

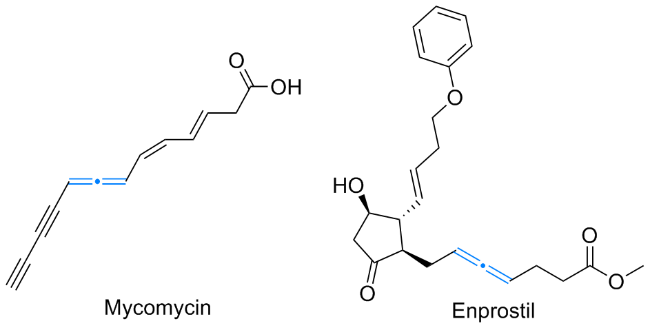
联烯类化合物因含有一组由两个相互垂直的π键组成的累积双键, 所以具有极为丰富的修饰潜力. 这种特殊结构广泛存在于天然产物和药物分子中, 例如菌霉素(Mycomycin)具有抑制结核分枝杆菌活性, 恩前列素(Enprostil)被用作制酸药和抗溃疡药[1](图1). 长久以来, 有机化学家们致力于开发制备多取代联烯类化合物的方法, 并取得了系列成果[2]. 然而, 在传统构建联烯类化合物的途径中, 所采用的联烯前体通常具有较高的 C—H键解离能, 这一特性使得在反应前往往需要对原料进行预官能化处理, 从而导致反应的原子经济性较低[3].
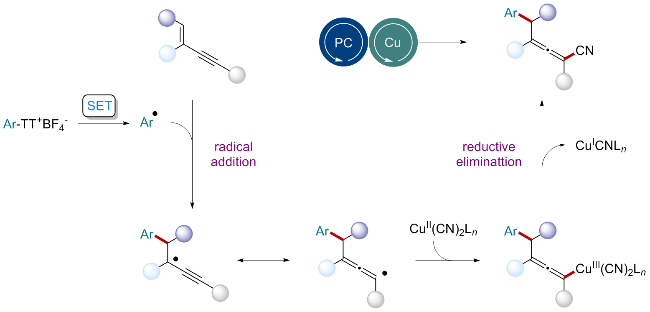
近年来, 1,3-烯炔官能化反应成为制备联烯类化合物的重要方法. 借助过渡金属催化策略, 可通过1,4-加成反应同时向1,3-烯炔中引入两个官能团. 然而, 该领域发展仍存在一些亟需攻克的难题. 首先在区域选择性控制方面, 现有反应表现欠佳, 难以实现对官能化位点的精准控制[4]. 其次, 适用的亲电试剂范围多集中于sp3杂化碳原子, 直接引入sp2杂化碳原子的方法有待开发[5]. 这些情况制约了四取代联烯类化合物的结构多样性与合成高效性. 因此, 如何高区域选择性地在联烯上引入sp2杂化碳原子是该研究领域的难点问题. 光/金属协同催化策略通过光氧化还原过程, 将含官能团的分子转化为活性自由基, 并促使其与金属催化中心结合, 进而实现非传统亲核试剂的直接交叉偶联[6]. 鉴于这一独特优势, 该策略在有机化学领域备受关注, 也为提升四取代联烯类化合物的结构多样性与合成效率提供了新思路.
近日, 青岛科技大学化学与分子工程学院杨道山课题组[7]利用芳基噻蒽盐作为芳基化试剂, 通过铜/光氧化还原协同催化体系, 成功实现了1,3-烯炔类化合物的1,4-芳基氰化反应, 高效合成了四取代联烯腈类化合物(Scheme 1). 值得注意的是, 该反应通过间接芳烃C—H官能化的方式构建了含芳基的四取代联烯腈类化合物. 这一策略避免了传统方法中对芳烃进行预活化的繁琐步骤, 具有步骤简洁、效率高的特点.
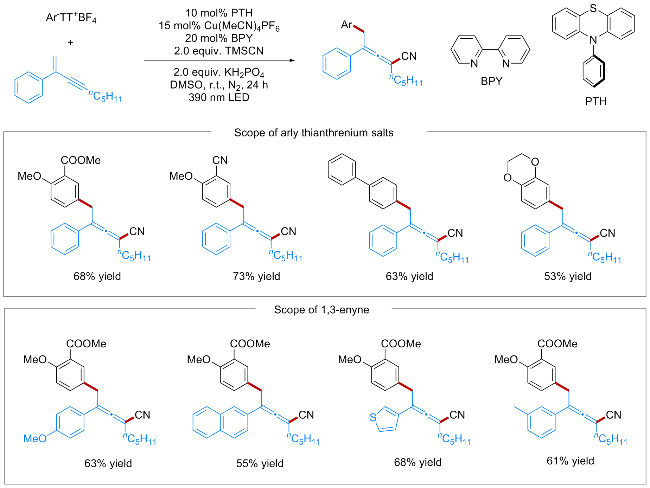
研究团队对关键参数进行了筛选与优化, 确定了最优反应条件: 以四乙腈合铜六氟磷酸盐[Cu(MeCN)4PF6]为铜盐, 2,2'-联吡啶(BPY)为配体, N-苯基吩噻嗪(PTH)为光催化剂, 磷酸二氢钾(KH2PO4)为碱, 二甲基亚砜(DMSO)为溶剂, 在390 nm紫色LED照射下反应24 h. 模型反应的分离产率可达68%.
该催化体系具备良好的底物普适性(Scheme 2). 对于芳基噻蒽盐底物而言, 无论是含有供电子基团(如甲氧基), 还是吸电子基团(如酯基、氰基), 均能在该反应体系中顺利反应并生成对应目标产物; 杂环芳香族噻蒽盐同样能完成转化并获得较高产率. 在1,3-烯炔底物方面, C(2)位被萘基、含卤素(如Cl、I)或甲氧基的芳基取代, 以及C(4)位为不同烷基链时, 均能顺利反应. 此外, 该方法可应用于吡丙醚(Pyriproxyfen)及吉非罗齐(Gem- fibrozil)等生物活性分子的后期官能化, 为复杂药物分子研发和天然产物合成等领域提供了潜在应用途径.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
在机理研究方面, 向体系中添加自由基捕获剂2,2,6,6-四甲基哌啶氧化物(TEMPO)后, 模型反应被完全抑制. 高分辨质谱(HRMS)检测到了TEMPO捕获产物, 表明芳基自由基可能是转化过程的关键中间体. Stern-Volmer荧光猝灭实验中, 芳基噻蒽盐与PTH溶液混合, 当逐渐增加芳基噻蒽盐用量时, 激发态PTH的荧光逐渐减弱, 证实芳基噻蒽盐能有效淬灭PTH. 紫外可见光谱分析(UV-Vis)确认了PTH为光活性物种. 循环伏安法(CV)测得了DMSO中芳基噻蒽盐的还原电位Ep为-1.51 V (vs SCE), PTH的氧化电位E1/2ox*为-2.10 V (vs SCE), 表明了激发态PTH具备还原芳基噻蒽盐的能力.
综上, 该研究开发了一例铜/光氧化还原协同催化的1,3-烯炔1,4-芳基氰基化反应, 以噻蒽盐作为芳基自由基前体合成官能化联烯腈类化合物. 该反应条件温和, 具有优异的区域选择性和化学选择性, 表现出广泛的官能团兼容性, 为构建含芳基联烯腈类化合物提供了高效途径. 机理研究表明, 芳基噻蒽鎓盐可作为芳基自由基前体, 为设计自由基介导的多组分反应提供了重要案例.
(Zhao, C.)