


自从1928年弗莱明发现青霉素以来, 抗生素经历了接近百年的发展历程, 从20世纪中后期的黄金发展时期, 到21世纪的缓慢发展时期[1]. 随着抗生素不断使用, 耐药性和毒性的问题日益突出, 世界卫生组织(WHO)预测到2030年将有1000万人死于耐药菌感染, 死亡人数将超过癌症; 联合国环境署公布, 到2030年微生物耐药将造成全球每年GDP损失3.4万亿美金. 上述突出的耐药菌感染问题不断催促新型抗生素的研发, 新型抗生素的开发主要基于经典抗生素的改造以及新结构抗生素的开发[2]. 抗生素(细菌)按类型主要分为β-内酰胺类和非β-内酰胺类, β-内酰胺类包括青霉素类、头孢类和碳青酶烯类, 占了整个抗生素的70%~80%, 而非β-内酰胺类包括四环素、大环内酯类、氨基糖苷类和糖肽类等. 抗真菌类药物包括唑类、两性霉素和棘白菌素等(表1). 为了解决天然抗生素的耐药、毒性及稳定性等不足, 化学半合成的抗生素被不断开发出来, 部分已经发展了多代的抗生素, 如头孢类、四环素类和大环内酯等. 随着多学科的交叉融合, 通过解析抗生素的作用机制、耐药机制以及毒性原理, 开发新一代的半合成药物[3], 如基于多黏菌素和两性霉素的结构优化, 可降低原有药物的毒性, 目前已有半合成药物进入临床研究, 有望推动新型抗生素的上市.
表1 抗菌、抗真菌药物分类Table 1 Classes of antibacterial and antifungal drugs |
| Classes of antibiotics | Natural (fermentation) | Semisynthetic | |
|---|---|---|---|
| Antibacterial drugs | Penicillins | Penicillin G | via semisynthesis from 6-APA |
| Cephalosporins | Cephalosporin C etc. | via semisynthesis from 7-ADCA, 7-ACA, D-7ACA | |
| Carbapenems | totally synthesized via 4-AA | ||
| Tetracyclines | Oxytetracycline, Tetracycline, Chlorotracycline | Doxycycline, Minocycline, Tigecycline, Omadacycline, Sareycline, Eravacycline (totally synthetic) | |
| Macrolides | Erythromycin, Tylosin | Clarithromycin, Azithromycin, Dirithromycin, Roxithromycin, Telithromycin, Tilmicosin, Tildipirosin, Tulathromycin | |
| Aminoglycosides | Streptomycin, Gentamicin, Apramycin, Neomycin | Plazomicin, Etimicin, Amikacin, Arbekacin | |
| Glycopeptides | Vancomycin, Teicoplanin | Telavancin, Oritavancin, Dalbavancin | |
| Lincosamides | Lincomycin | Clindamycin, Clindamycin Phosphate, Clindamycin Palmitate, Pirlimycin | |
| Pleuromutilin | Pleuromutilin | Tiamulin, Valnemulin, Retapamulin, Lefamulin | |
| Polymyxins | Polymyxin B, Colistin | Colistimethate Sodium, SPR206 (clinical trial), MRX-8 (clinical trial), QPX9003 (clinical trial, total trial) | |
| Other drugs such as quinolones (total synthesis) | |||
| Rifamycins | Rifampin, Rifapentine, Rifabutin, Rifaximin | ||
| Antifungal drugs | Azoles (totally synthetic) | ||
| Amphotericins | Amphotericin B | Am-2-19 (clinical trial) | |
| Echinocandins | Caspofungin, Micafungin, Anidulafungin, Rezafungin | ||
| Triterpenes | Ibrexafungerp | ||
| Other drugs |
由于天然抗生素可以通过生物发酵经济高效地获得, 当前大多数经过结构优化的新型抗生素通过化学半合成途径制备[4]. 由于天然产物中已存在关键骨架和手性中心, 且氧化态无需大幅调整, 因此大多数半合成抗生素的合成路线相对简短. 然而, 由于天然产物结构复杂、化学稳定性差、氧化态高以及官能团众多等特性, 基于复杂天然产物的半合成工艺仍面临诸多挑战: (1)化学和区域选择性大多依赖复杂的多步保护基操作; (2)氧化还原态的调整困难; (3)底物结构复杂, 导致可用的化学转化方法学严重受限; (4)分离纯化难度大, 如氨基糖苷类极性大, 导致分离困难.
我国在抗生素领域做出了重要的贡献, 抗生素原料药产能占全球60%以上, 掌控抗生素产业链上游的核心环节, 青霉素中间体中国产能占全球75%, 头孢中间体中国占据全球80%份额, 出口大量的四环素类、氯林可霉素及大环内脂类等原料药. 另外兽药抗生素也占了整个抗生素产能的半壁江山, 包括四环素类[土霉素、盐酸土霉素、盐酸多西环素(强力霉素)], 大环内酯类(硫氰酸红霉素、泰乐菌素、替米考星、泰万菌素)以及氨基糖苷类(硫酸链霉素、硫酸安普霉素、硫酸新霉素、硫酸庆大霉素), 每类抗生素的年产量从几千吨到上万吨不等, 价格在100~500元/公斤. 因此无论从成本控制还是环境影响等因素考虑, 开发绿色高效的半合成抗生素的工艺化学都是十分有意义的.
本综述将主要介绍半合成抗生素的工艺化学, 不包括喹诺酮和唑类等从头合成药物. 此外, 尽管β-内酰胺类药物是一类重要的半合成药物, 且发展较为成熟, 但本综述不对其进行讨论, 感兴趣的读者可参考其他相关综述以及辅助材料[5]. 本文主要聚焦于已上市半合成药物的工艺化学, 而对于尚处于临床研究阶段的半合成药物则涉及较少.
1 四环素类抗生素
1.1 四环素类抗生素简介
四环素类(Tetracycline)抗生素[6](图1)是一类具有广谱抗菌活性的化合物, 自20世纪40年代被发现以来, 被广泛应用于由革兰氏阳性菌、革兰氏阴性菌以及部分厌氧菌引起的各类感染性疾病. 四环素类药物的抗菌机制主要体现在其与细菌核糖体30S亚基的特异性结合[1]. 通过分子结构中的C(11)和C(12)的羰基/烯醇氧与Mg²⁺形成稳定的双齿螯合配位结构, 同时该Mg²⁺离子还与16S rRNA中高度保守的C1054、G1197和G1198核苷酸残基的磷酸氧原子配位. 这种多中心配位作用导致核糖体构象发生变构, 进而破坏tRNA的反密码子与mRNA密码子之间的碱基配对相互作用, 最终阻断氨 酰-tRNA与核糖体A位的结合, 实现对细菌蛋白质生物合成的可逆性抑制.
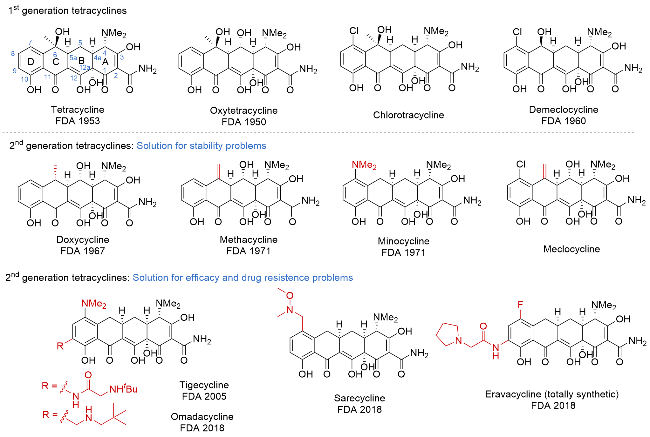
四环素类抗生素目前已发展至第三代. 第一代四环素类抗生素为链霉菌中提取出来的活性天然产物, 主要包括四环素(Tetracycline)、土霉素(Oxytetracycline)、金霉素(Chlorotetracycline)及地美环素(Demeclocycline). 然而, 这类天然四环素分子中C(6)位羟基的存在导致其化学稳定性较差, C环容易芳构化, 导致活性降低, 且在长期临床应用中引发了严重的细菌耐药性问题. 为克服上述局限性, 研究者通过半合成修饰策略开发了第二代四环素类抗生素, 主要为C(6)位去羟基化的定向修饰如多西环素(Doxycycline)和美他环素(Methacycline), 或C(7)位引入二甲氨基如米诺环素(Minocycline), 这种结构优化使得第二代衍生物展现出更优的药代动力学特性和更广谱的抗菌效果, 但其仍受外排泵(如TetA)和核糖体保护蛋白(如TetM)介导的耐药机制限制. 第三代四环素则在第二代基础上进一步改造, 通过在C(7)或C(9)位置引入官能团, 增强对核糖体靶点的亲和力, 并能有效规避外排泵介导的耐药机制, 不仅克服了传统的耐药机制, 还扩大了抗菌广谱性.
1.2 四环素类抗生素工艺
目前第一代的土霉素和金霉素以及第二代的强力霉素主要用于兽药, 前面两者通过发酵得到, 全球每年产量达到万吨以上, 中国是主要的供货商, 土霉素市场价格为80~100元/公斤, 而盐酸多西环素(强力霉素, 从土霉素半合成而来)目前价格为350~400元/公斤, 每年产量也有近万吨. 从结构来说, 强力霉素与土霉素的差别在于前者在C(6)位少了一个朝外的羟基, 最直接的工艺可能是直接C(6)位氢解脱羟基得到, 但是由于C(6)位C—O键的低活性, 目前为止还没有成熟工艺报道. 目前主流的工艺需要先在C(6)位构建双键, 利用底物的构象氢化得到目标构型的目标产物, 但是土霉素C(6)位直接脱水得到C环芳构化的副产物. 早期辉瑞公司Blackwood团队发展了经典的四环素化学, 由土霉素出发, 先经N-氯代丁二酰亚胺(NCS)或者氯代乙酰苯胺在C(11a)位置氯代, 再利用HF脱水构建C(6)位双键, 然后在Na2S2O3溶液中脱氯反应得到美他环素[7], 最后Pd/C氢化得到多西环素(强力霉素)(Scheme 1)[8-9]. 该工艺整体收率较高, 成本可控, 但需使用脱水剂HF, 而HF腐蚀性强, 因此具有对人体伤害大及设备要求高等缺点, 急需其他脱水剂替代, 虽然有其他脱水剂(H2SO4/ SOCl2/HCOOH)的报道, 但是从收率以及综合成本上考虑, 目前都无法替代HF.
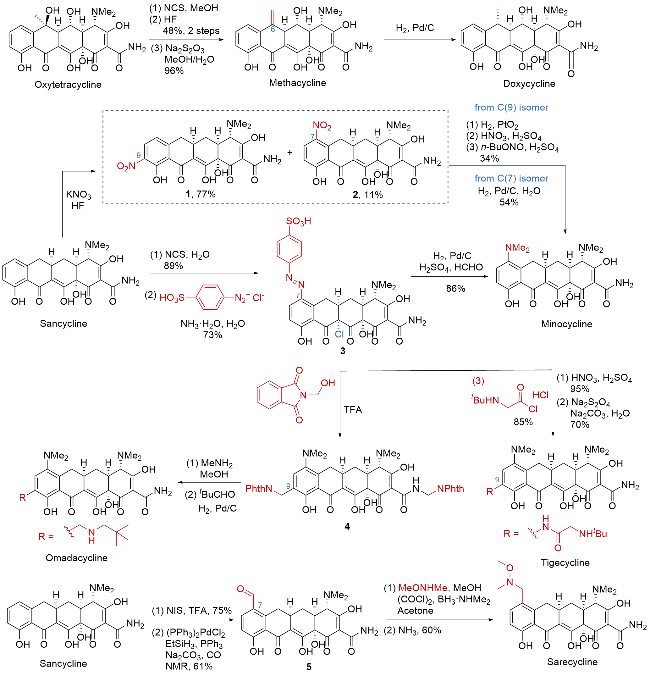
另一大类半合成四环素抗生素米诺环素目前主要用于人类(Scheme 1). 其结构是在经典四环素的基础上, 在C(6)和C(7)位进行了修饰, 由于区域选择性的难题, 在C(7)位引入二甲胺基的过程具有一定的挑战性. 化学家发展了不同的工艺路线, 基本上集中在山环素(San- cycline) C(7)位, 如何高效地引入二甲氨基, 而山环素则是从地美环素出发, 经C(7)位Pd/C脱氯及C(6)位Rh/C氢解羟基得到, 土霉素相比该结构, 在C(6)位多了甲基, 因此无法直接氢解得到强力霉素. 从山环素出发, Church团队[10a]首先完成了硝化-还原-还原胺化合成米诺环素的路线, 该方法不可避免地产生C(7)/C(9)位硝化的区域选择性问题, 虽然C(9)位反应产物能够通过C(9)位氢化、C(7)位硝化和C(9)位重氮化还原, 再次转化为C(7)位产物, 但是较长的反应路线造成成本过高. 改进后的合成工艺[10b]先在C(11a)位氯代以增强C(7)位的反应活性, 然后在C(7)位进行选择性重氮化反应, 最后经还原胺化高效获得目标化合物米诺环素.
第三代四环素类抗生素(Scheme 1)的结构修饰主要集中于C(9)及C(7)位的化学改造, 都可以从米诺环素出发获得. 替加环素[11](Tigecycline)经米诺环素C(9)位的硝化反应, 随后还原生成氨基中间体(该中间体不稳定), 最后通过酰胺化反应引入叔丁基甘氨酰氨基侧链. 奥玛环素[12](Omadacycline)的合成则需先在米诺环素C(9)位进行Friedel-Crafts烷基化反应, 继而通过胺解反应开环, 并经还原胺化得到最终抗生素. 相比之下, 沙雷环素[13](Sarecycline)的合成以山环素为起始原料, 其修饰策略聚焦于C(7)位: 首先通过选择性碘代反应引入碘原子, 随后在钯催化剂作用下进行羰基插入反应生成醛基衍生物; 该醛基与甲氧基甲基胺经缩合反应形成亚胺中间体, 并与草酸成盐后, 采用硼烷络合物进行还原胺化, 最终用氨水中和草酸盐, 经纯化获得目标化合物.
1.3 四环素类抗生素展望
半合成策略显著推动了四环素类抗生素的研发进程, 该方法通过定向结构修饰实现了多代四环素的上市. 然而目前的几个半合成工艺也存在不足, 比如强力霉素工艺中脱水剂HF的使用, 仍需寻求替代品. 另外米诺环素生产工艺中, 由于山环素的制备涉及到多步贵金属的使用, 最后米诺环素的价格居高不下(1~2万元/公斤). 因此, 利用新策略, 开发高效、可持续的绿色合成工艺仍是该领域的重要研究方向, 比如利用地美环素的C(7)位的氯原子直接一步引入二甲胺基, 将极大地缩短步骤, 提高合成工艺效率, 同时也对目前Pd催化或者Cu催化的C—N键构建效率提出更高的要求. 值得一提的是, Myers研究团队[14]通过Michael-Claisen/Dickmann环化策略, 制备了大量的四环素化合物库, 并最终推动依拉环素(Eravacycline上市, 体现了化学全合成在新药发现中的重要作用. 在依拉环素的全合成工艺[14d]中, 存在步骤较多及成本较高等缺点, 仍有较大的提升空间. 从结构上来说, 依拉环素相比类似的替加环素主要区别在于C(7)位氟原子, 因此有望在山环素的基础上进行依拉环素的半合成工艺开发.
2 大环内酯类抗生素
2.1 大环内酯类抗生素简介
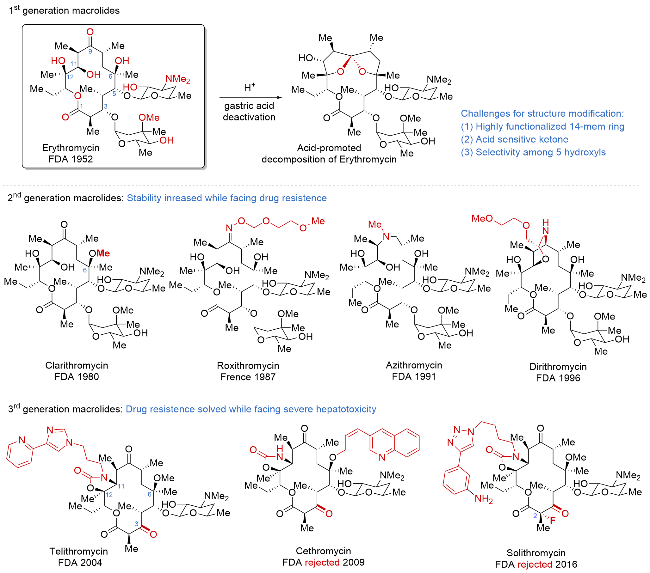
大环内酯类抗生素(Macrolides, Scheme 2)15]是临床上使用最为广泛的抗生素之一, 最早是从链霉菌中分离的红霉素(Erythromycin, 14元大环内酯)于1952年被美国食品药品监督管理局(FDA)批准上市. 红霉素对革兰氏阳性菌、某些革兰氏阴性球菌(如淋病奈瑟菌、流感嗜血杆菌、百日咳博德特氏菌和脑膜炎奈瑟菌等)和支原体/衣原体非常有效, 且与青霉素等抗生素无交叉耐药性. 大环内酯类抗生素主要作用机制是结合抑制细菌核糖体肽基转移酶中心(PTC), 且与真核细胞核糖体亲和力低, 具有良好的选择性, 所以毒性较低. 上市至今70余年, 红霉素仍在临床使用, 但已经不多, 主要是由于其存在两方面问题: 一方面是稳定性差, 导致口服利用度低, 并且其生物利用度差, 药代动力学不稳定; 另一方面是耐药性明显, 这限制了其在临床的应用. 尽管红霉素高度官能团化, 选择性后修饰难度大, 对红霉素结构进行改造, 开发出的第二代大环内酯和第三代酮内酯抗生素分别解决了稳定性(药代性质)和耐药性问题. 值得一提的是, 在红霉素的发酵过程中副产物多, 有效组分(红霉素A)比例低. 刘文团队[15d]通过改良菌株, 调整了后修饰酶的表达比例, 将无效组分几乎完全转化为有效组分红霉素A, 使产品质量显著提升(副产物基本消除), 同时产量提高约25%, 为国内红霉素半合成工艺提供了优质原料.
2.2 第二代大环内酯类抗生素合成工艺
在酸性条件下(如胃酸), 红霉素母核6,12-二羟基进攻9-羰基形成缩酮, 导致红霉素失活. 早期的开发策略自然围绕这一降解过程进行, 分别得到了二代大环内酯保护6-羟基的克拉霉素(Clarithromycin, FDA于1980年批准上市)、转化6-羰基的阿奇霉素(Azithromycin, FDA于1991年批准上市)、地红霉素(Dirithromycin, FDA于1996年批准上市)和罗红霉素(Roxithromycin, 1987年在法国上市, FDA尚未批准上市). 这些第二代半合成大环内酯解决了稳定性问题.
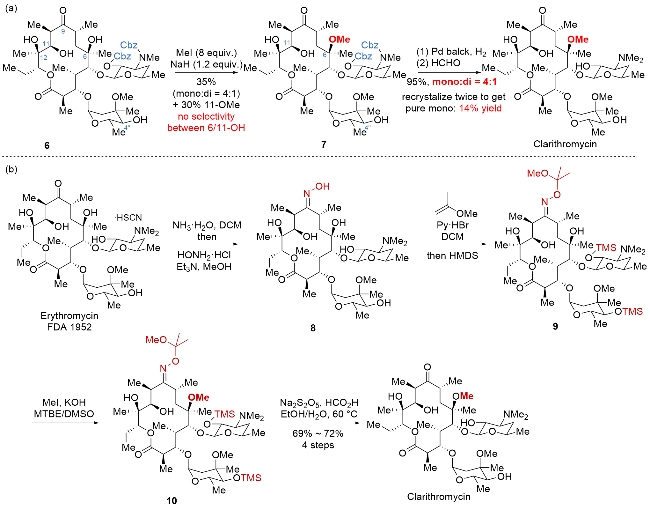
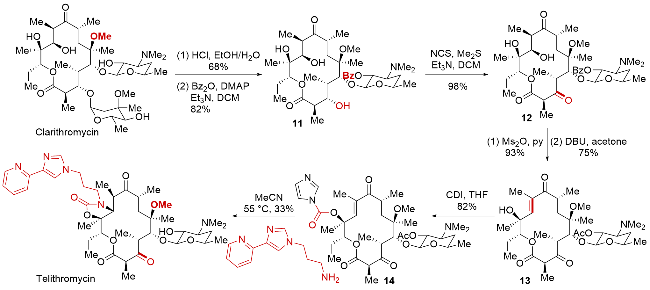
克拉霉素(Scheme 3)于1980年被FDA批准上市, 并且是第三代大环内酯类抗生素的合成前体(2000年以11.5亿美元销售额排名原料药第11名), 是非常重要的原料药. 日本大正制药在1984年报道了克拉霉素的合成(Scheme 3, a)[16]. 红霉素A经过苄氧羰基(Cbz)保护直接得到脱甲基产物6, 在NaH/MeI条件下对其进行甲基化反应得到6-OH甲基化产物7 (35%, 含有20%双甲基化产物)和11-OH甲基化产物(30%). 6中有4个裸露羟基, 从结果上看, 高度官能团化的骨架构象和分子内氢键导致的甲基化活性是12/4''<11=6, 即甲基化对6/11位羟基没有选择性. 后续经过钯黑脱Cbz、还原胺化甲基化胺基, 得到克拉霉素与11-甲基克拉霉素的混合物, 产率95%, 再经重结晶得到纯品克拉霉素, 产率14%. 日本大正制药又在1984年[16b]报道了对底物6中9-羰基进行肟化-保护可以调整分子构象, 增加6-OH甲基化选择性. 肟的保护基采用邻氯苄基, 可以以79%产率得到6-OH甲基化产物, 大大提高了选择性和工艺效率, 是第一个较为成熟的生产工艺. 不过, 该路线多次采用Pd/C催化剂, 路线成本依旧较高. 目前主流的工艺路线依旧采用类似策略, 但采用了2-甲氧基-2-丙基和三甲基硅基乙氧基(TMS)保护替代Cbz和邻氯苄基. 例如在韩美药品2000年报道的工艺路线中(Scheme 3, b)[16c], 硫氰酸红霉素与水合肼反应得到红霉素肟8, 8在2-甲氧基丙烯条件下保护肟-OH, 并一锅法对两个糖片段的羟基进行了TMS保护, 这两个羟基是中间体8中位阻最小的羟基, 其他羟基在该条件下不会连接大位阻的TMS保护基. 随后, C(12)叔醇羟基活性低, C(11)羟基在肟骨架中被构象钝化, C(6)羟基在MeI/KOH条件下选择性甲基化得到10; 最后, 在焦亚硫酸钠/甲酸条件下, 对整个分子进行脱保护得到克拉霉素. 上述均无需额外的纯化过程, 最后对克拉霉素进行结晶纯化即可得到纯品, 4步总收率69%~72%. 山东安信[16d]报道过类似的路线, 用二苯基磷酰基保护9-肟, 也可以实现克拉霉素高效的合成与脱保护.
2.3 第三代大环内酯类抗生素合成工艺
第三代大环内酯抗生素是基于克拉霉素的进一步改造, 一方面C(3)糖苷脱除并氧化成酮(故又称酮内酯); 另一方面, C(11)羟基取代为多样化取代的胺基, 并与C(12)羟基碳酰环化. 长期构效研究发现, C(3)糖苷对活性没有影响, 这与共晶显示的红霉素C(3)糖苷与细菌核糖体PTC无相互作用一致, 最后发现C(3)氧化为羰基效果最好. 而C(11)/(12)侧链结构与细菌核糖体PTC产生了新的作用(与A752产生π-π堆叠), 增强了活性, 克服了耐药性(A2058G耐药突变). 第三代大环内酯活性大大增强并能克服耐药性, 但严重受限于肝毒性问题. 目前, 第三代大环内酯抗生素有三个, 分别是泰利霉素(Telithromycin)、喹红霉素(Cethromycin)和索利霉素(Solithromycin). 其中, 泰利霉素于2004年上市, 在临床试验中未表现出肝毒性, 仅有肝酶轻微升高, 可是临床使用中发现了少见但严重的肝毒性问题(急性肝损伤、肝衰、死亡), 并于2009年被FDA限制使用. 后续喹红霉素和索利霉素虽然在临床试验中没有表现出肝毒性, 但被FDA认为有严重肝毒性风险, 并分别于2009年和2016年被拒绝上市.
泰利霉素是从克拉霉素出发进行进一步修饰获得的(Scheme 4). 克拉霉素中羟基活性的顺序是2'>11'>12'. 根据成都文氏2007年专利报道[20], 在酸性条件下水解克拉霉素C(3)糖苷, 随后Bz保护最活泼的C(2')羟基得到中间体11. 11在Me2S/N-氯代丁二酰亚胺(NCS)条件下可以实现C(3)羟基的选择性氧化, 得到中间体12. Ms2O选择性活化C(11')羟基, 再由1,8-二氮双环[5.4.0]十一-7-烯(DBU)消除得到双键中间体13. 最后, 只剩活性最低的C(12')羟基, NaH拔氢后与N,N'-羰基二咪唑(CDI)反应酰化得到14, 最后与侧链缩合, 并与分子内的双键发生Michael加成, 得到泰利霉素(Scheme 4). 2008年, 印Alembic制药公司[20b]报道了类似的工艺路线, 不过将保护基Bz换成了Ac.
2.4 大环内酯类半合成兽药
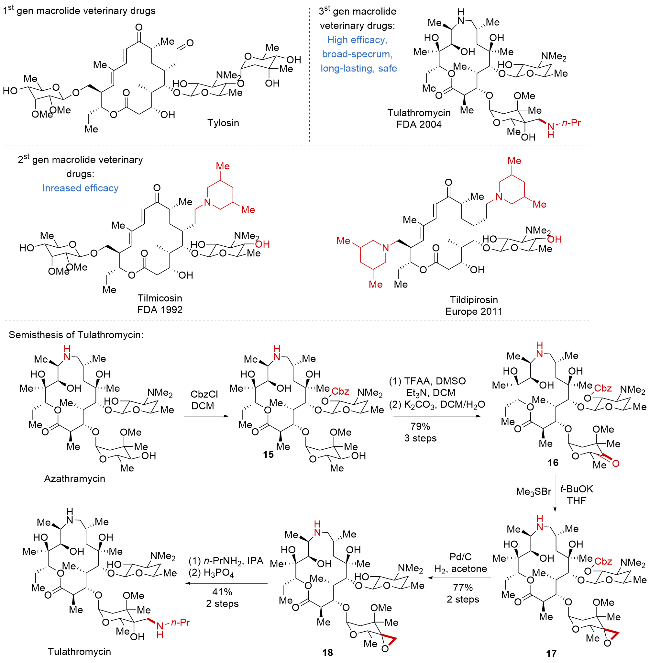
2004年美国辉瑞动物保健品公司开发的第三代大环内酯兽药泰拉霉素(Tulathromycin)上市. 由于泰拉霉素结构中有3个胺基, 对阴性菌透膜性好, 更加广谱高效, 且吸收快、用量少、药效持久(单次给药即能提供全程治疗). 目前, 其生产主要采用从去甲阿奇霉素(二氢高红霉素)出发的工艺路线. 例如江苏博瑞[24a]首先在苄氧基碳酰氯(CbzCl)条件下对活性最高的C(2')羟基进行Cbz保护, 随后在三氟乙酸酐(TFAA)/二甲基亚砜(DMSO)条件下对C(4'')羟基进行选择性氧化, 碳酸钾中和后得到中间体16. 16的C(4'')酮羰基与硫叶立德反应得到环氧化产物, 并在氢化脱Cbz后得到18. 中间体18即可与正丙胺反应进行环氧开环, 再与磷酸成盐后结晶得到泰拉霉素. 浙江海正制药[24b]和广东东阳光药[24c]等公司也报道了类似工艺路线. 泰拉霉素效果好, 前景广阔, 生产需求大, 进一步的工艺开发优化具有很大价值.
2.5 小结
3 氨基糖苷类抗生素
3.1 氨基糖苷类抗生素简介
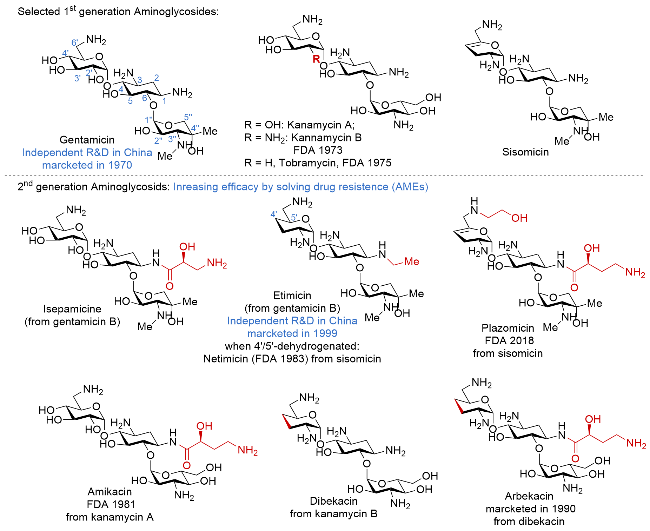
氨基糖苷类(Aminoglycosides)抗菌药物(图2)自问世以来一直在临床治疗中发挥重要作用[26], 虽然因毒性和耐药性问题使用受限, 但仍是治疗严重革兰阴性菌感染的首选药物. 氨基糖苷抗生素根据来源可分为链霉菌源和小单胞菌源; 从结构上可分为4,6-氨基糖苷[庆大霉素(Gentamycin)、卡那霉素(Kanamycin)和西索霉素(Sisomicin)等], 4,5-氨基糖苷[安普霉素(Apramycin)、巴龙霉素(Paromomycin)和新霉素(Neomycin)等]及4-氨基糖苷[链霉素(Streptomycin)和布鲁霉素(Bluensomycin)]; 而糖苷部分可分为吡喃糖和呋喃糖类型. 目前, 临床上使用的氨基糖苷类药物均为4,6-氨基糖苷类抗生素(新霉素因毒性问题使用严重受限), 用于需氧革兰氏阴性菌全身感染和结核病的治疗, 高效、广谱且无过敏反应. 氨基糖苷类抗生素作用靶点也是核糖体, 但是由于其对原核、真核核糖体选择性不好, 氨基糖苷抗生素都具有不可逆的严重耳毒性和肾毒性. 另外, 氨基糖苷类抗生素容易产生耐药性, 是氨基糖苷抗生素有效性的关键制约因素. 值得一提的是, 我国在氨基糖苷类抗生素领域有两大原创性突破, 分别是庆大霉素和依替米星(Etimicin), 陈代杰课题组[26d]对二者联产的绿色、高效关键技术创新及产业化做出了重大贡献.
3.2 第二代氨基糖苷抗生素合成工艺
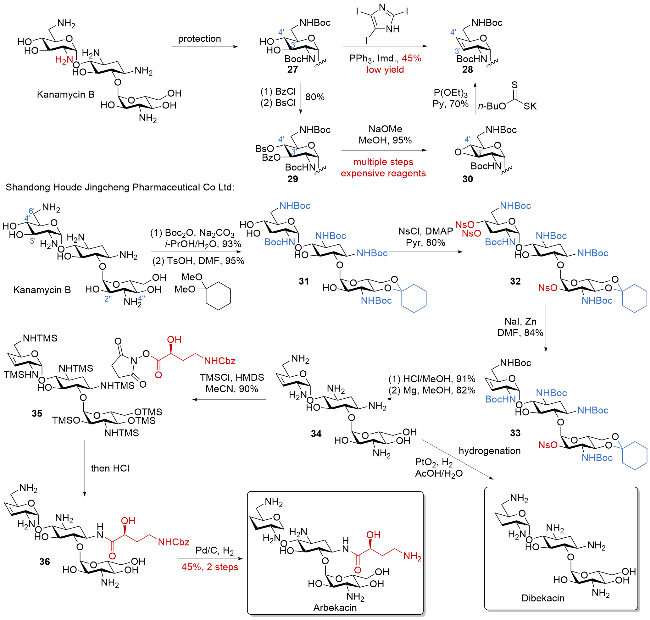
第二代氨基糖苷抗生素是半合成产物, 主要解决了耐药性问题从而增强了有效性[26]. 氨基糖苷抗生素耐药的主要机制是细菌氨基糖苷修饰酶(AMEs)修饰氨基糖苷分子致其失活. 半合成氨基糖苷抗生素通过羟基或胺基上的后修饰, 消除了该位点或相近位点AMEs的作用, 解决了对应的耐药性; 这样的修饰往往不能提高活性, 或在一定程度上降低抗菌活性和耳毒性. 此外, 已发现的耐药机制还有靶点修饰酶(RMTases)核糖甲基转移酶导致的核糖体RNA修饰, 目前的氨基糖苷抗生素都难以幸免此类耐药, 且与碳青霉烯交叉耐药. 半合成氨基糖苷主要在C(1)-NH2和C(6')-NH2进行修饰, 相对复杂的地贝卡星/阿贝卡星需要对卡那霉素B的C(3')/(4')进行脱氧. 而氨基糖苷的极端高氧化态结构以及其多达6个羟基和5个胺基的骨架, 导致其选择性后修饰具有巨大挑战性.
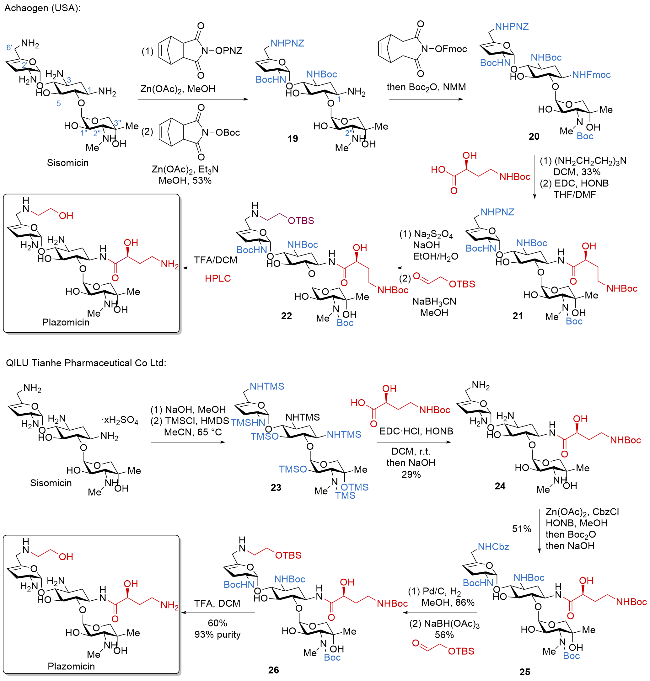
普拉佐米星(Plazomycin, Scheme 6)是目前唯一在两个位点带有侧链修饰的氨基糖苷抗生素, 于2018年被FDA批准上市, 用于复杂尿路感染. 原研工艺[27a]中, 所有的分离步骤都是通过柱层析或反相高效液相色谱(RPHPLC)实现的. 从西索霉素出发, 在Zn(OAc)2条件下, 1-NH2与3''-OH螯合Zn2+而被屏蔽, 2''-NH2与1''-OH螯合Zn2+而被屏蔽, 余下3/2'/6'-NH2具有亲和性, 对硝基苄氧羰基(PNZ)保护亲和性最强的6'-伯胺基. 随后, 在同样的锌离子螯合条件下, 用Boc保护余下的3/2'- NH2. 随后, 在无锌离子螯合下1-NH2亲和性强于位阻更大的2''-NH2, 于是优先被Fmoc保护, 随后一锅法Boc保护最后的2''-NH2. 这样, 整个西索霉素的胺基被选择性地保护起来, 可以依次暴露出1-NH2和6'-NH2分别修饰: 首先脱除Fmoc并缩合, 随后脱除PNZ并还原胺化; 最后, 在三氟乙酸(TFA)条件下脱除保护基, 即可得到普拉佐米星. 该路线不具有工业放大的可能性. 后续依次有不同的专利报道工艺路线, 相对成熟的是齐鲁天和惠世[27b]和山东安信制药[27c]的工艺路线. 在这一路线中, 首先仅需用TMS全保护西索霉素, 即可实现对1-NH2的选择性缩合. 随后, 采用类似的锌离子螯合策略屏蔽1/2''-NH2, 先用Cbz保护6'-伯胺基, 再用Boc保护余下的3/2'-NH2. 脱Cbz即可对6'-NH2还原胺化, 最后用TFA脱保护得到普拉佐米星. 该工艺路线快速选择性酰化1-NH2, 使整个路线效率大大提升. 异帕米星(Isepamicin)[28]、依替米星[29e]和奈替米星(Netimi- cin)[29a-29d]有着与上述原研路线类似的选择性保护工艺, 即先金属盐螯合屏蔽1/3''-NH2保护其它胺基, 随后修饰1-氨基. 阿米卡星(Amikacin)[30]是硅烷化工艺, 通过TMS对卡那霉素A全保护再缩合, 直接选择性修饰1-氨基.
对于地贝卡星(Arbekacin)和阿贝卡星(Debekacin, Scheme 7)的合成, 卡那霉素B的3'/4'脱氧是关键步骤. 目前的工艺路线类似, 在如何脱氧上有所差别, 主要包括环氧硫代热解消除[31a-31b]、碘代还原消除[31c]和磺酸酯还原消除[31d]. 以磺酸酯还原消除为例, 从卡那霉素B出发, 直接Boc保护所有胺基, 再用环己酮形成缩酮保护4''/6''羟基, 随后即可用邻硝基苯磺酰氯(NsCl)活化3'/4'-OH, 同时2''-OH也会与Ns形成磺酸酯. Zn/NaI条件下, 3'/4'-ONs可以被高效还原为碳碳双键. 随后HCl/MeOH脱Boc、Mg/MeOH脱2''羟基的Ns, 得到关键中间体, 该中间体再氢化得到地贝卡星. 合成阿贝卡星的工艺[31]是类似上述的1-NH2选择性酰化工艺, 即TMS全保护后进行缩合, 再经HCl脱TMS、Pd/C氢化脱Cbz得到阿贝卡星.
3.3 小结
总体上看, 半合成氨基糖苷抗生素在很大程度上解决了耐药性问题, 但是耳毒性问题仍然严重, 制约了氨基糖苷抗生素的使用. 值得一提的是, Crich小组[32]进行了一系列基于天然4,5-氨基糖苷抗生素(安普霉素和巴龙霉素)后修饰的构效关系研究, 发现了一些半合成4,5-氨基糖苷抗生素分子, 不仅在抗耐药性上有进一步的突破, 且在安全性上有很大改善, 耳毒性大大降低, 有望开发出新一代半合成氨基糖苷抗生素.
4 糖肽类抗生素
4.1 糖肽类抗生素简介
万古霉素(Vancomycin)[33]是最早应用于临床的糖肽类(Glycopeptides)抗菌药物(图3), 于20世纪50年代从头状无枝酸菌(Amycolatopsis orientalis)中分离得到, 对革兰氏阳性菌具有高度的有效性, 在1958年被FDA批准上市. 万古霉素主要用于治疗青霉素耐药的金黄色葡萄球菌引起的严重感染, 被称为治疗重症细菌感染的“最后一道防线”. 另一天然脂糖肽抗生素替考拉宁(Teicoplanin, 1988年意大利获首批上市)也具有相同靶点和类似的效用, 不过由于它在肽骨架上多出了脂肪侧链, 提高了亲脂性, 故更易渗透组织和细胞, 且替考拉宁肾毒性更低, 半衰期较长, 具有更强的体内抗菌活性.
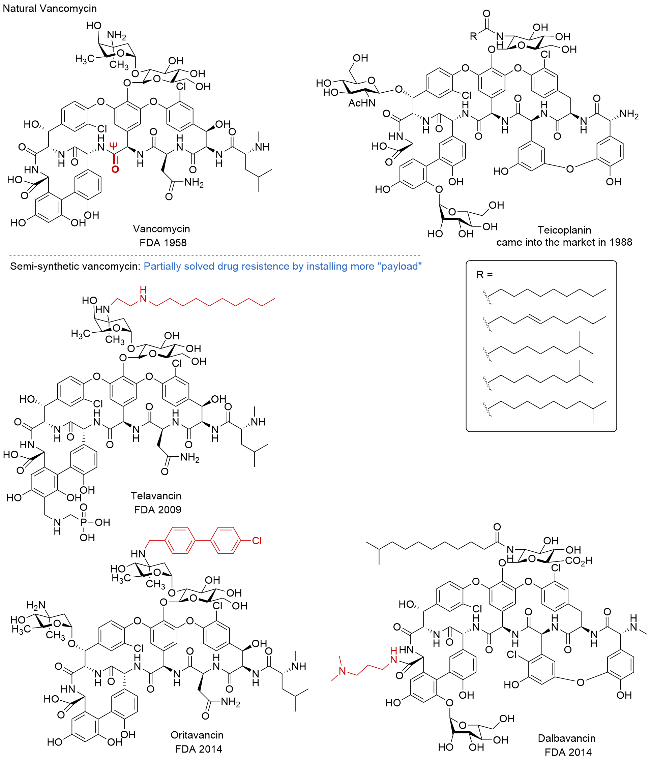
万古霉素可以与其中细菌细胞壁肽聚糖合成前体末端五肽的D-Ala-D-Ala高度结合, 从而抑制糖基转移酶和肽基转移酶催化糖肽链的聚合, 抑制细菌细胞壁的合成, 这一靶点高度保守, 故万古霉素不易产生耐药性. 经过长时间的临床使用, 在1988年才首次发现耐药肠球菌(VRE), 距上市间隔长达30年, 远超过青霉素5年和喹诺酮类10年. 其耐药机制是细胞壁肽聚糖末端残基D-Ala-D-Ala演变为D-Ala-D-Lac, 使万古霉素亲和力降低1000倍. 不过, 这需要多个基因协同表达才能促成, 且会使细胞壁合成效率显著降低, 故万古霉素耐药性难以产生. 该机制的突变类型分为VanA和VanB, 其中, VanB类型虽然万古霉素无效, 但替考拉宁有效, 且半合成抗生素特拉万星(Telavancin)和达巴万星(Dalbavancin)有效, 而VanA类型只有奥利万星(Oritavancin)有效. 这可能是由于VanB的突变程度不如VanA彻底.
4.2 糖肽类类抗生素半合成工艺
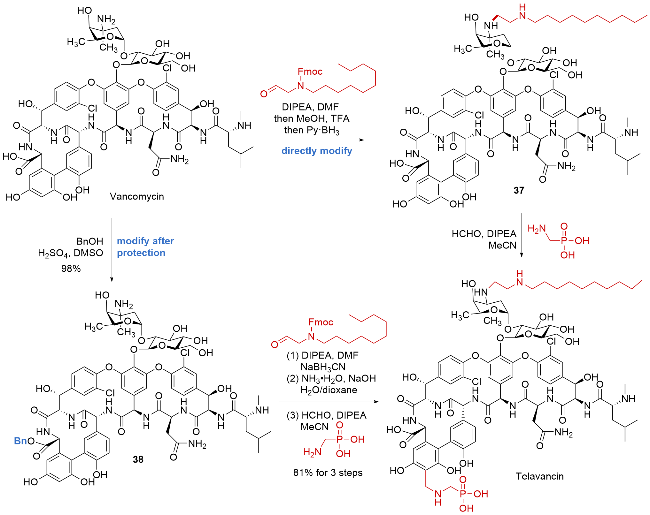
特拉万星(Scheme 8)[34](FDA于2019年批准其上市)是在万古霉素糖氨基部分连接了疏水基团增加了其与细胞膜相互作用, 在芳环引入亲水基团改善了药物的药代动力学性质, 增强了抗菌活性. 根据2002年原研公司Theravance的报道[34a], 万古霉素中唯一的伯胺可以与醛在碱性条件缩合成亚胺, 随后经还原、脱Fmoc引入疏水侧链. 接着, 采用甲醛-氨甲基磷酸曼尼希反应在芳环中亲核性最高的二酚邻位引入亲水侧链, 得到特拉万星. 中国医药[34b]和浙江医药[34c]对还原胺化及脱保护步骤进行了优化, 一定程度上提高了产率. 专利中并没有体现分离方法, 给出80% HPLC纯度产品. 2018年, 新北江制药[34d]报道了另一种特拉万星半合成工艺, 通过万古霉素羧基的苄酯保护, 增加反应选择性和收率(从50%以下提升到79%), 且仅需结晶就可以得到高纯度的产品(从C18分离96%纯度提升到>99%).
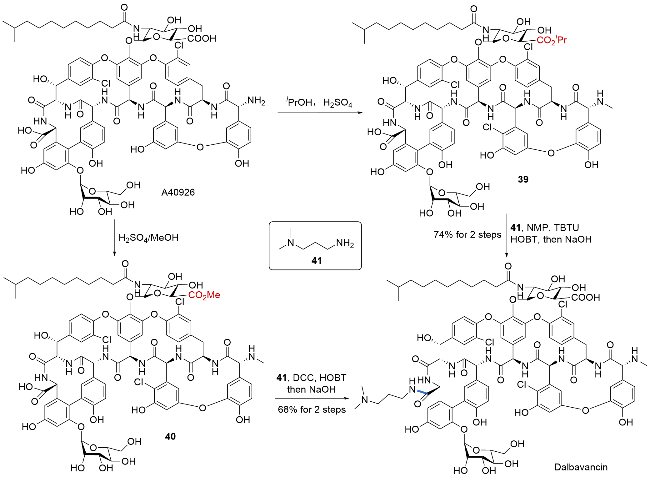
达巴万星(Scheme 9)[35](FDA于2014年批准其上市)是替考拉宁类似物A40926的衍生物, 亲脂侧链可增加其与细菌细胞膜的亲和力, 增强抗菌活性. 半合成引入的二甲胺基成盐形成阳离子, 可以增加药物对细菌细胞膜的穿透性, 从而增强活性. 其半合成的难点在于A40926具有两个羧基, 需要选择性缩合母核上的羧基. Vicuron公司[34e]报道了酸性条件下选择性甲酯化糖环上的羧基(86%产率, 74%纯度), 随后母核羧基与3,3-二甲氨基-1-丙胺进行缩合, 再水解甲酯得到达巴万星, 最终需要复杂的柱层析分离. 2023年, 福州福兴医药[35a]也使用类似的策略合成了达巴万星, 主要不同在于使用了异丙酯保护羧基, 虽然增加了选择性, 但同样需要柱层析分离除杂质.
奥利万星(FDA于2014年批准其上市)是唯一具有对VanA耐药菌抑制活性的糖肽类抗生素(Scheme 10). 其在结构上增加了糖基修饰4-(4-氯苯基)苄基(CBP), 增加了药物对糖基转移酶的结合力, 从而增强了抑制糖基转移酶的活性, 减少了对肽聚糖合成前体D-Ala-D-Ala结合力的依赖, 部分规避了D-Ala-D-Ala到D-Ala-D- Lac的影响, 从而实现了对VanA耐药菌的抑制. 特拉万星和达巴万星可能也有类似作用, 使得它们可以规避较弱的VanB的作用, 但可能由于它们侧链与糖基转移酶的结合力不足, 并不足以规避VanA的作用.
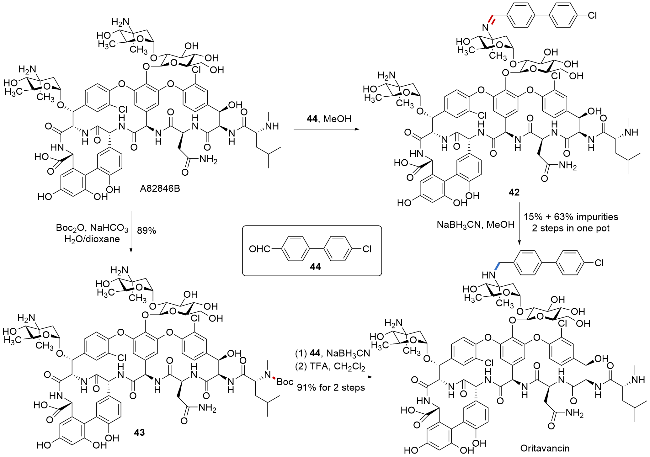
奥利万星[36]的直接前体为微生物发酵的次级代谢物A82846B, 在化学条件下引入CBP侧链实现其合成. 底物A82846B含有两个糖环伯胺和一个侧链仲胺, 存在区域选择性问题. 礼来公司[36a]于1995年首次报道了奥利万星工艺路线, 以A82846B为起始物, 与4'-氯联 苯-4-甲醛进行缩合形成席夫碱, 再通过氰基硼氢化钠进行还原胺化得到奥利万星. 反应基本不具有选择性, 总收率15%, 产生4种杂质(区域选择性和双取代). 2018年, 新北江制药[36b]报道了先保护再引入侧链的工艺路线. 在用Boc保护侧链仲胺后, 与4'-氯联苯-4-甲醛进行缩合与还原, 最后进行脱保护得到奥利万星, 总收率达到81%, 仅产生一种杂质(6%). 大大提高了产率和产品奥利万星的纯度.
4.3 小结
万古霉素作为革兰氏阳性菌抗感染的最后一道防线, 最大的特色是具有超高的有效性且不易产生耐药性. 在长时间使用后产生了VanA和VanB两种D-Ala- D-Ala突变为D-Ala-D-Lac的耐药菌株. 半合成糖肽类抗生素虽然在一定程度上对耐药菌株有效, 但有效性有限, 主要因为其半合成修饰没有对D-Ala-D-Lac恢复结合力, 只是通过其他机制回补了一定的抗菌活性. Boger小组[37]阐明了万古霉素对D-Ala-D-Lac失去结合力的机制: D-Ala-D-Ala中酰胺键转换成了D-Ala-D-Lac中的酯基O, 导致残基酰胺NH与Ψ-酰胺羰基O的氢键作用变成了酯O与Ψ-酰胺羰基O的静电排斥作用. 基于此, Boger小组通过全合成获得了Ψ-酰胺羰基O还原为H2或者转化成NH的万古霉素(图3中Ψ-O, 红色), 前者规避了静电排斥, 后者重建了氢键, 从解决了对D-Ala- D-Lac的结合力问题, 同时保留与D-Ala-D-Ala的结合力; Boger小组进一步引入CBP侧链和季铵盐阳离子侧链, 分别增强糖基转移酶结合力和细胞膜通透性. 三管齐下, 推出了“万古霉素3.0”, 与万古霉素相比对耐药菌活性提高了1万倍,与奥利万星相比提高了250倍. 不过, 这一药物全合成路线长达30步, 而选择性后修饰氢化Ψ-酰胺O来实现其半合成几乎是不可能的.
5 林可酰胺类抗生素
5.1 林可酰胺类抗生素简介
林可酰胺类(lincosamides)抗生素(Scheme 11)[38]主要包括人用的林可霉素(Lincomycin)、克林霉素(Clindamycin)以及兽用吡利霉素(Pirimycin), 对革兰氏阳性菌、厌氧菌及部分原虫具有活性, 在感染性疾病治疗中发挥重要作用[1]. 林可霉素是从林可链霉菌中分离出的天然抗生素, 活性中等, 而半合成克林霉素活性大大增强, 在临床上已经逐步替代掉林可霉素. 该类药物的核心机制是通过不可逆结合细菌核糖体50S亚基抑制蛋白质合成. 目前, 林可酰胺类药物耐药性常见, 主要源于erm基因介导的核糖体甲基化修饰[2]及lnu基因相关的药物外排或酶解机制, 与红霉素耐药高度关联. 目前, 耐药性挑战严峻, 而半合成林可酰胺类药物开发非常有限. 不过, 林克酰胺类抗生素对复杂感染(尤其是混合厌氧菌感染)的治疗具有不可替代性, 仍是临床抗感染治疗的重要工具.
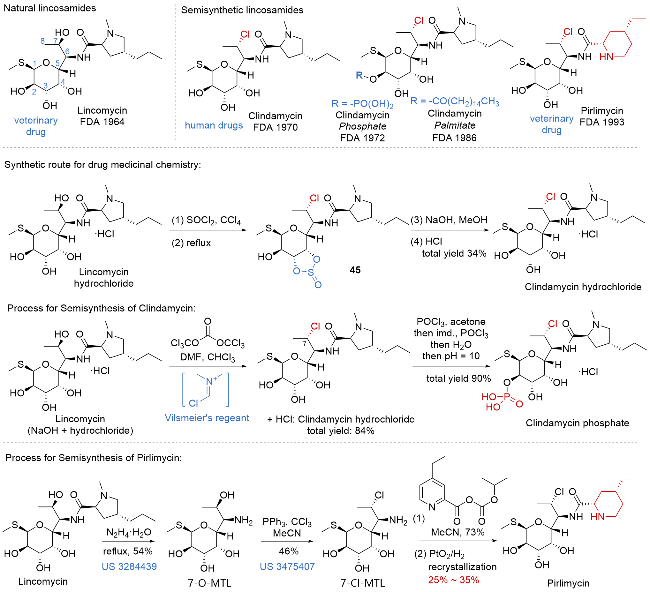
5.2 林可酰胺类抗生素半合成工艺
克林霉素是通过对林可霉素分子结构中7-OH进行氯代修饰得到的半合成药物分子, 增强了药物与核糖体50S亚基的结合力, 且增加了脂溶性, 具备更高的口服吸收率(达90%)和更强的组织穿透性, 尤其在骨组织中的浓度较高, 是目前临床应用最广泛的林可霉素类药物. 在早期的合成中(Scheme 11)[38b], 使用SOCl2保护3,4-顺式二羟基, 糖环上仅剩下亲核性弱的直立健5-OH; 随后加热, 构型翻转地氯代7-OH, 产率仅34%. 后来报道的Rydon试剂即可实现直接选择性氯代7-OH, 但需要使用PPh3. 目前的主流工艺是采用Vilsmeier试剂, 如天方药业股份有限公司[39a]报道的合成工艺中, 通过三光气与N,N-二甲基甲酰胺(DMF)原位制备Vilsmeier试剂, 该试剂具有高活性和选择性, 可以直接实现对7-OH的选择性氯代, 经历SN2过程发生构型翻转. 随后经酸化和重结晶得到克林霉素盐酸盐. 另外, 克林霉素也有前药克林霉素磷酸酯(Clindamycin Phosphate)[40]和克林霉素棕榈酸酯盐酸盐(Clindamycin Palmitate)[41], 改善了口服利用度和药代动力学, 后者也能去苦. 据天方药业股份有限公司[40]报道, 在克林霉素丙酮溶液中加入POCl3可以选择性保护邻二醇, 3/4-OH被丙酮缩酮保护; 随后加入咪唑和POCl3, 对2-OH膦酰化得到2-O磷酰氯; 再加入水, 水解丙酮缩酮保护; 最后调节pH到10, 水解2-O磷酰氯为磷酸, 一锅法得到磷酸克林霉素.
5.3 小结
天然林可霉素活性中等, 药代动力学差, 通过对将天然林可霉素的7-OH进行氯代大大提高了活性, 解决了口服吸收率问题, 其前药修饰可以进一步优化药代动力学, 提高药效. 不过目前, 林可霉素面临的耐药性问题仍未解决. Myers小组[43a]通过全合成对克林霉素进行了结构改造, 在侧链引入刚性结构七元环氧己烷环, 得到了新型合成抗生素Iboxamycin (IBX), 抗菌谱拓展到阴性菌和厌氧菌, 且有效对抗erm、cfr基因及靶标保护蛋白介导的耐药性. 随后Myers小组[43b]报道了C(7)与1-硫甲基成10元环的CRM分子, 其构象固定, 活性大增; 更进一步修饰得到环上含F原子的11元环结构BT- 33, 活性更强. 这二者对多重耐药的阳性菌、阴性菌都具有非常强效的抗菌活性, 不仅克服了林可酰胺类药物的耐药性问题, 更大大增强了活性和抗菌谱(尤其是对阴性菌), 是极具前景的潜在下一代林可酰胺类抗生素.
6 利福霉素类抗生素
6.1 利福霉素类抗生素简介
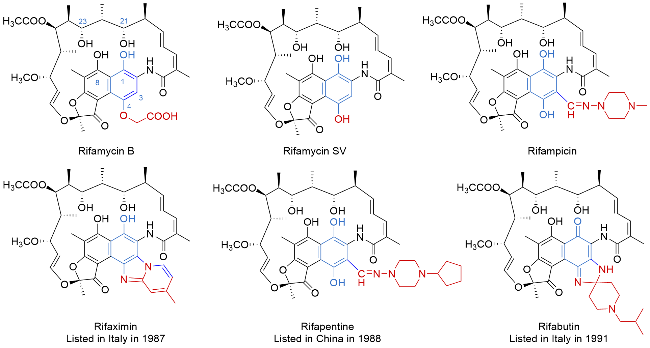
为进一步拓展抗菌谱, 研究团队针对3-甲酰基利福霉素SV进行定向结构优化, 最终获得关键衍生物利福平(Rifampicin). 该化合物不仅口服生物利用度良好, 而且对结核分枝杆菌(M. tuberculosis)表现出卓越的抗菌活性, 同时兼具较低的毒性特征.
在后续研究中, 基于利福平的进一步结构改造取得重要突破. 利福喷丁(Rifapentine)通过在C(3)位引入环戊基取代基, 显著提高其脂溶性和膜渗透性, 从而延长血浆半衰期[44], 该药物于1988年在我国率先获批上市. 另一重要衍生物利福布汀(Rifabutin)则采用螺哌啶基团修饰C(3)位, 不仅对鸟分枝杆菌复合体等非结核分枝杆菌具有优异活性, 其抗结核分枝杆菌和麻风分枝杆菌的效能亦优于利福平. 此外, 临床研究证实, 利福布汀可作为单一预防用药, 显著降低艾滋病(AIDS)患者播散性鸟分枝杆菌复合群(MAC)感染的发生率.
利福霉素类抗生素的抗菌机制主要是对细菌RNA合成的特异性抑制[44], 其作用靶点为细菌DNA依赖性RNA聚合酶(DDRP), 通过与该酶的β亚基不可逆结合, 阻断转录起始复合物的形成, 从而抑制mRNA的生物合成. 从分子水平分析, 利福霉素的萘氢醌核心结构通过π-π堆积作用与DDRP的Tyr残基发生相互作用, 同时其C(1)和C(8)位羟基精准嵌入酶活性中心的氨基酸疏水腔; 此外, 其脂肪链侧链的C(21)和C(23)位羟基可竞争性占据RNA链延伸位点, 导致DNA-RNA-聚合酶三元复合物稳定性破坏, 最终引发细菌死亡. 不同利福霉素衍生物的抗菌活性差异主要取决于其细胞壁穿透效率. 以利福霉素B为例, 其C(4)位羧基的高极性导致跨膜渗透性降低; 而经修饰的利福喷丁和利福布汀在C(4)位引入亚胺基团(CH=N), 显著增强了分子的脂溶性, 使其更易穿透细菌细胞壁的脂质双层结构, 从而大幅提升其抗菌效能.
6.2 利福霉素类抗生素的半合成工艺
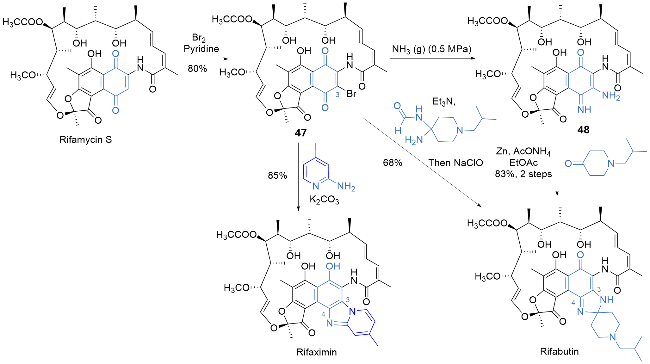
从结构上分析, 利福平和利福喷丁都是在利福霉素SV 3-位引入甲醛基之后, 与不同胺基缩合得到, 因此关键中间体3-甲酰基利福霉素SV的获得决定了该类工艺路线. 虽然有许多化学转化方法能够在苯酚的邻位引入甲酰基, 比如Gatermann-Koch反应、Reimer-Tiemann反应、醇镁反应以及Duf反应等, 但是由于底物的敏感性, 这些方法都不适用. 目前, 工业规模生产利福平(Scheme 12)主要采用以发酵获得的利福霉素SV为起始原料的多步合成路线, 依次经历氧化、环合、水解及缩合等关键反应步骤[45]. 具体合成路径如下: (1)在次氯酸钠氧化条件下将利福霉素SV转化为利福霉素S; (2)于酸性介质中与二羟甲基特丁胺进行环合反应, 构建利福霉素噁嗪骨架; (3)经水解反应生成关键中间体3-甲酰基利福霉素SV; (4)最终与1-氨基-4-甲基哌嗪缩合形成目标产物. 该工艺可采用“一锅法”连续操作, 并通过结晶纯化技术分离关键中间体及终产物. 利福喷丁[46]的合成策略与利福平类似, 均基于3-甲酰基利福霉素SV的衍生化. 由于该中间体3-甲酰基利福霉素SV的分离纯化具有一定的难度, Wessjohann课题组[45c]开发了固载的苄肼树脂用于该中间体的提纯和后续转化, 大大提高了利福平的分离和制备效率.
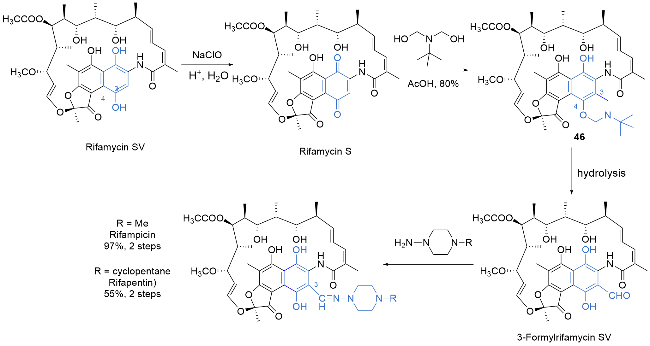
而利福布汀[47]因其独特的螺哌啶结构, 有两条工艺路线获得(Scheme 13). (1)邻二胺形式的天然产物与哌啶酮缩合获得, 以利福霉素S为起始物, 首先通过卤代-硝化/还原反应在C(3)位引入氨基, 随后将对二酚氧化为醌式结构, 并在C(4)位进行氨化反应, 最终经关环步骤构建螺环体系. 传统卤代-硝化法虽可行, 但存在合成步骤冗长、需使用锌粉(Zn)及亚硝酸钠(NaNO2)等高危试剂的缺陷. (2)改进后的工艺为3-溴对二醌形式的天然产物与哌啶酮偕二胺形式直接环化, 具体工艺采用“一锅法”策略, 以卤代利福霉素S为原料, 在三乙胺催化下直接与N-(2-甲基丙基)-4-氨基-4-氨甲酰基哌啶进行关环反应, 显著缩短了合成路线并提升了整体效率.利福昔明(Rifaximin)的结构与利福布丁相似, 以溴代利福霉素S为原料, 碱性条件下直接与氨基吡啶环合, 结晶后得到利福昔明[48].
6.3 小结
利福霉素作为一类重要的安莎霉素类抗生素, 其半合成修饰策略为结构优化与活性提升提供了关键途径. 科研人员通过靶向修饰利福霉素的芳香环和安莎链结构, 成功开发出具有更广抗菌谱和更强药理活性的衍生物. 然而, 随着利福霉素类药物的广泛使用, 其面临的耐药性问题日益突出, 同时复杂的合成工艺也导致了生产成本居高不下及发酵废料的环境负担等问题. 因此, 开发基于合成生物学和绿色化学的新型制备技术, 构建高效、可持续的利福霉素生产体系, 仍是当前研究的重点方向. 未来, 通过结合计算机辅助药物设计和生物催化等前沿技术, 有望突破现有局限, 推动利福霉素类药物的创新发展.
7 棘白菌素类抗生素
7.1 棘白菌素类抗生素简介
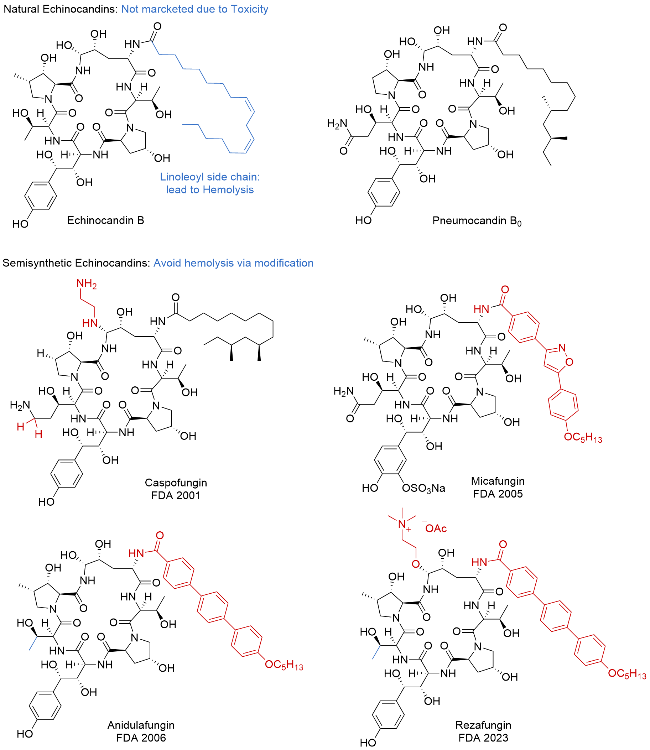
棘白菌素B (Echinocandin B)[49]是1974年从曲霉(Aspergillus rugulosus)中分离出的具有抗真菌活性的天然产物(图5), 尤其是对念珠菌具有优秀的活性. 棘白菌素B通过非竞争性抑制β-1,3-葡萄糖合成酶, 干扰真菌细胞壁β-1,3-葡萄糖的合成, 导致真菌细胞壁渗透性改变, 从而杀死或抑制真菌. 棘白菌素B存在的溶血毒性被认为来自于亚油酸侧链[49c], 此外, 其水溶性很差. 早期的结构改造降低了溶血毒性, 并改善药代动力学特性, 一系列棘白菌素类半合成抗生素得以上市, 包括卡泊芬净(Caspofungin)、米卡芬净(Micafungin)、阿尼芬净(Anidulafungin)和瑞扎芬净(Rezafungin). 值得一提的是, 棘白菌素类和后续三萜类艾瑞芬净是长期以来唯二的新型抗真菌药物, 大大改善了抗真菌药物匮乏的困境(此前主要是三唑类和两性霉素这两类).
7.2 棘白菌素类抗生素半合成工艺
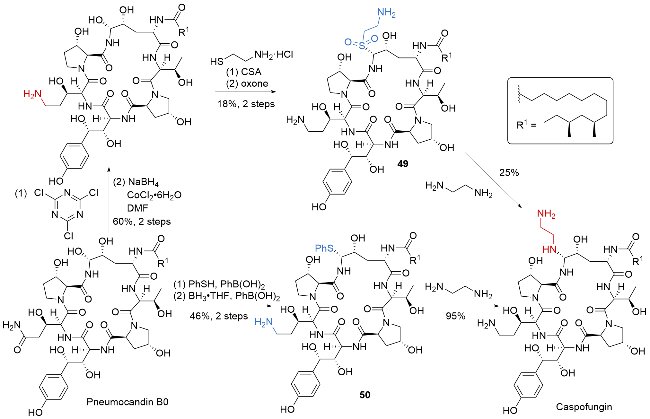
卡泊芬净(FDA于2001年批准其上市)是首个棘白菌素类药物, 由发酵产物纽莫康定(pneumocandin B0)出发半合成得到. 卡泊芬净引入了三个胺基, 提高了水溶性、稳定性以及抗菌活性. 除了水溶性提高, 分子中的侧链并非亚油酰基, 所以膜毒性低, 溶血毒性低.
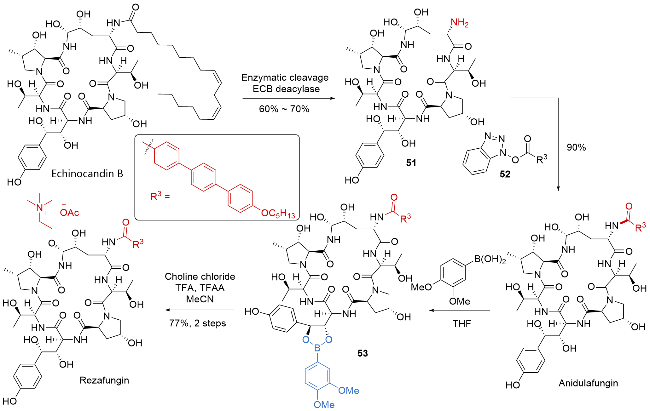
阿尼芬净(FDA于2006年批准其上市)最初由礼来公司研发, 后续由辉瑞公司推进其开发与上市(Scheme 15). 阿尼芬净是棘白菌素B的半合成衍生物, 用烷氧基三苯基侧链取代原本的烷基侧链, 可降低药物的溶血性, 并对细菌细胞膜夹层产生影响, 但阿尼芬净溶解性较差, 口服生物利用度低. 基于此, 为了提高阿尼芬净的稳定性及溶解度, 研发人员在阿尼芬净的基础上引入了一个胆碱醚结构, 得到了新型棘白菌素类药物瑞扎芬净(FDA于2023年批准其上市).
7.3 小结
棘白菌素B本身具有溶血毒性且溶解度差, 半合成解决了这方面的问题, 目前棘白菌素类抗生素凭借其独特的作用机制和较高的安全性, 已成为深部真菌感染的一线药物. 其局限性在于抗菌谱较窄(对烟曲霉作用有限)和耐药性迅速发展. 另外, 其给药方式只能静脉注射. 未来主要需要解决的是日益增加的耐药性问题, 或者通过进一步半合成修饰增强靶点亲和力. 另外, 几丁质合成酶抑制剂尼可霉素Z(抗球孢子菌临床II期)在一些小鼠模型中, 可以大幅增加棘白菌素类药物有效性(如烟曲霉), 且恢复棘白菌素对耐药菌株的活性[54].
8 截短侧耳素类抗生素
8.1 截短侧耳素类抗生素简介
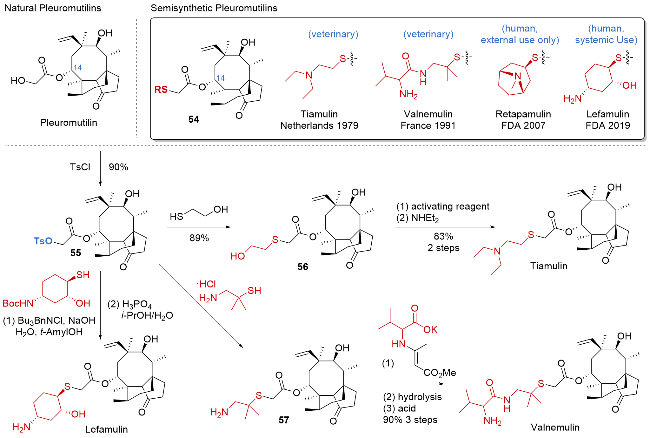
8.2 截短侧耳素类抗生素合成工艺
半合成截短侧耳素类药物以截短侧耳素为母核, 通过结构修饰开发而成, 包括泰妙菌素(Tiamulin)和沃尼妙林(Valnemulin), 作为兽用抗生素, 主要针对革兰氏阳性菌(如葡萄球菌、链球菌)和支原体, 对部分厌氧菌(如产气荚膜梭菌)也有效. 泰妙菌素口服吸收良好, 但半衰期较短(2~4 h), 需频繁给药; 沃尼妙林通过引入侧链延长半衰期至6~8 h, 代谢稳定性更高. 截短侧耳素经TsCl活化唯一伯醇羟基后, 与不同的侧链进行连接, 得到不同的半合成药物(Scheme 16). 先与β-巯基乙醇亲核取代, 再卤代羟基与二乙胺反应, 得到泰妙菌素[56];与二甲基半胱胺盐酸盐取代, 再与D-缬氨酸邓盐反应,经水解、盐酸酸化后得到盐酸沃尼妙林[57]; 与巯基托品烷反应直接得到瑞他莫林[58]; 与异亮氨酸酰胺侧链巯基反应, 再脱Boc得到来法莫林[59].
8.3 小结
半合成截短侧耳素药物主要是对天然截短侧耳素C(14)位进行结构改造, 从最开始的只能兽用(泰妙菌素和沃尼妙林), 到后来降低毒性可以治疗人的皮肤感染(瑞他莫林), 最终发展到现在的可以口服给药(来法莫林). 不过, 由于截短侧耳素结构复杂, 其进一步(其他位置)半合成修饰研究存在困难. 2017年, Herzon小组[60]实现了截短侧耳素的平台化高效全合成, 并在2022年进一步报道了其小分子库的构建. 通过核心骨架修饰、环大小调整、关键位的官能团引入及侧链优化, Herzon小组成功开发出多个高活性截短侧耳素衍生物, 尤其是针对耐药革兰氏阳性菌的化合物, 部分衍生物还展现出对革兰氏阴性菌的潜力, 有进一步开发的前景.
9 三萜类抗生素
9.1 三萜类抗生素简介
艾瑞芬净(Ibrexafungerp, 2021年FDA批准其上市)[60]是首个口服三萜类抗真菌药, 由真菌代谢产物三萜英夫马芬净(Enfumafungin)半合成改造得来(Scheme 17). 英夫马芬净本身具有良好的抗真菌活性, 但其稳定性和溶解性差, 导致其生物利用度低, 代谢稳定性差, 制剂困难. 经过半合成修饰, 其稳定性和脂溶性得到了大大提高, 且药代动力学得以改善, 生物利用度得到提高, 获得了第一个口服抗真菌药物艾瑞芬净. 艾瑞芬净作用机制是抑制真菌细胞壁合成酶β-(1,3)-D-葡聚糖, 具有广谱抗真菌活性, 且低毒[61], 与棘白菌素相同, 但更加高效且不交叉耐药, 具有显著优势.
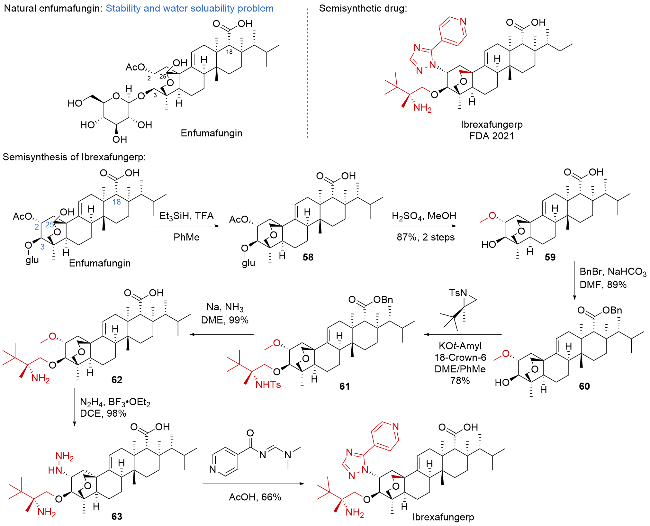
9.2 艾瑞芬净的合成工艺
德国默克公司[62]于2010年报道了艾瑞芬净的合成工艺(Scheme 17). 从天然产物英夫马芬净出发, 首先采用三乙基硅烷还原C(25)半缩醛生成稳定的醚键, 随后在硫酸/甲醇体系下水解C(3)糖苷键, 同时, 由于醚键氧原子的跨环邻基参与效应, C(2)位的乙酰基发生构型保持的SN1取代变为甲氧基. 苄酯保护C(18)羧酸后, 3-OH进攻氮杂环丙烷开环, 再经Birch还原脱除磺酰保护基和苄基. 在引入C(2)位的三唑时, 由于直接引入三唑基团时会生成1-/4-位异构体, 所以先引入C(2)肼基中间体(类似的SN1), 再与酰基脒环化, 专一性生成2-取代三唑. 终产物通过乙醇/水体系梯度结晶纯化, 半合成路线总收率15%.
9.3 小结
艾瑞芬净通过在C(2)和C(3)位进行结构改造, 解决了抗真菌药无法口服的问题. 虽然靶点都是葡聚糖合成酶(FKS1), 艾瑞芬净与棘白菌素类药物的交叉耐药性有限, 提示作用口袋重合度有限. 2022年于洪军小组[63]解析了FSK1蛋白结构, 如果可以进一步研究艾瑞芬净和棘白菌素药物与FSK1蛋白的结合口袋作用模式, 或许可以开发同时覆盖二者结合口袋的新型FSK1抑制剂, 进而开发新型抗真菌药物.
10 最新进展
10.1 抗革兰氏阴性菌药物
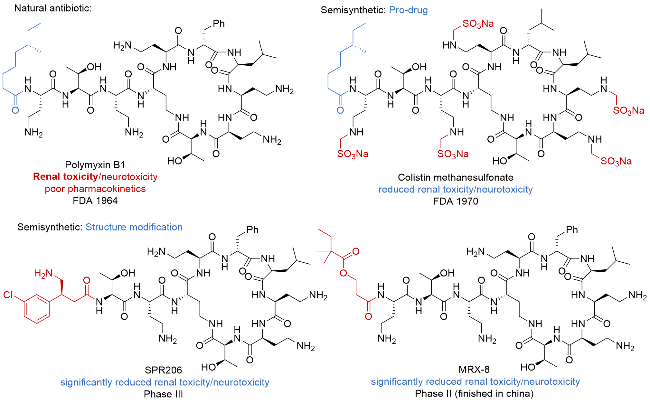
革兰氏阴性菌是对抗耐药菌感染中的重大挑战, 相比于革兰氏阳性菌, 其抗耐药新药研发更加困难. 近年来, 复杂天然产物半合成修饰、半合成工艺在对抗革兰氏阴性菌感染中起到关键性作用. 多黏菌素(Polymyxin B)[64]是抗阴性菌治疗的“最后一道防线”, 高效广谱, 不易耐药, 但受限于毒性问题(图6). 其半合成前药粘杆菌素甲基磺酸钠可以通过调节药代性质减弱毒性, 但没有从根本上解决毒性问题. 多黏菌素的半合成修饰同样具有挑战性, 分子中含有多个羟基和胺基, 且含有多个酰胺键. 不过, 在多黏菌素中可以采用酶催化这一定点精准修饰的工具, 切断特定的酰胺键, 再进行转化得到SPR206和MRX-8这两个毒性减弱的临床分子[64]. 目前, 二者均处于临床试验阶段.
10.2 抗真菌药物
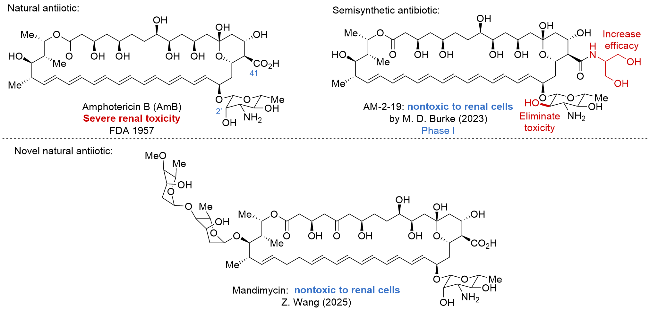
侵袭性真菌病(IFD)是重要的感染类疾病, 致死率高, 死亡人数多(200~400万/年). 复杂天然产物在抗真菌药物中占据重要地位, 尤其是两性霉素(Amphotericin B)[65], 它是抗真菌治疗的金标准, 具有高度的广谱性和有效性, 且不易产生耐药性, 但是具有严重肾毒性, 限制了其疗效(使用剂量低)(图7). 近年来, 半合成两性霉素的发现对改善这一现状起到了关键作用. Burke小 组[65b]长期致力于两性霉素的作用机制和构效关系研究, 基于其作用机制的关键进展, Burke小组[64]于2024年报道了低毒两性霉素类似物Am-2-19, 对两性霉素进行了两个关键后修饰. 其中, C(2')糖环羟基差向异构化可以消除肾毒性(增加“萃取”细胞膜麦角甾醇与胆固醇的选择性), 需11步; C(41)羧基转化为酰胺增加活性, 需1步. 总步骤共12步, 总收率0.26%, 但该路线分离困难, 无法工业化. 该工艺的关键挑战是在复杂官能团底物中实现特定羟基(2'-OH)的构型翻转, 原方法为逐步保护底物中各个官能团, 最后只暴露2'-OH, 再通过Mitsunobu反转构型. 显然, 直接修饰2'-OH可以大大提高合成效率, 但在化学转化上非常挑战, 需要开发新的精准化学方法. 实际上, 抗生素往往是高氧化态含复杂官能团的分子, 定点精准后修饰而非逐级分步保护通常都可以大幅提高效率, 但往往难以实现.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2025年, 王宗强小组[65c]基于系统发育策略发现了新型多烯类抗生素Mandimycin, 其与两性霉素B相比具有类似的结构, 并多出二糖侧链, 后者导致Mandi- mycin的靶点与两性霉素不同, 其并非靶向麦角甾醇而是靶向多种磷脂. Mandimycin表现出高效广谱抗耐药真菌活性, 是极具潜力的新型抗真菌药物先导化合物, 目前王宗强小组正与南京先声药业合作开展进一步的临床前研究. 对比两性霉素和Mandimycin, 或许可以发现对二者进行后修饰的新思路, 开发具有联合作用模式的新型低毒抗耐药多烯类抗真菌药物.
11 总结与展望
综上所述, 半合成抗生素能够解决天然抗生素的耐药性以及毒性等问题, 是新型抗生素的重要来源. 在抗生素被发现的近一百年时间后, 多类半合成抗生素在兽用、人类疾病中仍然发挥着重要作用. 为了解决环境污染、合成效率和成本控制等难题, 半合成抗生素合成工艺也在不断的优化改进, 以期达到绿色化学和精准合成的要求. 从上述的工艺化学路线可以总结如下策略: (1)合理使用保护基策略, 如四环素C(11a)氯代占位策略和棘白菌素苯硼酸保护邻二醇策略; (2)应用新的合成方法学, 如过渡金属参与的氢化和偶联反应; (3)利用生物酶催化策略, 如米卡芬净合成中通过酶水解天然产物的侧链获得关键中间体, 还有酶法水解/酰化用于β-内酰胺类(青霉素、头孢菌素)的母核(6-APA, 7-ACA, 7-ADCA)生产和侧链修饰; (4)发展新的分离纯化手段. 虽然大部分半合成的抗生素合成工艺不断地优化, 但是目前还存在着一些不足: (1)贵金属的大量使用, 如米诺环素的成本主要在于前两步的贵金属催化剂Pd/C和Rh/C的使用; (2)为了解决选择性的问题而必须使用保护基; (3)分离纯化难题, 需要使用树脂分离等.
另外基于目前半合成工艺化学的不足和当代合成化学的发展, 我们提出如下展望: (1)发挥化学合成与生物合成的优势, 利用基因工程改造的微生物菌株直接发酵生产更复杂的中间体或关键砌块, 减少后续化学合成步骤的复杂度和废物量. 如在氨基糖苷类药物中的生物合成过程中, 直接实现1位胺基选择性酰基化, 从而避免化学半合成过程中区域选择性难题. (2)充分应用新合成方法学, 使用金属催化、光/电自由基化学等新合成方法学. 一方面可以设计全新的合成策略, 从而减少反应步骤, 提升整个合成工艺效率; 另一方面可以解决合成工艺中反应活性和选择性等难题, 从而提高原子利用率(如发展“一锅法”串联反应), 最大限度减少废物产生(提高E-因子). (3)生物催化(酶催化)的进一步应用, 利用酶的高效性、区域/立体选择性和温和反应条件(常温常压、水相)替代传统化学步骤. (4)发挥化学全合成与半合成各自的优势. 在早期新药开发研究中, 利用化学全合成或者半合成的方式制备大量的类似物进行活性筛选, 尽快推动新药的上市. 而在后期工艺开发中, 尽量使用半合成工艺, 结合生物合成和化学合成的优势, 针对单一的底物进行生物合成的优化, 从而提高整个药物的合成效率. 如依拉环素的发现和上市都是基于化学全合成, 但是成本较高, 目前也有报道通过半合成策略合成依拉环素: 在山环素的C(7)位直接引入氟原子, 从而降低成本.
(Zhao, C.)