MHz, CDCl3) δ: 7.77 (s, 1H), 7.24 (t, J=7.5 Hz, 1H), 7.14 (d, J=7.2 Hz, 1H), 7.03~6.98 (m, 4H), 6.90 (s, 1H), 6.81 (s, 1H), 4.20 (t, J=6.6 Hz, 2H), 2.33 (s, 3H), 2.19 (s, 3H), 1.69~1.60 (m, 2H), 1.43~1.31 (m, 2H), 0.92 (t, J=7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 168.0, 139.9, 138.0, 137.6, 135.9, 134.6, 132.7, 131.6, 130.2, 129.7, 128.4, 128.3, 127.9, 127.5, 126.7, 64.9, 30.6, 21.4, 21.2, 19.2, 13.7; HRMS (ESI) calcd for C21H25O2 [M+H]+ 309.1849, found 309.1846.
Butyl (E)-2,3-di-p-tolylacrylate (4b): Yellow oil (46.9 mg, 76% yield). 1H NMR (300 MHz, CDCl3) δ: 7.78 (s, 1H), 7.17~7.08 (m, 4H), 6.96 (s, 4H), 4.19 (t, J=6.9 Hz, 2H), 2.38 (s, 3H), 2.26 (s, 3H), 1.68~1.59 (m, 2H), 1.38~1.32 (m, 2H), 0.92 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 168.1, 139.8, 139.1, 137.2, 133.1, 132.0, 131.8, 130.5, 129.6, 129.2, 128.9, 64.9, 30.7, 21.33, 21.28, 19.1, 13.7; HRMS (ESI) calcd for C21H25O2 [M+H]+ 309.1849, found 309.1845.
Butyl (E)-2,3-bis(4-(tert-butyl)phenyl)acrylate (4c): Yellow oil (63.6 mg, 81% yield). 1H NMR (300 MHz, CDCl3) δ: 7.77 (s, 1H), 7.39 (d, J=8.4 Hz, 2H), 7.18~7.14 (m, 4H), 7.01 (d, J=8.4 Hz, 2H), 4.19 (t, J=6.6 Hz, 2H), 1.68~1.59 (m, 2H), 1.36 (s, 9H), 1.34~1.32 (m, 2H), 1.25 (s, 9H), 0.90 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 168.2, 152.3, 150.5, 139.6, 133.1, 131.9, 131.7, 130.5, 129.2, 125.4, 125.1, 64.9, 34.7, 34.6, 31.4, 31.1, 30.7, 19.2, 13.7; HRMS (ESI) calcd for C27H37O2 [M+H]+ 393.2788, found 393.2783.
Butyl (E)-2,3-bis(2-fluorophenyl)acrylate (4d): Yellow oil (46.8 mg, 74% yield). 1H NMR (300 MHz, CDCl3) δ: 8.01 (s, 1H), 7.20~7.13 (m, 1H), 7.07~7.00 (m, 1H), 6.97~6.94 (m, 2H), 6.90 (d, J=7.5 Hz, 1H), 6.84 (d, J=8.7 Hz, 1H), 6.70~6.62 (m, 2H), 4.09 (t, J=6.6 Hz, 2H), 1.54~1.45 (m, 2H), 1.28~1.16 (m, 2H), 0.77 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 166.3, 160.9 (d, 1JC-F=250.5 Hz), 160.1 (d, 1JC-F=245.3 Hz), 133.7 (d, 3JC-F=5.3 Hz), 131.3 (d, 4JC-F=3.0 Hz), 130.8 (d, 3JC-F=9.0 Hz), 129.8 (d, 2JC-F=27.8 Hz), 129.7 (d, 2JC-F=21.0 Hz), 128.8, 124.0 (d, 4JC-F=3.8 Hz), 123.5 (d, 4JC-F=3.8 Hz), 123.4 (d, 3JC-F=16.5 Hz), 122.4 (d, 3JC-F=12.0 Hz), 115.5 (d, 2JC-F=21.8 Hz), 115.4 (d, 2JC-F=21.8 Hz), 65.0, 30.4, 18.9, 13.4; HRMS (ESI) calcd for C19H19F2O2 [M+H]+ 317.1348, found 317.1344.
Butyl (E)-2,3-bis(3-chlorophenyl)acrylate (4e): Yellow oil (59.4 mg, 85% yield). 1H NMR (300 MHz, CDCl3) δ: 7.77 (s, 1H), 7.31~7.23 (m, 4H), 7.14 (d, J=8.1 Hz, 1H), 7.06 (d, J=6.9 Hz, 2H), 6.87 (d, J=7.8 Hz, 1H), 4.20 (t, J=6.6 Hz, 2H), 1.67~1.58 (m, 2H), 1.41~1.29 (m, 2H), 0.90 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 166.5, 138.9, 136.9, 135.8, 134.2, 134.0, 132.6, 130.2, 129.6, 129.5, 129.3, 128.9, 128.1, 127.9, 127.7, 65.1, 30.4, 18.9, 13.5; HRMS (ESI) calcd for C19H19Cl2O2 [M+H]+ 349.0757, found 349.0755.
Butyl (E)-2,3-bis(3-bromophenyl)acrylate (4f): Yellow oil (71.9 mg, 82% yield). 1H NMR (300 MHz, CDCl3) δ: 7.75 (s, 1H), 7.47 (d, J=7.5 Hz, 1H), 7.38 (s, 1H), 7.32 (d, J=7.2 Hz, 1H), 7.23 (s, 1H), 7.20 (d, J=7.8 Hz, 1H), 7.11 (d, J=7.5 Hz, 1H), 7.00 (t, J=7.8 Hz, 1H), 6.91 (d, J=7.5 Hz, 1H), 4.21 (t, J=6.6 Hz, 2H), 1.68~1.59 (m, 2H), 1.42~1.30 (m, 2H), 0.91 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 166.6, 138.9, 137.2, 136.1, 133.3, 132.6, 132.4, 131.9, 131.0, 130.0, 129.6, 128.5, 128.2, 122.4, 122.2, 65.2, 30.4, 19.0, 13.6; HRMS (ESI) calcd for C19H19Br2O2 [M+H]+ 436.9746, found 436.9746.
Butyl (E)-2,3-bis(4-fluorophenyl)acrylate (4g): Yellow oil (55.1 mg, 87% yield). 1H NMR (300 MHz, CDCl3) δ: 7.81 (s, 1H), 7.19~7.15 (m, 2H), 7.05~7.00 (m, 2H), 6.81 (t, J=8.7 Hz, 2H), 4.20 (t, J=6.6 Hz, 2H), 1.67~1.58 (m, 2H), 1.41~1.29 (m, 2H), 0.90 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.5, 162.8 (d, 1JC-F=249.8 Hz), 162.4 (d, 1JC-F=245.3 Hz), 139.2, 132.4 (d, 3JC-F=8.3 Hz), 131.55, 131.53 (d, 3JC-F=7.5 Hz), 130.7 (d, 4JC-F=3.0 Hz), 115.7 (d, 2JC-F=24.0 Hz), 115.4 (d, 2JC-F=24.8 Hz), 65.2, 30.6, 19.2, 13.7; HRMS (ESI) calcd for C19H19F2O2 [M+H]+ 317.1348, found 317.1342.
Butyl (E)-2,3-bis(4-chlorophenyl)acrylate (4h): Yellow oil (57.9 mg, 83% yield). 1H NMR (300 MHz, CDCl3) δ: 7.68 (s, 1H), 7.21 (d, J=8.4 Hz, 2H), 7.03~7.00 (m, 4H), 6.85 (t, J=8.4 Hz, 2H), 4.09 (t, J=6.6 Hz, 2H), 1.57~1.47 (m, 2H), 1.30~1.20 (m, 2H), 0.80 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 166.9, 139.0, 134.9, 133.8, 133.7, 132.7, 132.0, 131.5, 131.0, 128.7, 128.4, 65.1, 30.4, 19.0, 13.5; HRMS (ESI) calcd for C19H19Cl2O2 [M+H]+ 349.0757, found 349.0755.
Butyl (E)-2,3-bis(4-bromophenyl)acrylate (4i): Yellow oil (72.7 mg, 83% yield). 1H NMR (300 MHz, CDCl3) δ: 7.76 (s, 1H), 7.49 (d, J=8.4 Hz, 2H), 7.32 (d, J=8.4 Hz, 2H), 7.07 (d, J=8.4 Hz, 2H), 6.91 (d, J=8.4 Hz, 2H), 4.20 (t, J=6.6 Hz, 2H), 1.68~1.59 (m, 2H), 1.42~1.29 (m, 2H), 0.92 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.1, 139.2, 134.4, 133.2, 132.3, 131.84, 131.82, 131.6, 131.4, 123.5, 122.2, 65.3, 30.6, 19.1, 13.7; HRMS (ESI) calcd for C19H19Br2O2 [M+H]+ 436.9746, found 436.9747.
Butyl (E)-3-phenyl-2-(p-tolyl)acrylate and butyl (E)-2- phenyl-3-(p-tolyl)acrylate (4k+4k'): Yellow oil (47.7 mg, 81% yield). 1H NMR (300 MHz, CDCl3) δ: 7.80 (s, 1H), 7.79 (s, 1H), 7.36~7.33 (m, 2H), 7.25~7.06 (m, 12H), 6.97~6.91 (m, 4H), 4.22~4.17 (m, 4H), 2.37 (s, 3H), 2.26 (s, 3H), 1.67~1.60 (m, 4H), 1.43~1.33 (m, 4H), 0.95~0.88 (m, 6H); 13C NMR (75 MHz, CDCl3) δ: 168.03, 167.96, 140.1, 139.7, 139.2, 137.4, 136.2, 134.9, 132.84, 132.81, 131.8, 130.6, 130.5, 129.7, 129.6, 129.2, 128.9, 128.8, 128.5, 128.1, 127.6, 65.0, 64.9, 30.7, 21.32, 21.28, 19.1, 13.7; HRMS (ESI) calcd for C20H23O2 [M+H]+ 295.1693, found 295.1692.
Butyl (E)-2-phenylbut-2-enoate (4l): Yellow oil (19.7 mg, 45% yield). 1H NMR (300 MHz, CDCl3) δ: 7.69 (s, 1H), 7.40~7.31 (m, 5H), 4.22 (t, J=6.6 Hz, 2H), 2.12 (d, J=0.9 Hz, 3H), 1.76~1.66 (m, 2H), 1.49~1.41 (m, 2H), 0.97 (t, J=7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 168.7, 138.6, 136.0, 129.6, 128.7, 128.3, 128.2, 64.8, 30.8, 19.3, 14.0, 13.8; HRMS (ESI) calcd for C14H19O2 [M+H]+ 219.1380, found 219.1376.
4.2.2 Procedure for the synthesis of products 6
A mixture of 2,3-diphenylcyclopropenone (1a) (0.2 mmol), nucleophiles 5 (0.2 mmol), PPh3 (5.2 mg, 0.02 mmol) and K2CO3 (55.3 mg, 0.4 mmol) was stirred in DMSO (3.0 mL) at room temperature for 0.5 h. Once the reaction was complete (monitored by TLC), the resulting mixture was extracted with EtOAc (30 mL×2) and washed with saturated NaCl solution (30 mL). The collected organic phase was then dried over anhydrous Na2SO4 and filtered. The solvent was removed under reduced pressure to give a residue, which was purified through silica gel column chromatography (petroleum ether/ethyl acetate, V∶V=10∶1) to afford products 6.
(E)-N-Cyclohexyl-2,3-diphenylacrylamide (6a): White solid (50.1 mg, 82% yield). m.p. 57~58 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.84 (s, 1H), 7.43~7.42 (m, 3H), 7.26~7.23 (m, 2H), 7.14~7.11 (m, 3H), 6.98 (d, J=6.9 Hz, 2H), 5.35 (d, J=6.9 Hz, 1H), 3.93~3.84 (m, 1H), 1.88-1.84 (m, 2H), 1.58~1.55 (m, 3H), 1.42~1.30 (m, 2H), 1.17~0.98 (m, 3H); 13C NMR (75 MHz, CDCl3) δ: 166.0, 136.7, 136.3, 135.0, 134.6, 130.2, 129.8, 129.5, 128.4, 128.3, 128.0, 48.4, 32.7, 25.4, 24.5; HRMS (ESI) calcd for C21H24NO [M+ H]+ 306.1852, found 306.1848.
(E)-N,2,3-Triphenylacrylamide (6b): White solid (46.7 mg, 78% yield). m.p. 142~143 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.97 (s, 1H), 7.50~7.43 (m, 5H), 7.36~7.30 (m, 4H), 7.25~7.14 (m, 4H), 7.09 (d, J=7.5 Hz, 1H), 7.03 (t, J=5.7 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ: 164.9, 138.1, 137.7, 135.7, 134.7, 134.6, 130.3, 129.9, 129.8, 128.8, 128.7, 128.1, 124.3, 119.8; HRMS (ESI) calcd for C21H18NO [M+H]+ 300.1383, found 300.1381.
Phenyl (E)-2,3-diphenylacrylate (6c): White solid (48.1 mg, 80% yield). m.p. 134~135 ℃; 1H NMR (300 MHz, CDCl3) δ: 8.04 (s, 1H), 7.38~7.30 (m, 7H), 7.22~7.07 (m, 8H); 13C NMR (75 MHz, CDCl3) δ: 166.3, 151.1, 141.9, 135.4, 134.3, 131.9, 130.7, 129.8, 129.33, 129.26, 128.7, 128.2, 128.0, 125.6, 121.5; HRMS (ESI) calcd for C21H17O2 [M+H]+ 301.1223, found 301.1222.
S-(p-Tolyl) (E)-2,3-diphenylprop-2-enethioate (6d): White solid (50.2 mg, 76% yield). m.p. 98~99 ℃; 1H NMR (300 MHz, CDCl3) δ: 7.73 (s, 1H), 7.45~7.42 (m, 3H), 7.38~7.35 (m, 2H), 7.29 (d, J=8.1 Hz, 2H), 7.21~7.11 (m, 5H), 7.04 (d, J=6.9 Hz, 2H), 2.35 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 192.0, 139.3, 138.4, 136.8, 135.0, 134.5, 134.1, 131.0, 130.7, 129.9, 129.4, 128.8, 128.7, 128.2, 125.1, 21.3; HRMS (ESI) calcd for C22H19OS [M+H]+ 331.1151, found 331.1150.
4.2.3 Gram-scale experiment
A 100 mL round-bottom flask was charged with 2,3-di- phenylcyclopropenone (1a, 825.0 mg, 4.0 mmol) and 1- bromobutane (2a, 822.1 mg, 6.0 mmol), PPh3 (104.9 mg, 0.4 mmol), K2CO3 (1.11 g, 8.0 mmol), and DMSO (50 mL). The reaction underwent continuous agitation at ambient temperature for 2 h. After complete consumption of starting materials (verified by TLC analysis), 100 mL of water was then added, and the mixture was extracted using EtOAc (80 mL×3). The combined organic phases underwent sequential washing with saturated NaCl solution (100 mL×2), dried over anhydrous Na2SO4, and filtered. The solution was concentrated under vacuum to give a crude residue, which underwent purification through silica gel column chromatography (petroleum ether/ethyl acetate, V∶V=10∶1) to afford the desired product 3a as a yellow oil (0.87 g, 78% yield).
4.2.4 Procedure for the synthesis of product 7
An oven-dried 10 mL vessel was charged with butyl (E)- 2,3-diphenylacrylate (3a, 56.0 mg, 0.2 mmol) and DDQ (54.5 mg, 0.24 mmol). After flushing the vessel with alternating vacuum and nitrogen purge cycles, a solution of anhydrous dichloromethane (DCM, 2.0 mL) and methane- sulfonic acid (0.2 mL) was added at 0 ℃, the reaction was maintained at ambient temperature with continuous agitation. On completion (monitored by TLC), the resulting mixture was neutralized using 10% NaHCO3 solution (20 mL) followed by extraction with EtOAc (30 mL×2). The combined organic phases were washed with saturated NaCl solution (30 mL), dried over anhydrous Na2SO4, and filtered. Concentration of the solution under reduced pressure gave a residue, which was purified through silica gel column chromatography (eluting with petroleum ether/ethyl acetate, V∶V=10∶1) to afford butyl phenanthrene-9- carboxylate (7). Yellow oil (40.6 mg, 73% yield). 1H NMR (300 MHz, CDCl3) δ: 8.79 (dd, J=7.2, 2.1 Hz, 1H), 8.48 (dd, J=7.2, 1.8 Hz, 1H), 8.42 (d, J=8.4 Hz, 1H), 8.27 (s, 1H), 7.75 (d, J=7.8 Hz, 1H), 7.54~7.45 (m, 3H), 7.41 (t, J=7.5 Hz, 1H), 4.30 (t, J=6.6 Hz, 2H), 1.73~1.64 (m, 2H), 1.45~1.32 (m, 2H), 0.87 (t, J=7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 167.6, 132.0, 131.9, 130.5, 129.9, 129.7, 129.0, 128.6, 127.2, 126.8, 126.7, 126.51, 126.45, 122.7, 122.5, 65.0, 30.8, 19.3, 13.7; HRMS (ESI) calcd for C19H19O2 [M+H]+ 279.1380, found 279.1382.
4.2.5 Procedure for the synthesis of product 8
An oven-dried 10 mL vessel was charged with butyl (E)- 2,3-diphenylacrylate (3a, 84.1 mg, 0.3 mmol). After flushing the vessel with alternating vacuum and nitrogen purge cycles, anhydrous tetrahydrofuran (THF, 2.0 mL) and LiAlH4 (0.18 mL, 2.5 mol/L in THF) were added under N2, and the resulting mixture was stirred at 0 ℃. After 1 h, the reaction was quenched with saturated NH4Cl (20 mL) and extracted with EtOAc (30 mL×2). The collected organic phases were dehydrated with anhydrous Na2SO4 and subsequently concentrated via rotary evaporation. Final purification of the crude residue through silica gel column chromatography (eluting with petroleum ether/ethyl acetate, V∶V=10∶1) afforded (E)-2,3-diphenylprop-2-en-1-ol (8). Yellow oil (51.5 mg, 82% yield). 1H NMR (300 MHz, CDCl3) δ: 7.34~7.28 (m, 3H), 7.23~7.20 (m, 2H), 7.13~7.09 (m, 3H), 7.00~6.97 (m, 2H), 6.68 (s, 1H), 4.44 (s, 2H), 1.82 (s, 1H); 13C NMR (75 MHz, CDCl3) δ: 141.4, 138.5, 136.4, 129.2, 128.8, 128.7, 127.9, 127.5, 126.8, 126.4, 68.5; HRMS (ESI) calcd for C15H13O [M-H]- 209.0972, found 209.0971.
Supporting Information 1H NMR and 13C NMR spectra of products 3, 4 and 6~8, and X-Ray crystal structure data of compound 3h. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Zhao, C.)