

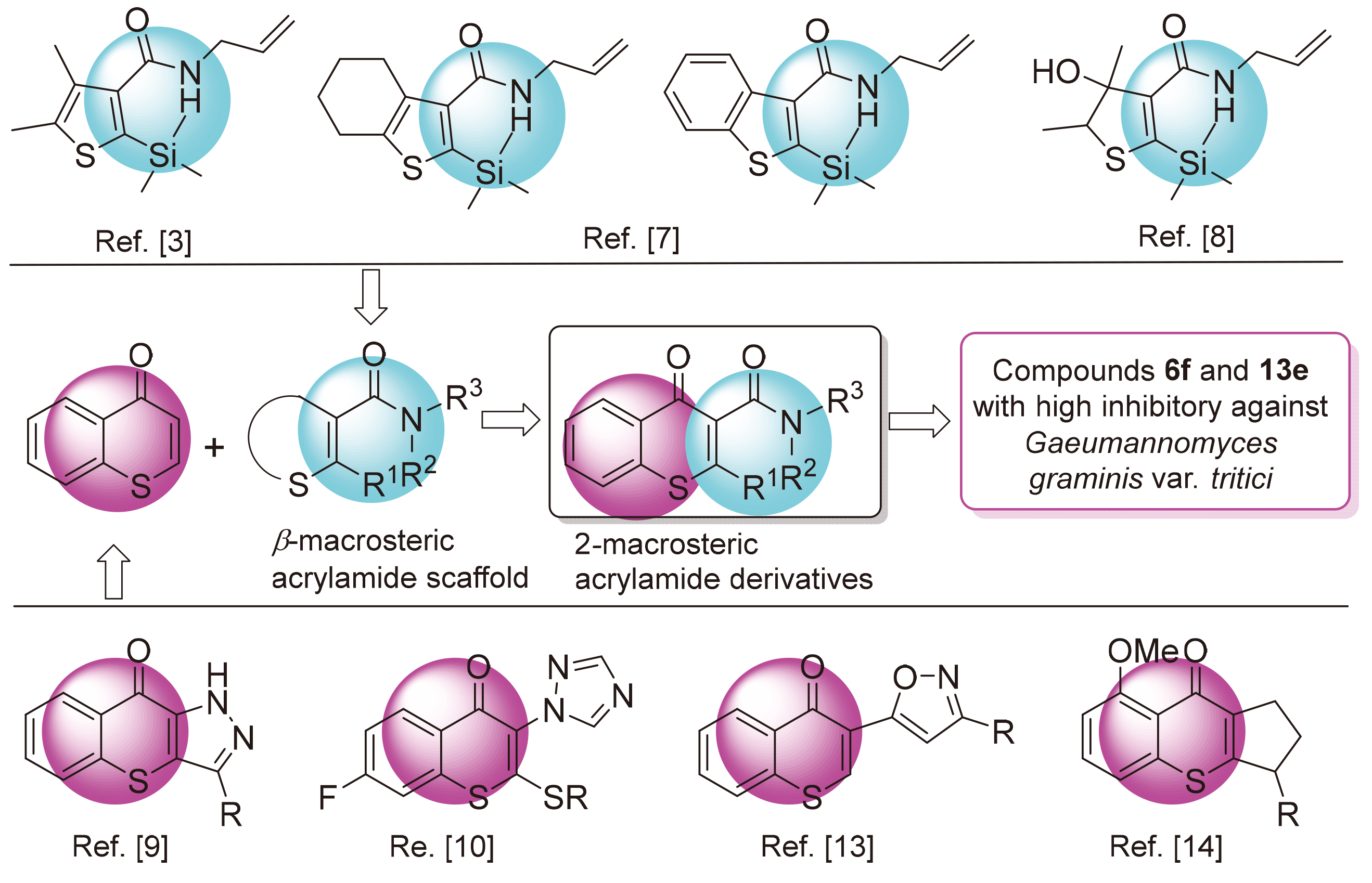
全蚀病是一种顽固土传病害[1], 严重威胁着小麦的生产安全. 戊唑醇、苯醚甲环唑和氟硅唑[2]等三唑类杀菌剂对小麦全蚀病的防治具有一定效果, 但不够理想. 硅噻菌胺(Silthiopham)[3]是目前已登记杀菌剂品种中能够有效防治小麦全蚀病的杀菌剂, 但品种单一, 抗性风险增加[4]. 我们课题组近来的研究发现具有不同结构的噻吩甲酰胺衍生物对小麦全蚀病菌表现出较好抑制活性[5-6], 同时苯并噻吩甲酰胺、环己基并噻吩甲酰胺[7]及其它噻吩甲酰胺[8]等类似物对小麦全蚀病菌也具有较高活性, 这些化合物均具有β-大位阻丙烯酰胺结构单元(图1蓝色部分). 因此, 在新活性化合物设计时, 该结构单元可作为活性化合物的基本骨架.
为此, 我们根据活性化合物骨架迁移策略, 将β-大位阻丙烯酰胺结构特征与硫色酮骨架进行巧妙结合, 设计如图1所示的2-取代硫色酮甲酰胺衍生物, 研究化合物的构效关系, 以期开发出具有优异活性防治小麦全蚀病的新型化合物, 为小麦全蚀病防治以及抗性治理提供技术支持.
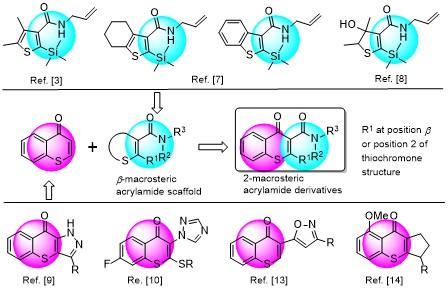
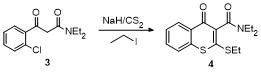
针对所设计2-位为杂原子的硫色酮甲酰胺衍生物, 课题组设计了如Scheme 1所示的合成路线. 该路线以2-氯苯乙酮(1)为原料, 通过其与碳酸二甲酯的缩合, 缩合产物与二乙胺的酰胺化得到中间体3-(2-氯苯基)-N,N-二乙基-3-氧代丙酰胺(3), 中间体3在氢化钠的存在下先后与二硫化碳和碘乙烷反应, 然后再用双氧水氧化得到中间体5, 中间体5与各种含氮杂环及取代胺反应, 分别得到6a~6d, 6a分别与异丙胺及取代的苯胺反应得到6e~6h, 共合成了8个2-位为杂原子的硫色酮甲酰胺衍生物.
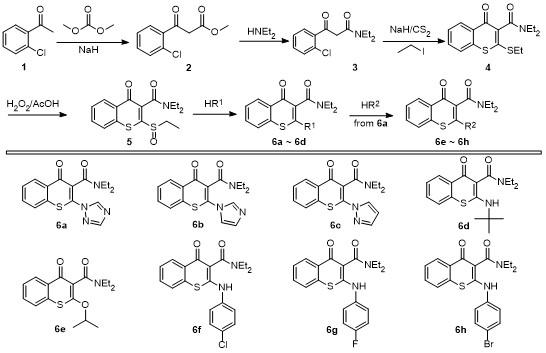
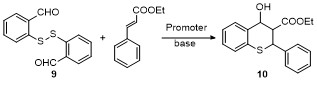
2-位为苯基硫色酮甲酰胺衍生物的合成具有一定的挑战, 为此本课题组又设计了如Scheme 2所示的合成路线. 该路线以巯基苯甲酸为原料, 通过还原、选择性氧化以及与肉桂酸乙酯的缩合得到关键中间体10, 中间体10通过羟基氧化、酯的水解以及羧酸与各种取代胺的酰胺化反应等, 分别得到目的产物13a~13e. 共合成了5个2-位为苯基的硫色酮甲酰胺衍生物.
所合成硫色酮甲酰胺衍生物都进行了分离与纯化, 并在此基础上分别评估了化合物对小麦全蚀病致病菌(Gaeumannomyces graminis var. tritici)的抑制活性, 分析了其结构活性关系等.
1 结果与讨论
1.1 目标化合物6的合成
表1 N,N-二乙基-2-乙硫基-4-氧代硫色酮-3-甲酰胺(4)的合成条件优化Table 1 Optimization of conditions for synthesizing N,N-dieth- yl-2-ethylthio-4-oxo-4H-thiochromene-3-carboxamide |
| Entry | Base | T/℃ | Time/h | Solvent | Yield/% |
|---|---|---|---|---|---|
| 1 | NaOH | 130 | 8 | DMF | 13 |
| 2 | NaOH | 130 | 8 | DMSO | 15 |
| 3 | NaOH | 40 | 8 | DMSO | 35 |
| 4 | NaH | 130 | 8 | DMSO | 15 |
| 5 | NaH | 40 | 8 | DMSO | 57 |
通过对碱、温度和溶剂的优化, 发现以NaH (2 equiv.)为碱、二甲亚砜(DMSO)为溶剂, 在40 ℃下分别与CS2 (1.5 equiv.)和碘乙烷(1.5 equiv.)反应8 h, 以57%的中等收率获得中间体4 (Entry 4), 为目标化合物6的合成奠定了基础.
中间体4以乙酸为介质在双氧水氧化下, 以44%的收率转化为中间体5; 中间体5以N,N-二甲基甲酰胺(DMF)为反应介质, 在110 ℃条件下分别与相应的亲核试剂反应12 h, 以42~67%的收率得到6a~6d. 然而类似的方法不适用于6e~6h的合成, 进一步研究发现6e~6h可以通过6a分别与相应的醇或取代苯胺发生交换反应获得, 收率在41%~87%之间.
1.2 化合物13的合成
表2 4-羟基-2-苯基-3,4-二氢硫色烷-3-甲酸乙酯(10)的合成条件优化Table 2 Optimization of conditions for ethyl 4-hydroxy-2- phenyl-3,4-dihydro-2H-thiochromene-3-carboxylate (10) |
| Entry | Promoter (1.6 equiv.) | Base (1 equiv.) | T/℃ | Time/h | Solvent | Yield/% |
|---|---|---|---|---|---|---|
| 1 | — | K2CO3 | r.t. | 7 | Toluene | 0 |
| 2 | PPh3 | — | r.t. | 7 | Toluene | Trace |
| 3 | PPh3 | CS2CO3 | r.t. | 7 | Toluene | Trace |
| 4 | PPh3 | K2CO3 | r.t. | 7 | Toluene | Trace |
| 5 | K2CO3 | TMG | r.t. | 7 | Toluene | 0 |
| 6 | PPh3 | TMG | r.t. | 7 | Toluene | 51 |
| 7 | PPh3 | TMG | r.t. | 7 | DMF | Trace |
| 8 | PPh3 | TMG | r.t. | 7 | DMSO | Trace |
| 9 | PPh3 | TMG | 50 | 7 | Toluene | 73 |
| 10 | PPh3 | TMG | 70 | 7 | Toluene | 57 |
将肉桂酸乙酯(1 equiv.)与2-巯基苯甲醛二聚体(1.3 equiv.)分别在碱(K2CO3、CS2CO3)及解聚剂(PPh3)的存在下, 以甲苯为溶剂室温下反应7 h, 没有得到相应的环合产物(Entries 1~4). 尝试在相似条件下, 使用K2CO3和四甲基胍(TMG)组合, 也没有得到环合产物(Entry 5). 于是尝试PPh3和TMG的组合[19], 在常温下反应7 h, 以51%的收率获得了环合产物(Entry 6). 为进一步提高收率, 考察了以强极性溶剂DMF和DMSO取代甲苯时的反应效果, 收率明显降低(Entries 7, 8). 接着继续以甲苯为溶剂提高反应温度至50 ℃, 环合收率提高到73% (Entry 9). 进一步提高温度, 收率没有进一步改善. 该反应较优惠的条件是: 在促进剂三苯基膦的存在下, 以TMG为碱, 甲苯为溶剂, 50 ℃条件下反应7 h, 可以获得73%的收率(以肉桂酸乙酯计).
中间体10在氧化剂2,3-二氯-5,6-二氰基苯醌(DDQ)的存在下, 以94%的收率转化为中间体11, 中间体11水解为相应的酸, 再在缩合剂N,N'-二环己基碳二亚胺(DCC)的存在下, 分别与不同的取代胺缩合, 以38%~59%的收率得到目标产物13a~13e.
1.3 硫色酮甲酰胺衍生物的结构与活性关系
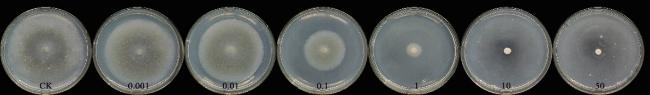
受噻吩甲酰胺氮原子上的取代基为简单烷基时, 表现出较高活性[5]的启发, 在结构设计时首先保持N,N-二乙基酰胺的基本结构, 2-位引入不同的杂原子取代基, 获得了具有6a~6h结构的化合物. 为了评估其结构活性关系, 采用平皿菌丝生长速率法[20]测定了对小麦全蚀病致病菌(Gaeumannomyces graminis var. tritici)的抑制活性, 结果如表3所示. 正如预期, 所设计化合物对小麦全蚀病菌都表现出不同程度的抑制活性. 6b~6d和6f~6h在200 mg/L的浓度下对小麦全蚀病菌的抑制率超过了80%, 6c和6f在此浓度下达到了100%的抑制水平. 2-位取代基对化合物活性具有重要影响, 当2-位取代基为4-氯苯胺时(6f), 化合物活性更为突出, 在25 mg/L的浓度下抑制率达到了70.6%, 超过了阳性对照硅噻菌胺的活性水平.
表3 2-取代的硫色酮甲酰胺衍生物的抑菌活性Table 3 Inhibitory activities of 2-substituted thiochromone formamide derivatives |
| Compd. | Percent inhibitiona/% (in vitro) | ||||
|---|---|---|---|---|---|
| 200 mg/L | 100 mg/L | 50 mg/L | 25 mg/L | 12.5 mg/L | |
| 6a | 71.6 | 39.2 | 22.6 | 18.0 | 10.5 |
| 6b | 87.0 | 80.9 | 67.3 | 50.4 | 41.3 |
| 6c | 100 | 51.6 | 32.7 | 26.5 | 20.5 |
| 6d | 99.1 | 62.9 | 39.8 | 20.7 | 3.8 |
| 6e | 56.9 | 32.8 | 15.5 | 12.1 | 10.1 |
| 6f | 100 | 100 | 100 | 70.6 | 29.2 |
| 6g | 89.8 | 80.2 | 63.6 | 37.6 | 17.3 |
| 6h | 97.4 | 90.8 | 88.6 | 86.6 | 57.6 |
| 13a | 99.5 | 91.7 | 51.6 | 23.6 | 20.1 |
| 13b | 58.3 | 44.9 | 28.9 | 23.3 | 17.5 |
| 13c | 93.5 | 93.0 | 90.3 | 90.9 | 86.3 |
| 13d | 98.6 | 93.0 | 87.0 | 87.8 | 80.6 |
| 13e | 100 | 100 | 100 | 100 | 100 |
| Silthiopham | 100 | 92.3 | 66.7 | 40.5 | |
a Mean value of three replicates. |
受到以上结果的鼓舞, 课题组又对化合物结构进行了新的设计, 并合成了化合物13a~13e. 其活性结果表明, 当2-位为苯基、酰胺胺基为取代的苯胺时, 其活性有提高的趋势, 如化合物13c~13e在25 mg/L的浓度下的抑制率均超过了80%, 其中13e的活性表现优异, 甚至在12.5 mg/L的较低浓度下的抑制率也达到了100%. 进一步活性评估表明, 化合物13e的EC50达到了0.07 mg/L, 比硅噻菌胺的EC50 (26.7 mg/L)几乎降低了2个数量级(图2). 因此, 化合物的结构设计取得了重要进展, 对小麦全蚀病新型防治剂的开发具有一定借鉴意义.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2 结论
设计了以2-氯苯乙酮为原料合成化合物6a~6h及以2-巯基苯甲酸和肉桂酸乙酯为原料合成化合物13a~13e的路线, 探索优化了N,N-二乙基-2-乙硫基-4-氧代硫色酮-3-甲酰胺(4)和4-羟基-2-苯基-3,4-二氢硫色烷-3-甲酸乙酯(10)两个重要中间体的反应条件. 通过多步合成, 顺利获得了13个具有代表性的目标化合物. 活性研究表明, 所设计化合物对供试小麦全蚀病菌均表现出不同程度的抑制活性, 其中化合物6f在50 mg/L浓度下抑制率可以达到100%, 化合物13e的抑制活性更高, 在12.5 mg/L的较低浓度下, 抑制率仍然可以达到100%, EC50达到了0.07 mg/L的较低水平, 比硅噻菌胺的EC50几乎降低了2个数量级. 此结果一定程度上印证了活性化合物的设计思想, 对活性化合物的进一步设计和新农药开发具有一定的参考价值.
3 实验部分
3.1 仪器与试剂
反应及目标化合物纯度使用HPLC (Agilent-1400)检测和分析; 使用Agilent-NMR-vnmrs 400核磁共振仪获得化合物的1H NMR、13C NMR谱, 以DMSO-d6或CDCl3为溶剂, TMS为内标; 使用Bruker micrOTOF-QⅡ质谱仪获得HRMS; 使用WRS-1A数字熔点仪测试熔点; 病原菌使用HPG-280H人工气候箱培养. 所有国产或进口AR或CP级试剂未经纯化直接使用.
3.2 病原菌菌株
供试的小麦全蚀病病原菌由河南农业大学植物保护学院病理实验室提供.
3.3 中间体及目标化合物的合成
3.3.1 3-(2-氯苯基)-N,N-二乙基-3-氧代丙酰胺(3)的合成
中间体2-氯苯甲酰乙酸甲酯(2)依照文献[15]合成, 收率83%.
将合成的中间体2 (2.12 g, 10 mmol)与二乙胺(0.146 g, 20 mmol)投入到盛有20 mL甲苯的三口反应瓶中, 加热至90 ℃反应10 h. 反应溶液冷却至室温后, 用水(10 mL×2)洗涤, 将洗涤后的有机相用无水硫酸钠干燥, 真空浓缩, 残余物用硅胶柱分离(石油醚/乙酸乙酯, V∶ V=5∶1), 得2.18 g (8.61 mmol)化合物3, 淡黄色液体, 收率86%. 该化合物在测试条件下同时呈现出烯醇式和二酮式结构, 1H NMR和13C NMR能够清晰地表明烯醇式和二酮式结构比例大约为4∶1. 1H NMR (400 Hz, CDCl3) δ: 15.44 (s, 1H), 7.68~7.29 (m, 5H), 5.66 (s, 1H), 3.48~3.32 (m, 5H), 1.23~1.10 (m, 7H); 13C NMR (100 Hz, CDCl3) δ: 196.59, 171.02, 169.96, 165.67, 138.30, 135.03, 132.20, 131.83, 130.98, 130.51, 130.45, 130.16, 130.08, 127.06, 126.81, 90.89, 48.71, 42.75, 42.33, 40.57, 40.15, 14.40, 14.23, 13.32, 12.85; HRMS (ESI) calcd for C13H17ClNO2 [M+H]+ 254.0948, found 254.0941.
3.3.2 N,N-二乙基-2-乙硫基-4-氧代-4H-硫色酮-3-甲酰胺(4)的合成
将化合物3 (2.53 g, 10 mmol)投入盛有15 mL DMSO的三口反应瓶中, 然后投入60% NaH (0.8 g, 20 mmol), 搅拌下升温至40 ℃. 然后将含有CS2 (1.14 g, 15 mmol)的DMSO (4 mL)溶液缓慢滴入反应瓶中(40 ℃). 反应完毕后, 冷却至室温, 边搅拌边滴加碘乙烷(2.34 g, 15 mmol)的DMSO (5 mL)溶液, 滴毕, 室温反应至反应完全. 将反应液倒入50 mL水中, 调节pH值至中性, 然后用乙酸乙酯(30 mL)萃取, 有机相水洗(15 mL×2), 干燥后浓缩, 残余物用硅胶柱分离(石油 醚/乙酸乙酯, V∶V=10∶1), 得1.82 g (5.67 mmol)化合物4, 淡黄色液体, 收率57%. 1H NMR (400 Hz, CDCl3) δ: 8.44 (d, J=8 Hz, 1H), 7.60~7.28 (m, 3H), 3.70~3.63 (m, 1H), 3.55~3.48 (m, 1H), 3.24~3.10 (m, 4H), 1.41 (t, J=7.4 Hz, 3H), 1.28 (t, J=7 Hz, 3H), 1.13 (t, J=7 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 175.66, 164.98, 149.59, 136.98, 133.34, 131.59, 130.34, 129.13, 127.97, 125.33, 42.85, 39.11, 27.95, 14.55, 14.15, 12.65.
3.3.3 N,N-二乙基-2-(乙基亚磺酰基)-4-氧代-4H-硫色酮-3-甲酰胺(5)的合成
将化合物4 (0.963 g, 3 mmol)放入盛有3 mL AcOH的三口瓶反应中, 然后加入二氯甲烷(3 mL). 在60 ℃滴加3 mL 1 mol/L H2O2的AcOH溶液, 反应完毕后将反应液倒入水(15 mL)中并用二氯甲烷(15 mL×2)萃取. 将有机相依次用水(15 mL)、饱和NaHCO3溶液(15 mL)和盐水(15 mL)洗涤, 然后干燥、浓缩. 残余物用硅胶柱分离(石油醚/乙酸乙酯, V∶V=2∶1), 得到0.48 g (1.43 mmol)化合物5, 淡黄色固体, m.p. 91.5~93.4 ℃; 收率44%. 1H NMR (400 MHz, CDCl3) δ: 8.49 (d, J=8 Hz, 1H), 7.76~7.61 (m, 3H), 3.72~3.64 (m, 2H), 3.58~3.54 (m, 1H), 3.49~3.46 (m, 1H), 3.21~3.18 (m, 2H), 1.41 (t, J=7.4 Hz, 3H), 1.27 (t, J=7.2 Hz, 3H), 1.15 (t, J=7.2 Hz, 3H).
3.3.4 目标化合物6a~6d的合成
将化合物5 (337 mg, 1 mmol)和各种唑杂环化合物(2 mmol)加入到盛有5 mL DMF的50 mL三口瓶中, 110 ℃加热12 h. 反应混合物冷却至室温后, 加入30 mL二氯甲烷稀释, 用水(20 mL×2)洗涤, 将洗涤后的有机相用无水硫酸钠干燥, 真空浓缩, 用薄层色谱分离(石油醚/乙酸乙酯, V∶V=1∶1), 得到目标化合物6a~6d.
N,N-二乙基-4-氧-2-(1H-1,2,4-三唑-1-基)-4H-硫色酮-3-甲酰胺(6a): 淡黄色粉末(纯度98%), 收率67%. m.p. 130.6~133.0 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.92 (s, 1H), 8.55 (d, J=7.6 Hz, 1H), 8.17 (s, 1H), 7.74~7.63 (m, 3H), 3.57~3.52 (m, 2H), 3.20~3.15 (m, 2H), 1.21 (dq, J=2.7, 7 Hz), 0.90 (dq, J=2.5, 7 Hz); 13C NMR (100 Hz, CDCl3) δ: 178.50, 163.88, 153.34, 145.06, 142.73, 134.78, 132.91, 129.71, 129.30, 128.59, 127.21, 126.54, 43.29, 39.76, 13.79, 12.48; HRMS (ESI) calcd for C16H17N4O2S [M+H]+ 329.1072, found 329.1064.
N,N-二乙基-2-(1H-咪唑-1-基)-4-氧代-4H-硫色酮 基-3-甲酰胺(6b): 白色粉末(纯度97%), 收率42%. m.p. 127.5~129.0 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.54 (dd, J=1, 8.2 Hz), 8.00 (s, 1H), 7.75~7.71 (m, 1H), 7.65~7.60 (m, 2H), 7.44 (t, J=1.4 Hz, 1H), 7.21 (s, 1H), 3.64~3.58 (m, 1H), 3.36~3.31 (m, 1H), 3.16~3.05 (m, 2H), 1.12 (t, J=7.2 Hz, 3H), 0.91 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 178.52, 163.42, 142.12, 137.16, 134.14, 132.86, 131.10, 129.91, 129.51, 129.44, 128.72, 126.26, 120.01, 43.01, 39.28, 13.84, 12.38; HRMS (ESI) calcd for C17H18N3O2S [M+H]+ 328.1120, found 328.1111.
N,N-二乙基-4-氧-2-(1H-吡唑-1-基)-4H-硫色酮-3-甲酰胺(6c): 白色粉末(纯度99%), 收率46%. m.p. 127.4~129.3 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.52 (dd, J=8.2, 1 Hz, 1H), 8.32 (d, J=2.4 Hz, 1H), 7.82~7.56 (m, 4H), 6.51 (q, J=2.4 Hz, 1H), 3.64~3.48 (m, 2H), 3.15 (q, J=7.2 Hz, 2H), 1.23 (t, J=7.2 Hz, 3H), 0.84 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 178.94, 164.85, 146.54, 143.30, 135.36, 132.47, 131.75, 129.58, 128.99, 128.04, 126.35, 124.06, 109.58, 43.18, 39.57, 13.52, 12.42; HRMS (ESI) calcd for C17H18N3O2S [M+H]+ 328.1120, found 328.1112.
2-(叔丁基氨基)-N,N-二乙基-4-氧代-4H-硫色酮基- 3-甲酰胺(6d): 红色粉末(纯度96%), 收率53%. m.p. 132.6~134.7 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.43~8.41 (m, 1H), 7.52~7.29 (m, 3H), 6.34 (s, 1H), 3.59~3.54 (m, 2H), 3.36~3.13 (m, 2H), 1.49 (s, 9H), 1.26 (t, J=7.2 Hz, 3H), 1.07 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 175.28, 167.58, 157.06, 131.91, 130.76, 130.35, 128.38, 127.19, 125.38, 109.83, 54.07, 43.39, 39.82, 29.90, 14.56, 13.07; HRMS (ESI) calcd for C18H25- N2O2S [M+H]+ 333.1637, found 333.1632.
3.3.5 目标化合物6e~6h的合成
将化合物6a (328 mg, 1 mmol)和异丙醇(120 mg, 2 mmol)加入到装有5 mL DMF的50 mL三口瓶中, 110 ℃加热12 h. 反应混合物冷却至室温后, 加入30 mL二氯甲烷稀释, 用水(20 mL×2)洗涤, 将洗涤后的有机相用无水硫酸钠干燥, 真空浓缩, 用薄层色谱分离(石油醚/乙酸乙酯, V∶V=1∶1), 得到131 mg (0.41 mmol) N,N-二乙基-2-异丙氧基-4-氧代-4H-硫色酮基-3-甲酰胺(6e), 淡黄色固体(纯度97%), 收率41%. m.p. 42.3~44.2 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.50~8.48 (m, 1H), 7.60~7.49 (m, 3H), 4.90~4.87 (m, 1H), 3.76~3.71 (m, 1H), 3.46~3.41 (m, 1H), 3.27~3.22 (m, 2H), 1.46~1.44 (m, 6H), 1.26 (t, J=7 Hz, 3H), 1.12 (t, J=7 Hz, 3H); 13C NMR (400 Hz, CDCl3) δ: 178.75, 164.51, 164.34, 132.11, 131.59, 130.37, 128.91, 127.63, 126.34, 119.92, 76.62, 42.89, 39.05, 22.61, 22.36, 14.24, 12.72; HRMS (ESI) calcd for C17H22NO3S [M+H]+ 320.1320, found 320.1314.
将化合物6a (328 mg, 1 mmol)和取代苯胺(2 mmol)加入100 mL的封管反应器中, 加入5 mL二甲苯, 在密闭状态下165 ℃加热12 h. 反应混合物冷却至室温, 加入10 mL乙酸乙酯稀释, 用水(20 mL×2)洗涤, 将洗涤后的有机相用无水硫酸钠干燥, 真空浓缩, 用薄层色谱分离(石油醚/乙酸乙酯, V∶V=1∶1), 得到目标化合物6f~6h.
2-[(4-氯苯基)氨基]-N,N-二乙基-4-氧代-4H-硫色酮基-3-甲酰胺(6f): 白色粉末(纯度98%), 收率87%. m.p. 144.5~146.4 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.46 (d, J=7.6 Hz, 1H), 8.36 (d, J=6.4 Hz, 1H), 7.56~7.47 (m, 2H), 7.42~7.37 (m, 3H), 7.30~7.27 (m, 2H), 3.87~3.82 (m, 1H), 3.46~3.27 (m, 3H), 1.32 (t, J=7.2 Hz, 3H), 1.18 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 176.39, 166.98, 156.87, 136.00, 132.48, 131.69, 131.29, 130.20, 129.79, 128.57, 127.49, 126.35, 125.83, 111.67, 43.33, 39.70, 14.50, 12.98; HRMS (ESI) calcd for C20H20Cl- N2O2S [M+H]+ 387.0934, Found 387.0926.
2-[(4-氟苯基)氨基]-N,N-二乙基-4-氧代-4H-硫色酮基-3-甲酰胺(6g): 淡黄色粉末(纯度98%), 收率78%. m.p. 71.1~71.8 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.44 (d, J=7.6 Hz, 1H), 8.29 (s, 1H), 7.50~7.46 (m, 2H), 7.35~7.27 (m, 3H), 7.13~7.09 (m, 2H), 3.86~3.79 (m, 1H), 3.43~3.25 (m, 3H), 1.31 (t, J=7.2 Hz, 3H), 1.17 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 176.23, 167.05, 161.39 (d, J=246.3 Hz), 157.90, 133.26 (d, J=2.9 Hz), 131.71, 131.15, 130.21, 128.56, 127.79 (d, J=8.6 Hz), 127.39, 125.78, 116.59 (d, J=22.6 Hz), 110.85, 43.32, 39.70, 14.53, 13.00; HRMS (ESI) calcd for C20H20FN2O2S [M+H]+ 371.1230, found 371.1222.
2-[(4-溴苯基)氨基]-N,N-二乙基-4-氧代-4H-硫色酮基-3-甲酰胺(6h): 淡黄色粉末(纯度97%), 收率81%. m.p. 146.1~148.7 ℃; 1H NMR (400 Hz, CDCl3) δ: 8.44 (dd, J=7.8, 1.4 Hz, 1H), 8.35 (s, 1H), 7.54~7.45 (m, 4H), 7.37~7.35 (m, 1H), 7.21~7.19 (m, 2H), 3.84~3.79 (m, 1H), 3.43~3.26 (m, 3H), 1.30 (t, J=7.2 Hz, 3H), 1.16 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, CDCl3) δ: 176.41, 166.99, 156.60, 136.60, 132.74, 131.71, 131.29, 130.21, 128.58, 127.49, 126.50, 125.83, 120.17, 111.87, 43.35, 39.72, 14.48, 12.96; HRMS (ESI) calcd for C20H20Br- N2O2S [M+H]+ 431.0429, found 431.0424.
3.3.6 中间体8~10的合成
中间体8和9参考文献[18]合成, 收率分别为98%和72%.
2-巯基苯甲醛二聚体(9): 褐色固体. m.p. 140.1~142.8 ℃; 1H NMR (400 Hz, CDCl3) δ: 10.23 (s, 2H), 7.88~7.77 (m, 4H), 7.52~7.37 (m, 4H); 13C NMR (100 Hz, CDCl3) δ: 191.83, 140.07, 134.68, 134.29, 133.85, 126.74, 126.33.
将肉桂酸乙酯(2.64 g, 0.015 mol)、2-巯基苯甲醛二聚体(9, 5.48 g, 0.02 mol)和三苯基膦(6.55 g, 0.025 mol)投入到盛有50 mL甲苯的三口反应瓶中, 搅拌, 升温至50 ℃后, 滴加四甲基胍(1.725 g, 0.015 mol), 7 h后降温, 将反应溶液倒入100 mL水中, 并用乙酸乙酯(30 mL×2)萃取, 有机相干燥浓缩后, 硅胶柱分离(石油醚/乙酸乙酯, V∶V=10∶1)得到3.44 g (0.011 mol) 4-羟基-2-苯基硫代色烷-3-甲酸乙酯(10), 淡黄色粉末, 收率73%. m.p. 96.7~98.2 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 7.49~7.33 (m, 6H), 7.27~7.12 (m, 3H), 5.87 (d, J=6.0 Hz, 1H), 5.05 (dd, J=5.6 Hz, 1H), 4.92 (d, J=12.0 Hz, 1H), 4.04~3.88 (m, 2H), 3.47 (dd, J=12.0 Hz, 1H), 1.01 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, DMSO-d6) δ: 170.92, 140.26, 135.17, 133.50, 131.54, 128.89, 128.83, 128.79, 128.05, 124.99, 124.63, 68.93, 60.42, 50.80, 40.52, 14.26; MS(ESI) calcd for C18H18NaO3S [M+Na]+ 337.0874, found 337.0870.
3.3.7 中间体11和12的合成
将化合物10 (3.14 g, 0.01 mol)和DDQ (9.08 g, 0.04 mol))加入到盛有80 mL二氧六环的三口反应瓶中, 搅拌并加热至90 ℃, 反应结束后降温, 加入100 mL质量分数为8%的亚硫酸钠水溶液, 用乙酸乙酯(30 mL×2)萃取, 有机相干燥浓缩后得2.9 g (9.38 mmol) 4-氧代-2-苯基-4H-硫色酮-3-甲酸乙酯(11), 收率94%. 淡黄色固体, m.p. 95.4~97.5 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 8.40 (dd, J=8, 1.2 Hz, 1H), 7.97 (d, J=8 Hz, 1H), 7.86~7.82 (m, 1H), 7.73~7.69 (m, 1H), 7.59~7.56 (m, 5H), 4.06 (q, J=7 Hz, 2H), 0.97 (t, J=7.2 Hz, 3H); 13C NMR (100 Hz, DMSO-d6) δ: 176.90, 165.38, 152.53, 136.91, 135.02, 133.30, 131.29, 130.60, 130.05, 129.56, 129.21, 128.49, 128.30, 127.38, 61.59, 14.07; HRMS (ESI) calcd for C18H15O3S [M+H]+ 311.0742, found 311.0743.
将上述中间体11 (3.1 g, 0.01 mol)投入到盛有30 mL无水乙醇的三口反应瓶中, 然后加入8 mL质量分数为10%的氢氧化钠水溶液, 加热至40 ℃水解2 h. 反应液减压浓缩除去大部分无水乙醇, 加入20 mL二氯甲烷, 用水(20 mL×2)萃取, 水相用质量分数为10%的盐酸酸化至pH<3, 再用二氯甲烷(20 mL×2)萃取. 有机相干燥、浓缩得2.5 g (0.009 mol) 4-氧代-2-苯基-4H-硫色酮- 3-甲酸(12), 白色固体, 收率90%. m.p. 220.4~221.4 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 13.25 (s, 1H), 8.41 (dd, J=8.2, 1 Hz, 1H), 7.96 (d, J=8 Hz, 1H), 7.85~7.81 (m, 1H), 7.73~7.69 (m, 1H), 7.63~7.55 (m, 5H); 13C NMR (100 Hz, DMSO-d6) δ: 177.16, 166.86, 137.09, 135.37, 133.16, 132.01, 131.16, 130.11, 129.52, 129.14, 129.05, 128.51, 128.44, 127.31.
3.3.8 目标化合物13a~13e的合成
将化合物12 (564 mg, 2 mmol)和DCC (494.4 mg, 2.4 mmol))投入到盛有20 mL二氯甲烷的三口反应瓶中, 在0 ℃下滴加含有2.2 mmol各种取代胺的5 mL二氯甲烷溶液, 在此温度搅拌1 h后升温至30 ℃. 反应结束后过滤, 滤液倒入20 mL水中, 分液后取有机相浓缩, 残余物用薄层色谱分离(石油醚/乙酸乙酯, V∶V=2∶1), 得到目标化合物13a~13e.
4-氧代-N,N-二乙基-2-苯基-4H-硫色酮基-3-甲酰胺(13a): 淡红色粉末(纯度99%), 收率46%. m.p. 105.3~107.9 ℃; H NMR (400 Hz, DMSO-d6) δ: 8.40 (dd, J=8.2, 1 Hz, 1H), 7.95 (d, J=8 Hz, 1H), 7.84~7.80 (m, 1H), 7.71~7.67 (m, 1H), 7.60~7.49 (m, 5H), 3.55~3.47 (m, 1H), 3.21~3.12 (m, 1H), 2.99~2.87 (m, 2H), 0.83 (t, J=7.2 Hz, 3H), 0.72 (t, J=7 Hz, 3H); 13C NMR (100 Hz, DMSO-d6) δ: 177.39, 164.68, 149.36, 137.38, 135.09, 132.98, 130.91, 130.14, 129.17, 128.88, 128.68, 128.59, 127.23, 42.56, 38.11, 14.00, 12.25; HRMS (ESI) calcd for C20H20NO2S [M+H]+ 338.1215, found 338.1207.
N-烯丙基-4-氧代-2-苯基-4H-硫代色氨酸-3-甲酰胺(13b): 淡黄色粉末(纯度97%), 收率59%. m.p. 184.0~186.5 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 8.42~8.34 (m, 2H), 7.93 ((d J=8 Hz, 1H), 7.83~7.79 (m, 1H), 7.71~7.67 (m, 1H), 7.60~7.48 (m, 5H), 5.56~5.48 (m, 1H), 4.91~4.85 (m, 2H), 3.63~3.60 (m, 2H); 13C NMR (100 Hz, DMSO-d6) δ: 177.58, 164.63, 150.32, 137.07, 135.64, 134.95, 134.26, 132.92, 130.73, 130.45, 129.14, 128.85, 128.66, 128.59, 127.19, 115.47, 41.28; HRMS (ESI) calcd for C19H16NO2S [M+H]+ 322.0902, found 322.0896.
N-(4-氯苄基)-4-氧代-2-苯基-4H-硫色酮基-3-甲酰胺(13c): 红棕色粉末(纯度97%), 收率38%. m.p. 255.1~258.0 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 8.73 (t, J=6 Hz, 1H), 8.42 (dd, J=8, 1.2 Hz, 1H), 7.94 (dd, J=8, 0.8 Hz, 1H), 7.82~7.48 (m, 7H), 7.23~7.20 (m, 2H), 6.90 (d, J=8.4 Hz, 2H), 4.21 (d, J=6 Hz, 2H); 13C NMR (100 Hz, DMSO-d6) δ: 177.62, 164.96, 150.58, 138.36, 137.10, 135.46, 134.06, 132.99, 131.52, 130.79, 130.41, 129.25, 129.10, 128.92, 128.72, 128.61, 128.43, 127.22, 41.82; HRMS (ESI) calcd for C23H17ClNO2S [M+H]+ 406.0669, found 406.0663.
N-(4-氯苯基)-4-氧代-2-苯基-4H-硫色酮基-3-甲酰胺(13d): 红色粉末(纯度98%), 收率54%. m.p. 123.5~127.4 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 10.48 (s, 1H), 8.44 (dd, J=8.2, 1 Hz, 1H), 7.99 (dd, J=8, 1 Hz, 1H), 7.85~7.61 (m, 4H), 7.50~7.46 (m, 5H), 7.33~7.31 (m, 2H); 13C NMR (100 Hz, DMSO-d6) δ: 177.62, 163.55, 151.44, 138.08, 137.04, 135.33, 133.87, 133.18, 130.98, 130.44, 129.35, 129.14, 129.10, 128.62, 128.44, 127.64, 127.32, 121.09; HRMS (ESI) calcd for C22H15ClNO2S [M+H]+ 392.0512, found 392.0504.
4-(4-氧代-2-苯基-4H-硫色酮基-3-甲酰胺基)苯甲酸异丙酯(13e): 红色粉末(纯度98%), 收率53%. m.p. 126.5~128.5 ℃; 1H NMR (400 Hz, DMSO-d6) δ: 10.71 (s, 1H), 8.45 (dd, J=8, 1.2 Hz, 1H), 7.99 (d, J=8 Hz, 1H), 7.88~7.84 (m, 3H), 7.75~7.71 (m, 1H), 7.64~7.58 (m, 4H), 7.50~7.48 (m, 3H), 5.13~5.07 (m, 1H), 1.29 (d, J=6.4 Hz, 6H); 13C NMR (100 Hz, DMSO-d6) δ: 177.65, 165.22, 163.99, 151.65, 143.33, 137.05, 135.26, 133.75, 133.21, 131.01, 130.67, 130.41, 129.37, 129.12, 128.61, 128.43, 127.33, 125.43, 118.91, 68.29, 22.16; HRMS (ESI) calcd for C26H22NO4S [M+H]+ 444.1270, found 444.1262.
3.4 硫色酮酰胺衍生物的生物活性测试
合成的硫色酮酰胺衍生物的生物活性采用菌丝生长速率法测试[20]. 首先, 制取马铃薯葡萄糖琼脂(PDA)培养基. 然后, 以DMSO为助溶剂, 并加入适量吐温80配制成适当浓度梯度的供试样品备用. 将熔化并冷却至50 ℃的PDA培养基(9 mL)与适当浓度的供试样品溶液(1 mL)充分混合, 然后倒入无菌培养皿中冷却至室温. 从培养7 d的小麦全蚀病菌PDA平板上取直径5 mm的菌饼, 以倒扣的方式接种在含药的PDA平板中央, 以不含药的平板培养基为对照, 每个药液浓度设4个重复. 培养箱中避光培养5 d (20~25 ℃), 测量菌落直径, 计算相对抑制率.
相对抑制率(%)=[(空白对照菌落直径-药剂处理菌落直径)/(空白对照菌落直径-菌饼直径)]×100%
辅助材料(Supporting Information) 关键中间体及目的产物的1H NMR、13C NMR和HRMS谱图. 这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(Zhao, C.)