


自19世纪Fischer开创不对称合成研究领域以来, 不对称催化技术已取得显著进展, 并发展成为手性医药、农药及精细化学品合成的核心方法[1]. 从作用机制来看, 不对称催化通过手性催化剂与前手性底物之间的立体专一性相互作用实现反应控制, 其本质在于催化剂与前手性中心/面通过精确的空间位阻和电子效应匹配, 从而实现对产物立体构型的高效调控.
20世纪中叶以来, 该领域相继取得若干里程碑式突破, 代表性成果有Sharpless不对称环氧化反应[2]、Noyori不对称氢化反应[3]及Knowles手性膦配体设计[4]等, 不仅推动了有机合成方法学的革新, 更在制药工业与功能材料等领域实现了规模化应用. 需特别指出的是, 现有成功的不对称催化体系通常依赖于底物结构中显著的立体或电子效应差异, 这些差异为手性识别提供了明确的相互作用位点, 进而通过空间位阻或静电作用实现立体控制.
然而, 传统手性催化剂在应对取代基立体差异较小的底物(如烷基同系物)时, 往往难以有效区分其细微结构差异, 导致对映选择性显著下降. 尽管已有研究尝试通过引入氢键、π-π堆积或静电相互作用等次级分子作用力来增强识别能力, 但对于仅存在诱导效应差异的烷基取代基(如甲基与乙基), 这些相互作用通常难以产生足够的能垒差异以实现有效区分(Scheme 1). 因此, 开发能够精确识别微小立体差异的新型催化策略, 仍是不对称催化领域亟待解决的关键科学问题.
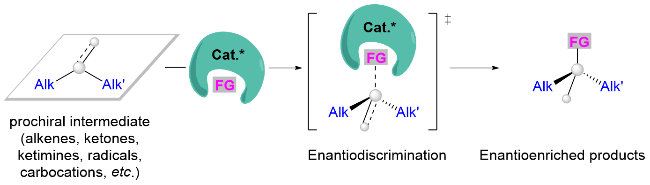
与人工催化体系相比, 生物酶在底物识别方面展现出卓越的精确性. 其通过活性中心氨基酸残基网络构建的立体控制微环境, 整合了氢键作用、静电/偶极相互作用、π-π堆积及范德华力等多重协同效应, 实现了近乎完美的立体选择性控制[1,5]. 值得注意的是, 即便在酶催化体系中, 区分结构高度相似的底物同样面临显著挑战. Arnold团队[6]近期的重要研究突破表明, 通过定向进化获得的P450酶突变体P411-TEA-5274, 可高效实现含甲基/乙基的外消旋叔碳底物的C—H胺化反应, 选择性构建手性α-叔碳胺类化合物. 这一进展证实, 通过合理的蛋白质工程改造与动态位点调控, 酶催化策略可为解决微妙手性识别难题提供创新解决方案.
本文将系统总结近年来化学催化及酶催化策略在实现细微差别取代化合物的手性识别及不对称催化反应的研究进展. 首先, 综述了传统化学催化策略在实现细微差别取代底物的高选择性不对称转化中的进展; 进而综述生物催化面对细微差别取代底物的精准识别及不对称催化挑战的研究现状; 最后总结两类策略的互补性与融合潜力, 并对未来发展方向提出展望, 以期为开发新型高精度手性催化体系提供参考.
1 化学催化微小差异取代化合物的不对称转化
1.1 酮的不对称催化反应
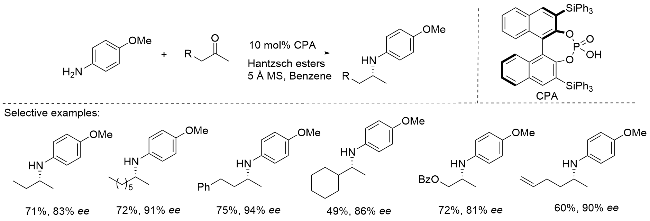
还原胺化反应是现代化学合成中广泛使用的重要转化方法之一. 作为一种多功能偶联反应, 它能够实现多种酮类与胺类化合物的化学选择性结合, 并可高效构建天然产物和药物分子中普遍存在的立体C—N键结构单元. 2006年, MacMillan课题组[7]报道了有机催化还原胺化反应, 该体系通过手性氢键催化剂与Hantzsch酯的协同作用, 实现了复杂片段的不对称仿生偶联. 研究人员系统考察了多种双烷基取代酮在该有机催化体系中的适用性, 其中2-丁酮转化生成的2-氨基丁烷产物获得了83% ee值. 这一结果表明, 具有相似空间位阻和电子效应的二烷基酮类底物能够在该催化体系中实现有效的基团识别(Scheme 2).
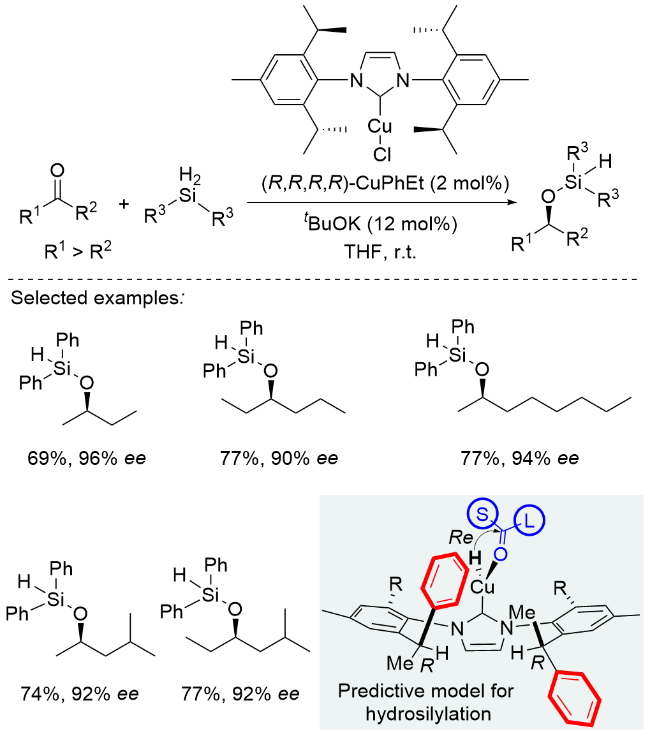
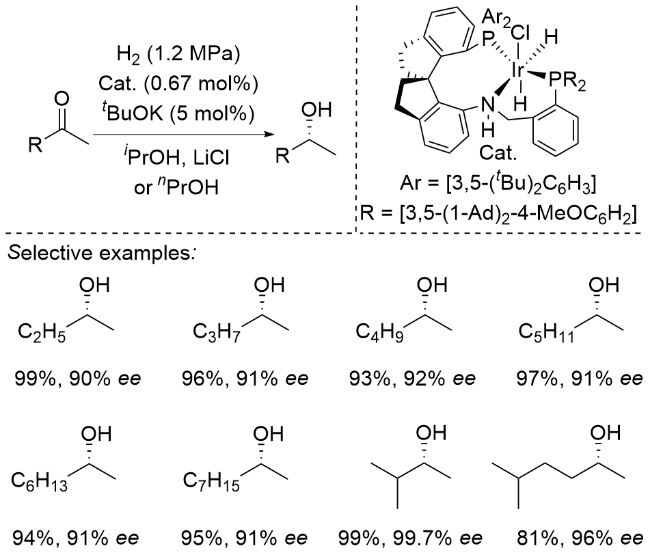
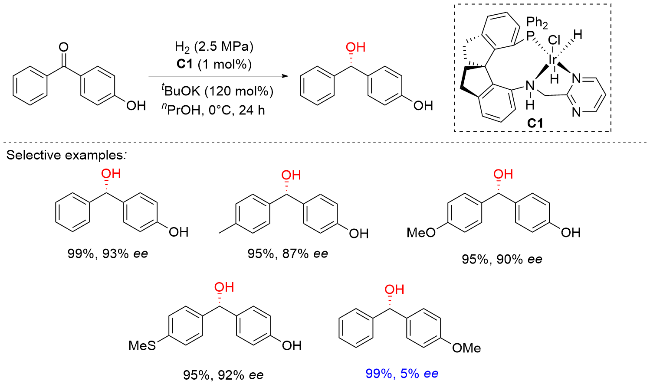
2025年, 周其林课题组[10]开发了一种基于螺环骨架的新型膦-嘧啶胺多齿配体体系. 该体系以氢气为还原剂, 仅需1 mol%铱催化剂即可高效催化4-羟基二苯甲酮的不对称氢化反应, 以99%的产率和95% ee值获得手性二芳基醇. 底物普适性研究表明, 该催化体系对卤素、烷基、芳基及烷氧基等多种官能团均表现出良好的兼容性, 并能精准识别电性及位阻差异较小的芳环. 通过控制实验发现, 对位羟基的存在对反应立体选择性至关重要, 当羟基被保护或移至间位/邻位时, ee值显著降低. 密度泛函理论(DFT)计算揭示, 在碱性条件下苯酚基团可通过异构化为苯醌负离子参与反应, 其与催化剂之间通过π-π堆积和C—H-π相互作用形成不同过渡态, 从而产生R/S构型间的能垒差异, 实现对相似芳基的精准识别(Scheme 5). 值得注意的是, 所得手性二芳基醇类化合物可经简单转化高效合成重要手性药物, 如降血脂药非诺贝特(Fenirofibrate)和镇咳药左氯哌斯汀(Levocloperastine).
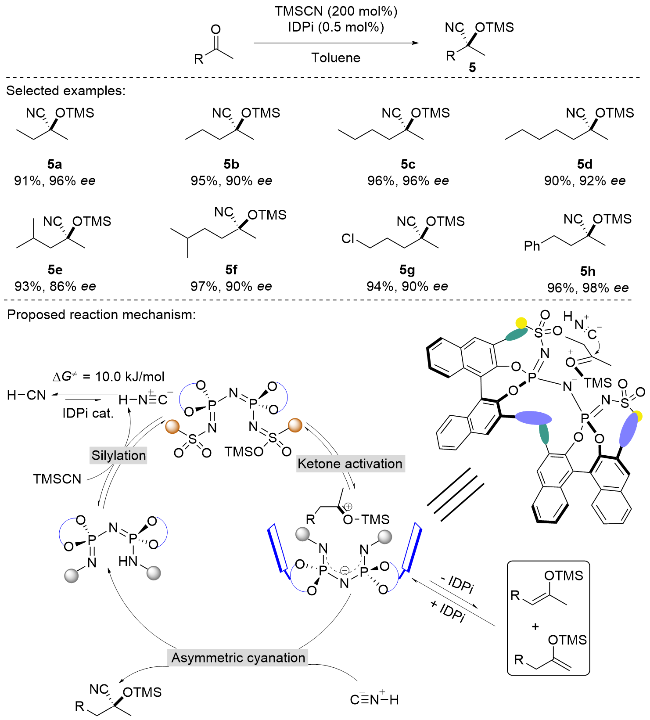
在过去的几十年里, 不对称硅氰化反应得到了广泛研究, 并发展得较为成熟[11]. 然而, 对于2-丁酮这类具有甲基和乙基等相似取代基的底物, 其不对称反应研究仍存在显著挑战. 目前文献报道的成功案例中, 经官能化修饰的酶催化剂最高可获得87% ee值[12], 而金属催化剂和有机小分子硫脲催化剂仅能达到11% ee[13]. 2022年, List课题组[14]突破性地设计了一种仿酶穴状空腔结构的有机超强酸IDPi催化剂, 成功实现了2-丁酮的高效不对称硅氰化反应(Scheme 6). 该催化剂通过其结合位点的受限手性微环境, 实现了对底物对映面的精确识别, 获得了高达98%的ee值. 值得注意的是, 对于甲基/乙基、甲基/丙基等取代基差异细微的双烷基取代酮, 该催化体系同样表现出优异的催化性能, 且产物收率良好. 基于实验数据、光谱分析和理论计算, 提出了IDPi催化酮类化合物硅氰化的反应机理(Scheme 6): 首先, 催化剂与三甲基硅氰(TMSCN)反应生成硅基化催化剂; 随后, 该活性物种活化酮底物形成硅氧基羰基中间体. 在反应体系中, 该中间体易被催化剂的手性阴离子去质子化, 转化为相应的烯醇硅烷. 酸性IDPi物质可高效再生烯醇硅烷, 生成氧羰基离子中间体, 后者经HNC捕获后形成最终产物, 同时完成IDPi催化剂的循环. 该反应过程在动力学上有利于通过Si—H交换生成烯醇硅烷, 在热力学上有利于硅氧羰基离子与HNC反应生成硅基氰丙烷.
1.2 酮亚胺的不对称催化反应
手性胺是生物活性分子、天然产物和农用化学品的重要结构单元, 迄今为止化学家发展了多种方法来构建手性胺, 其中最具代表性的是亚胺的不对称氢化反应及不对称加成反应[15]. 然而, 二烷基酮亚胺的不对称催化反应极具挑战性, 这是因为E-和Z-立体异构体之间存在快速平衡, 而且它们在常规反应条件下极易互变异构为4种可能的烯胺中间体, 从而引发复杂的反应途径并导致产物的对映选择性较低. 经过科学家的不断努力, 手性Ti, Rh, Ir, Pd, Ru, Fe等[16]的配合物被开发, 并用于催化各种亚胺的加氢, 具有高对映选择性. 但是, 大多数催化剂的适用范围相当狭窄, 特别是相似基团取代酮亚胺的不对称加氢反应仍然极具挑战.
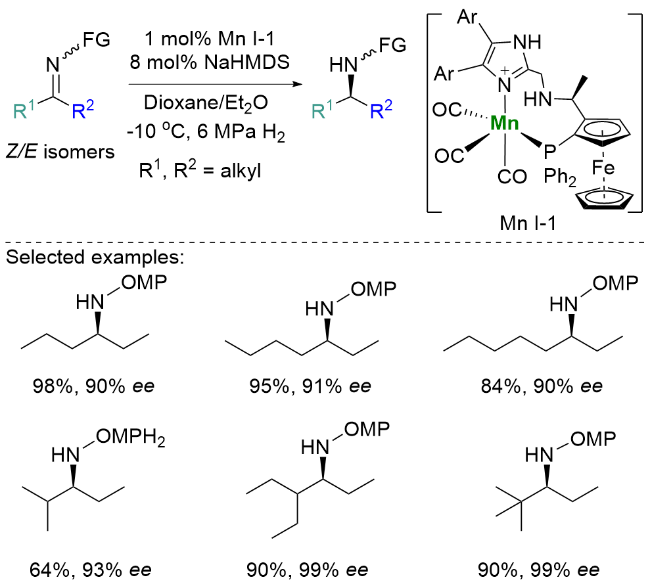
2024年, 刘强和蓝宇等[17]成功开发了一类结构可精准调控的阴离子型锰氢配合物催化剂. 该催化剂在二烷基酮亚胺不对称氢化反应中表现出卓越的立体选择性识别能力, 可有效区分仅存在一个亚甲基差异的烷基取代基(如甲基/乙基、乙基/正丙基以及正丙基/正戊基等), 最终以>99%的对映选择性获得系列手性胺类化合物, 其催化转化数(TON)达到107800的优异水平. 机理研究表明, 优异的立体选择性源于手性配体、金属阳离子及其溶剂化壳层的模块化组装, 这种模块化结构构建了尺寸可精准调控的限域手性微环境, 显著提升了催化剂的对映体识别能力. 研究进一步揭示, 通过形成阴离子型锰氢催化活性物种, 不仅增强了催化剂与底物间的氢键作用和配位相互作用, 还通过σ-活化与π-活化的协同效应, 显著提高底物的亲电性, 并优化了催化剂的手性诱导效率. 值得注意的是, 阴离子配体的引入使锰氢中间体表现出更强的负氢亲核性和电子富集特性, 这一特性有效抑制了富电子烯胺中间体发生竞争性氢化副反应的路径(Scheme 7).
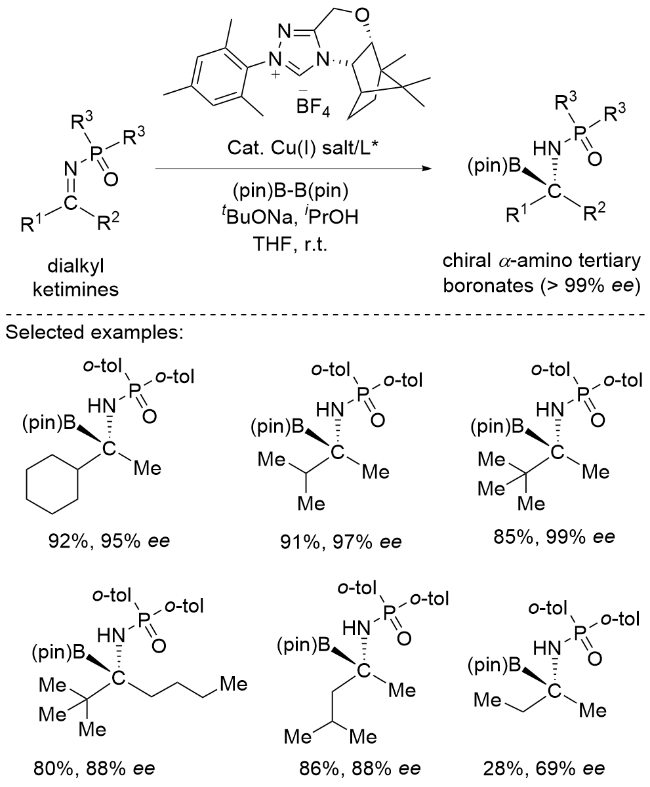
α-氨基硼酸衍生物是α-氨基酸的生物电子等排体, 能够表现出不同的生物活性模式, 因此越来越多的氨基硼酸衍生物被用于许多疾病的治疗干预. 这种不断增长的需求增加了开发高效和立体选择性的方法来制备新型对映异构富集α-氨基硼酸衍生物的需求. 2021年, Ito课题组[18]报道了酮胺的催化对映选择性亲核硼化反应(Scheme 8). 一系列链状二烷基酮亚胺在铜(I)/手性氮杂环碳催化体系下与双(pinacolato)二硼反应, 以高对映选择性制备一系列α-氨基叔硼酸盐. 但是, 该体系在处理甲基/乙基这一最小差异的底物时, 仅能以69% ee获得目标产物, 这也凸显了甲基/乙基这一细微差别取代体系的精准识别极度困难. 该系列产物可转化为含有大量脂肪取代基的肽基硼酸衍生物, 这是其他方法难以合成的化合物. DFT计算表明, 对映体选择性决定步骤涉及识别前手性二烷基酮亚胺的非共价相互作用, 从而实现高效的对映体识别.
1.3 手性辅剂诱导的细微差别化合物合成
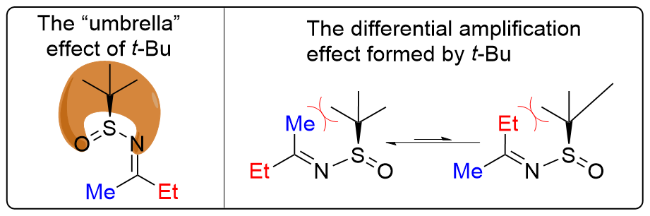
助剂与细微差别取代的酮脱水缩合成手性的酮亚胺, 其不对称还原和加成反应主要由底物本身手性诱导. 由于亚砜基团的极化效应, 其O原子存在一定的配位能力, 其关键的过渡态是由亚砜O原子与金属氢化物(或烷基/芳基锂化物)的络合作用形成的稳定六元环椅式过渡态, 从而实现高效的立体选择性控制. 对于烷基的酮亚胺, 由于金属氢化物和亲核试剂存在一定的碱性, 会导致底物的烯醇化, 对于细微差别基团, 烯醇化后双键的E/Z构型也较难控制, 因此会降低立体选择性和产率. 在酮亚胺与甲基锂试剂的加成反应中[20-21], 对于细微差别基团2-MeC6H4和苯基, 其非对映选择性可高达100%. 在对Ellman酮亚胺的不对称还原反应 中[22-24], 以NaBH4为还原剂, 对甲基和丁基识别效应良好, 其非对映选择性可达92:8, 当使用大位阻的还原剂L-selectride时, dr值可提高至96:4 (Scheme 10).
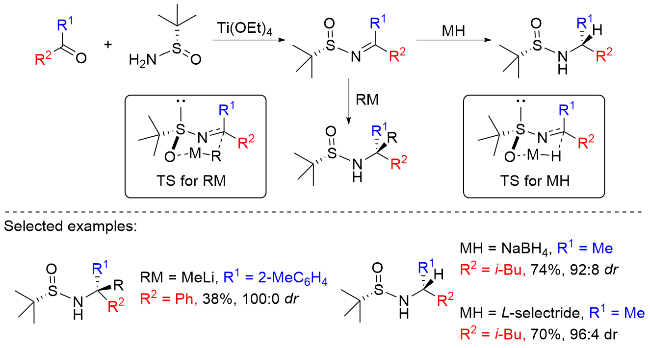
可烯胺化的Ellman酮亚胺可作为一种手性亲核试剂, 由于大位阻的叔丁基的存在, 其Re面和Si面的空间位阻效应差异较大, 若酮亚胺的α位连有两个相似基团, 其不对称反应的立体选择性与烯胺双键的顺反构型有关, 因此, 手性助剂对相似基团的区分有助于稳定的E/Z构型形成, 从而提高整个反应的非对映选择性. α位含氢的Ellman酮亚胺和烯胺在碱的作用下去质子后作为亲核试剂, 实现了亚胺α位不对称羟化[25]、氯化[26]和氰基化[27]反应. 以靛红衍生的酮亚胺为亲电试剂, 经不对称Mannich反应, 连续构建了两个季碳中心[28]. 并且, 以最小的H+为亲电试剂, 也能以优异的非对映选择性实现烯胺-酮亚胺的异构化反应(de值高达99%)[29]. 值得一提的是, 这些反应的de值普遍>20:1, 产率优异, 对相似基团的区分效应很高, 如对甲基和乙基、丙基和烯丙基、乙基和丙基等细微差别基团皆有非常高的区分度, de值高达99%. 此外, 细微差别取代亚胺α位高非对映选择性氟化[30]、羟甲基化[31]、硫化[32]、烯基化[33]、烯丙基化[34]、胺甲基化[35]和环丙烷化[36]等相继被报道(Scheme 11).
1.4 细微差别取代的其他类型不对称催化反应
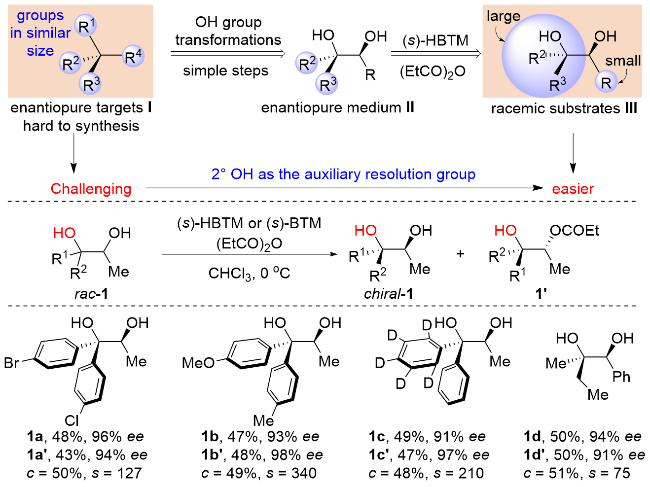
直接合成高光学纯的细微差别取代化合物需要催化体系对两个相似取代基位阻和电性的精准识别, 因此存在较大的挑战, 2021年, 房新强课题组[38]提出了“拆分辅基”策略: 目标分子I可以通过羟基的转化从II制备而来, 而II可以从消旋的III拆分得到; 所以合成手性I的困难最终被转变成制备消旋III的单一非对映异构体, 这又是很容易实现的. 所以, 这一方法就将直接构建四取代碳中心的难点进行了转移, 是一种能够实现多种类型手性四取代碳立体中心分子获取的方法, 二级醇在整个过程中起到了“拆分辅基”的作用. 底物普适性研究发现, 这一拆分体系对多种细微差别取代的芳基和烷基都具有卓越的区分度, 其ee值高达99%, 拆分因子s值高达340. 并且, 通过羟基的后期转化, 可高对映选择性地构筑一系列细微差异取代的季碳类化合物(Scheme 13).
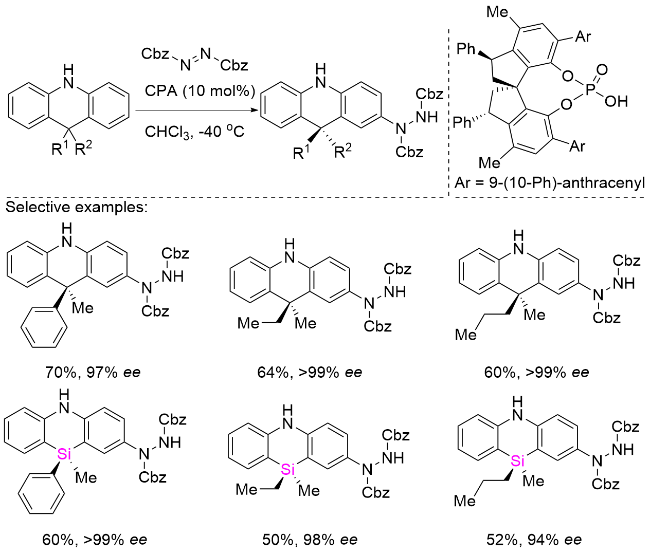
光学纯的9,10-二氢吖啶是手性N-杂环功能分子, 在新型有机光催化剂、生物活性分子等方面有着广泛的应用, 特别是这些杂环已被广泛应用于有机发光二极管(OLED)器件中, 因为它们可以作为典型的供体(D)-受体(A)型热激活延迟荧光(TADF)发射器骨架中的电子供体段[39]. 近年来, 圆偏振发光(CPL)因其在三维光学领域的潜在应用而受到广泛关注[40]. 然而, 它们的不对称催化合成相当具有挑战性, 因为立体中心远离官能团. 2022年, 杨晓瑜课题组[41]开发了一种基于新型螺环手性磷酸(CPA)催化剂的高效远程对映选择性去对称化策略, 成功实现了不对称芳族胺化反应(Scheme 14). 该催化体系能够高效构建一系列带有9,9-二取代基的手性二氢吖啶衍生物, 其底物适用范围极其广泛, 可兼容芳基、烷基以及二烷基等多种取代基类型, 并展现出优异的立体选择性控制能力. 值得注意的是, 该催化体系对空间位阻相近的甲基/乙基等细微差异的底物也能实现出色的对映区分效果(高达>99% ee). 此外, 该方法学还具有广泛的合成应用价值, 不仅适用于立体异构硅中心体系的不对称构建, 还可实现二氢吖啶衍生物的高效动力学拆分.
过渡金属催化硫醚的不对称氮宾转移反应是高效合成手性硫亚胺的方法之一[42]. 在过去几十年里, 尽管芳基烷基硫醚的对映选择性胺化反应取得了诸多进展, 但双烷基硫醚的高立体选择性胺化进展缓慢. 由于双烷基硫醚分子取代基电性相似, 存在更多的构象异构体, 与催化剂缺乏较强的相互作用, 精准识别硫原子上的两对孤对电子难度高. 2024年, 冯小明课题组[43]利用手性双氮氧配体和廉价易得的铁盐作为催化剂, 以亚胺碘苯或二噁唑酮作为氮宾前体, 实现了双烷基硫醚和芳基烷基硫醚的高效高选择性胺化反应. 该方法底物普适性广, 具有优异的区域选择性、广泛的官能团兼容性. 值得提及的是, 上述体系还能用于氨基酸及肽类等复杂分子的后期修饰, 利用上述反应为关键步骤, 成功实现了手性杀虫剂氟啶虫胺腈和相关生物活性化合物中间体的高效合成. 结合机理实验和密度泛函理论计算, 提出了水绑定的高自旋铁氮宾活性物种是反应的关键中间体, 且该物种的生成是反应的决速步, 硫醚进攻铁氮宾中间体是反应的立体选择性控制步骤, 硫醚分子中较大取代基与配体的酰胺部分之间的空间排斥作用是反应立体选择性控制的关键. 该研究成功验证了手性双氮氧化合物可以作为高效配体与铁盐形成配合物实现不对称氮宾转移, 进一步拓展了手性双氮氧/金属配合物的应用, 该合成方法也为含硫手性中心分子及其类似物的高效简洁合成提供了新路径(Scheme 15).

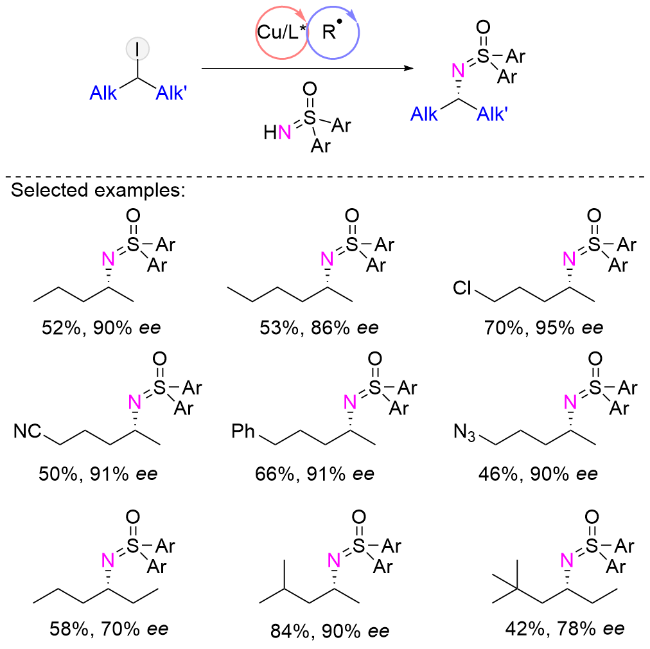
2025年, 刘心元课题组[44]报道了手性阴离子多齿配体对未活化的前手性仲烷基进行铜催化的不对称胺化. 该类催化剂具有一个圆台状半开放式的手性口袋, 其靠近催化中心的区域空间较为狭窄, 远离催化中心的区域则相对开阔. 在反应过程中, 底物中位阻较小的烷基优先进入催化口袋内部狭窄区域, 而位阻较大的烷基则被排斥至外部较宽敞的空间. 与此同时, 催化口袋边壁上的大位阻基团还能与底物中的烷基形成多种非共价弱相互作用, 这不仅有助于在一定程度上稳定高活性的纯烷基中间体, 也进一步提升了对两个结构相近烷基取代基的立体识别能力. 值得一提的是, 对于区分度极小的甲基/丙基(2-碘戊烷)和乙基/丙基(3-碘己烷)等底物, 该催化体系依然表现出优异的选择性. 此外, 该反应还首次实现了以简单烷烃为底物的不对称碳-氢胺化反应, 为推动烷烃这类大宗工业原料向高附加值产物的转化奠定了坚实基础. 通过该策略快速制备的手性伯胺可高效转化为抗癌“明星”药物伊布替尼、降糖药阿格列汀与曲格列汀等活性药物成分或其关键中间体, 为药物学前沿领域的创新发展提供有力支撑(Scheme 16).
2 酶催化微小差异取代化合物的不对称转化
酶是自然界的高效生物催化剂, 在生命体系的各类生理过程中发挥着核心作用. 其催化机制主要体现为: 通过对底物构象的预组织作用, 酶能够精确识别并结合底物分子, 确保催化活性中心与反应位点的特异性匹配, 从而表现出优异的催化效率. 此外, 酶分子空腔周围的氨基酸残基等功能单元可通过多种非共价相互作用, 有效稳定反应中间体或过渡态. 基于其精准的选择性控制、可定向进化改造以及环境友好等显著优势, 酶催化剂日益受到不对称催化领域的广泛关注[45].
近年来, 人工智能技术的快速发展为生物催化领域带来了革命性突破. 随着酶结构解析技术的进步和催化机理研究的深入, 研究者能够通过理性设计对目标酶进行分子改造, 进而实现具有更高底物耐受性、更优区域选择性和立体选择性的生物转化过程. 这一进展不仅推动了多个重磅药物中间体的绿色合成工艺开发, 更为我国医药、农药和信息材料等领域关键化学品的规模化生产提供了重要技术支撑. 目前, 已有诸多重要药物及其关键中间体能够通过酶催化反应实现工业化绿色合成.
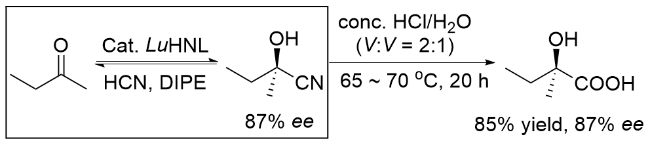
四取代立体碳中心的对映选择性构建是有机化学领域的重要挑战之一. 其中, 手性叔醇类化合物的合成备受关注, 例如作为关键结构单元的2-羟基-2-甲基丁酸, 该结构单元存在于多种上市药物(如Beclobrate、Clinofibrate和Paramethadione)的药效基团中. 这类化合物的合成难点主要在于2-丁酮的对映选择性氰化反应需要实现对甲基和乙基的立体区分, 这一过程具有显著挑战性, 传统化学催化方法在该转化中效果有限. 2008年, Sheldon研究团队[12]利用羟腈裂解酶(HNL)催化2-丁酮与HCN的不对称氰化反应, 成功实现了甲基/乙基的立体选择性识别, 以87% ee值获得了(R)-2-羟基-2-甲基丁腈(Scheme 17).
2022年, 我们课题组[46]开发了一种“一锅一步”生物催化级联系统, 该系统通过高对映选择性还原酶甲硫氨酸亚砜还原酶(MsrA)与低对映选择性氧化酶苯乙烯单加氧酶(SMO)的协同作用, 实现了亚砜化合物的循环去消旋化. 其中, MsrA作为一种(S)-选择性亚砜还原酶, 展现出优异的对映选择性、高底物耐受性以及广泛的底物适用范围; 而SMO则是一种还原型辅酶II (NADPH)依赖性单加氧酶, 具有硫化物氧化活性但表现出较低的对映选择性.
在该循环去消旋系统中, 外消旋亚砜的一种对映异构体首先被MsrA选择性地还原为硫醚, 随后SMO将硫醚非选择性地氧化生成两种亚砜对映异构体. 通过这种“选择性还原-非选择性氧化”的循环过程, 可高效积累目标对映异构体, 最终实现亚砜的高产率、高对映选择性去消旋化. 基于MsrA和SMO的催化机制, 我们进一步构建了包含硫氧还蛋白(Txr)、硫氧还蛋白还原酶(Trx-R)和葡萄糖脱氢酶(GDH)的多酶辅因子再生系统, 利用廉价的D-葡萄糖实现了NADPH等辅因子的原位再生(Scheme 18).
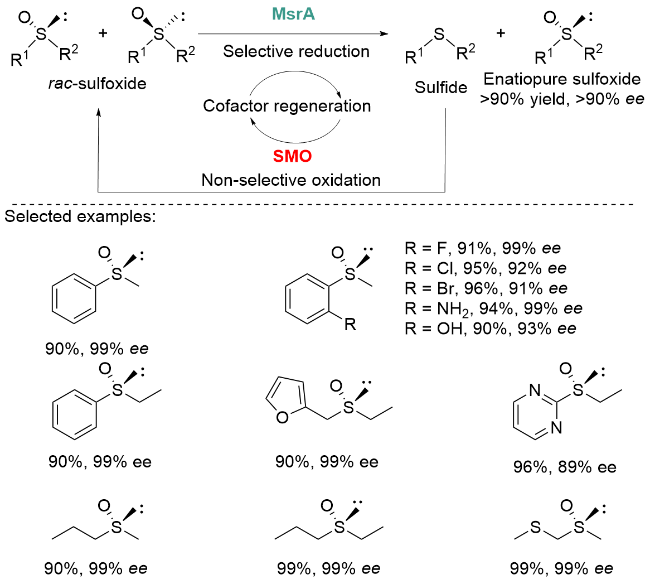
通过MsrA-SMO与三种辅助酶的级联催化, 成功制备了一系列芳基、杂芳基、烷基及硫代烷基亚砜化合物, 其产率和ee值均超过90%. 特别值得注意的是, 即使当底物含有甲基/丙基、乙基/丙基或甲基/硫代烷基等空间性质相近的取代基时, 该体系仍能保持优异的选择性控制能力, 以高产率和高对映选择性获得目标产物. 该系统有效突破了传统MsrA动力学拆分50%的产率限制, 同时弥补了SMO氧化过程对映选择性不足的缺陷. 辅因子再生系统的引入避免了化学计量辅助试剂的浪费, 且该级联反应可顺利放大至1 L规模而不影响产率与ee值.
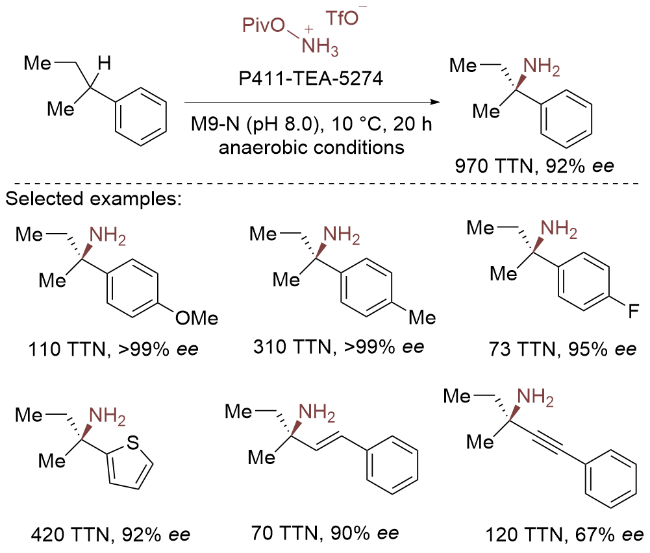
C(sp³)—H键的直接对映选择性官能团化因其优异的原子经济性和步骤经济性, 成为合成高附加值手性分子的理想策略. 近年来, 科学家们已开发出多种高效催化体系, 在二级C—H键的对映选择性转化领域取得显著突破. 然而, 外消旋叔碳C—H键的分子间不对称官能团化仍面临严峻挑战, 特别是在构建四取代立体碳中心方面. 这一难题使得生物催化体系中叔碳中心的不对称修饰成为极具价值的研究目标.
2024年, Arnold团队[6]在酶催化细微差别取代化合物的不对称合成上取得突破性进展, 他们开发的工程酶P411-TEA-5274实现了三级C—H键的高效、高选择性直接胺化, 可精准制备手性α-三级伯胺. 与小分子催化剂相比, 该生物催化剂展现出显著优势: 不仅具有卓越的对映选择性和催化活性, 还表现出广泛的底物普适性. 系统研究表明, 该酶能专一性地识别各类三级C—H键, 并展现出优异的区域选择性和立体选择性. 特别值得注意的是, 该体系成功构建了带有修饰甲基/乙基取代的立体碳中心, 这充分证明了酶催化剂独特的立体识别和控制能力. 结合实验与计算研究揭示了其出色的对映选择性源于活性位点对底物对映体的精准识别以及催化过程中稳定的立体控制机制(Scheme 19).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 总结与展望
综上所述, 本文阐述了不对称催化中空间与电子性质高度相似的取代底物在立体选择性控制方面所面临的重大挑战. 传统手性催化剂通过刚性骨架的空间位阻效应和定向相互作用, 在众多不对称转化中展现了卓越的立体控制能力, 为手性分子的高效构建奠定了重要基础. 然而, 当面对仅存在微弱诱导效应差异的烷基取代基时, 现有催化体系尽管已取得一些突破性的进展, 但实现类型丰富的反应选择性调控仍有一定局限. 相比之下, 自然界中的酶通过活性位点的多重协同作用展现出卓越的立体选择性, 为人工催化体系提供了重要启示. 未来研究有望从以下方向突破. (1)仿生催化策略: 借鉴酶的协同作用机制, 设计更多兼具刚性骨架与柔性调控位点的催化剂, 通过动态相互作用增强对细微差异结构的识别能力; (2)动态共价化学: 利用可逆共价键构建自适应催化体系, 实时响应底物细微差异, 优化过渡态能垒; (3)计算辅助设计: 结合机器学习与量子化学计算, 预测催化剂-底物复合物的立体控制模式, 加速高选择性催化剂的开发; (4)发展绿色生物催化: 结合人工智能技术加快生物催化技术的发展, 通过对酶进行催化功能分子改造, 使其实现更高区域选择性和更高立体选择性精准控制的生物转化反应. 随着对立体控制机理的深入理解和新技术的交叉应用, 不对称催化有望突破“细微差异精准识别”难题, 在药物不对称合成及手性功能材料制备等领域实现精准构筑, 最终推动绿色合成化学向精准分子编辑的阶段发展.
(Zhao, C.)