

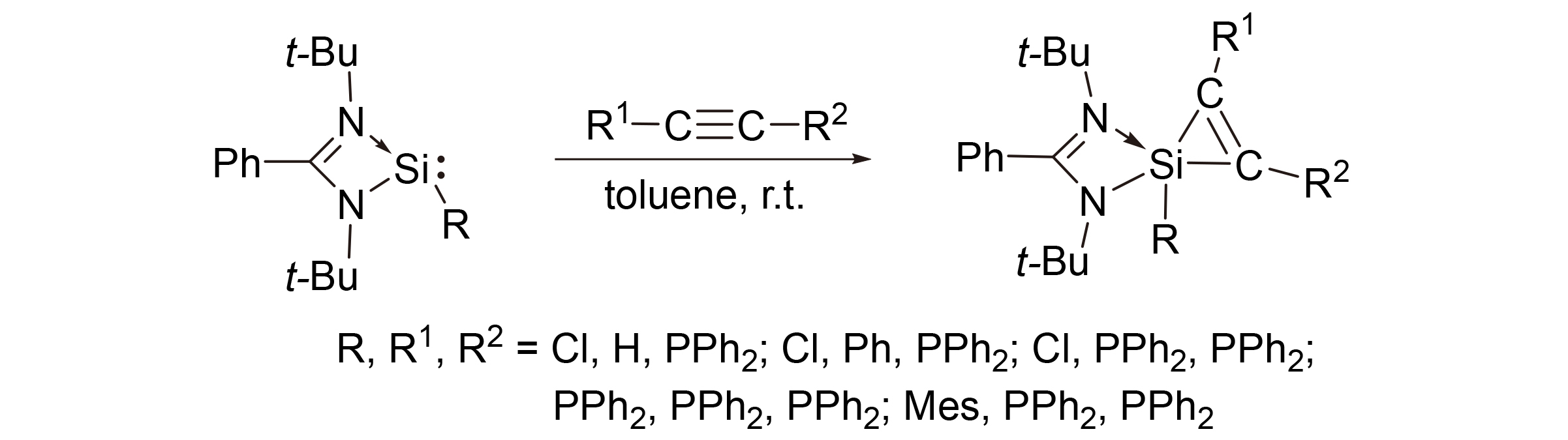
1 结果与讨论
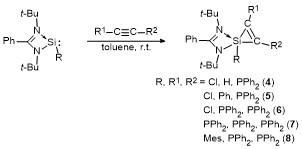
表1 化合物4~6中Si(η2-C2)环的13C和29Si以及PPh2 基团31P 的NMR 数据Table 1 Key NMR data of the 13C and 29Si for the Si(η2-C2)- cycle and the 31P for the PPh2 group from compounds 4~6 |
| Compd. | SiC2 | SiC2 | PPh2 |
|---|---|---|---|
| 4 | -134.6 | 170.52, 194.60 | -6.47 |
| 5 | -139.58 | 174.38, 189.23 | -12.33 |
| 6 | -139.32 | 189.66 | -7.41 |
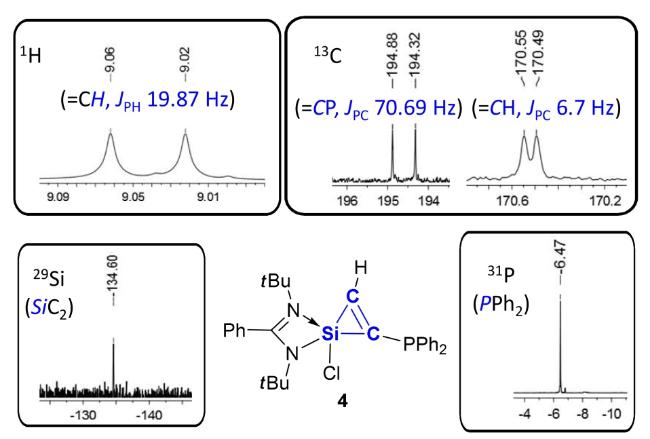
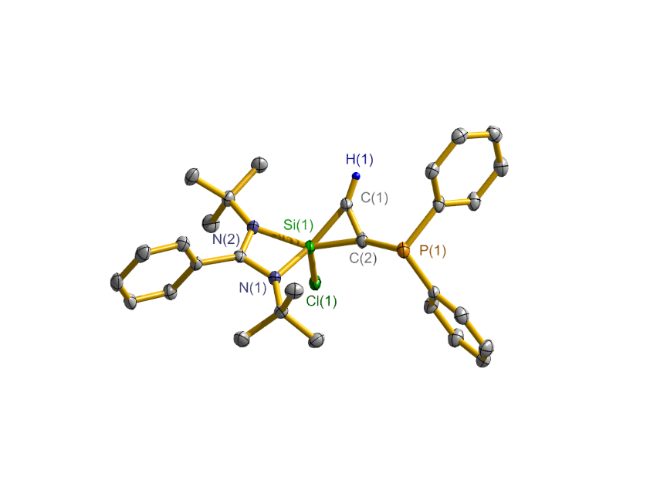
图2 化合物4的50%热椭圆球体晶体结构图Figure 2 X-ray crystal structure of compound 4 with thermal ellipsoids at 50% probability |
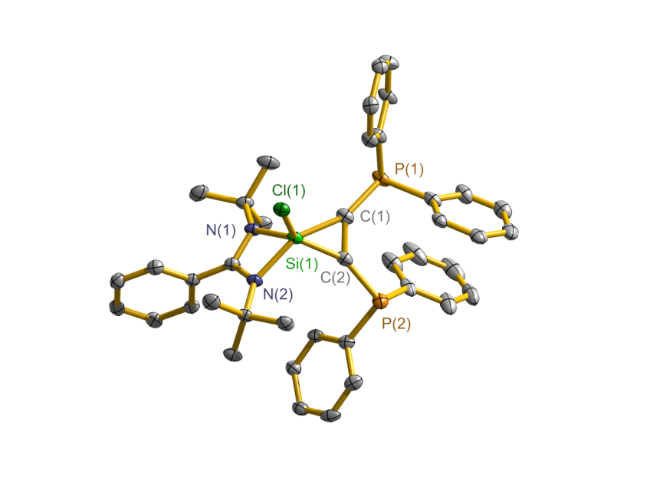
图3 化合物6的50%热椭圆球体晶体结构图Figure 3 X-ray crystal structure of compound 6 with thermal ellipsoids at 50% probability |
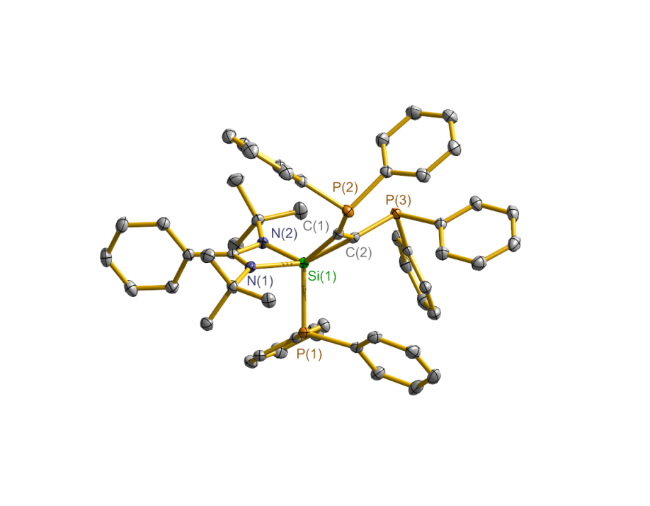
图4 化合物7的50%热椭圆球体晶体结构图Figure 4 X-ray crystal structure of compound 7 with thermal ellipsoids at 50% probability |
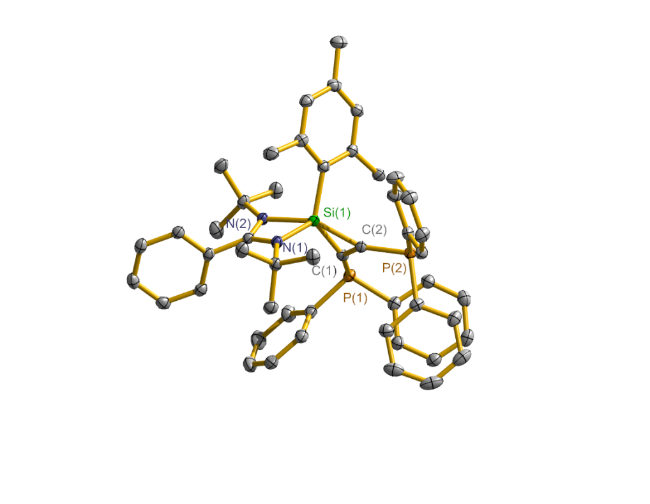
图5 化合物8的30%热椭圆球体晶体结构图Figure 5 X-ray crystal structure of compound 8 with thermal ellipsoids at 30% probability |
表2 化合物4和6~8中Si(η2-C2)环的重要键长(nm)和键角(°)Table 2 Important bond length (nm) and angle (°) for the Si(η2-C2)-cycle of compounds 4 and 6~8 |
| Comp. | Si—C/nm | C—C/nm | Si—C—C/(°) | C—Si—C/(°) | ||
|---|---|---|---|---|---|---|
| 4 | 0.1781(2) 0.1842(2) | 0.1351(3) | 65.76(10) 70.50(11) | 43.74(8) | ||
| 6 | 0.1788(3) 0.1830(3) | 0.1371(5) | 66.12(18) 69.40(20) | 44.52(15) | ||
| 7 | 0.1806(2) 0.1871(2) | 0.1365(2) | 65.71(7) 70.77(7) | 43.52(5) | ||
| 8 | 0.1806(2) 0.1880(2) | 0.1361(2) | 65.44(9) 71.27(9) | 43.29(7) | ||

图式4 硅宾与取代炔烃反应生成硅杂环丙烯的可能作用机理Scheme 4 Proposed interaction mechanism of the silylene and substituted alkyne to silacyclopropene |
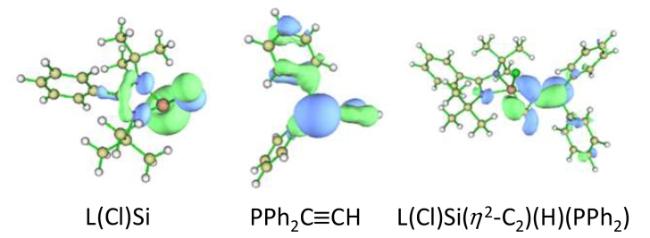
表3 NBO分析法计算的不同取代基炔烃分子中C≡C键C原子的电荷分布数值Table 3 Charge distribution calculated for the C≡C carbon atoms of the substituted alkynes by the NBO analysis method |
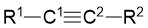
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | C1 | C2 |
|---|---|---|---|
| H | SiMe3 | -0.187 | -0.487 |
| H | OEt | -0.383 | 0.327 |
| H | nBu | 0.005 | 0.249 |
| H | PPh2 | -0.113 | -0.342 |
| H | Ph | -0.192 | -0.033 |
| Et | CO2Et | 0.062 | -0.151 |
| Ph | SiMe3 | 0.008 | -0.451 |
| Ph | Me | -0.078 | 0.025 |
| Ph | Et | -0.074 | 0.012 |
| Ph | Pr | -0.073 | 0.018 |
| Ph | CO2Et | 0.040 | -0.107 |
| Ph | PPh2 | 0.135 | -0.359 |
| PPh2 | SiHMe2 | -0.300 | -0.407 |
| PPh2 | SiHPh2 | -0.279 | -0.418 |