

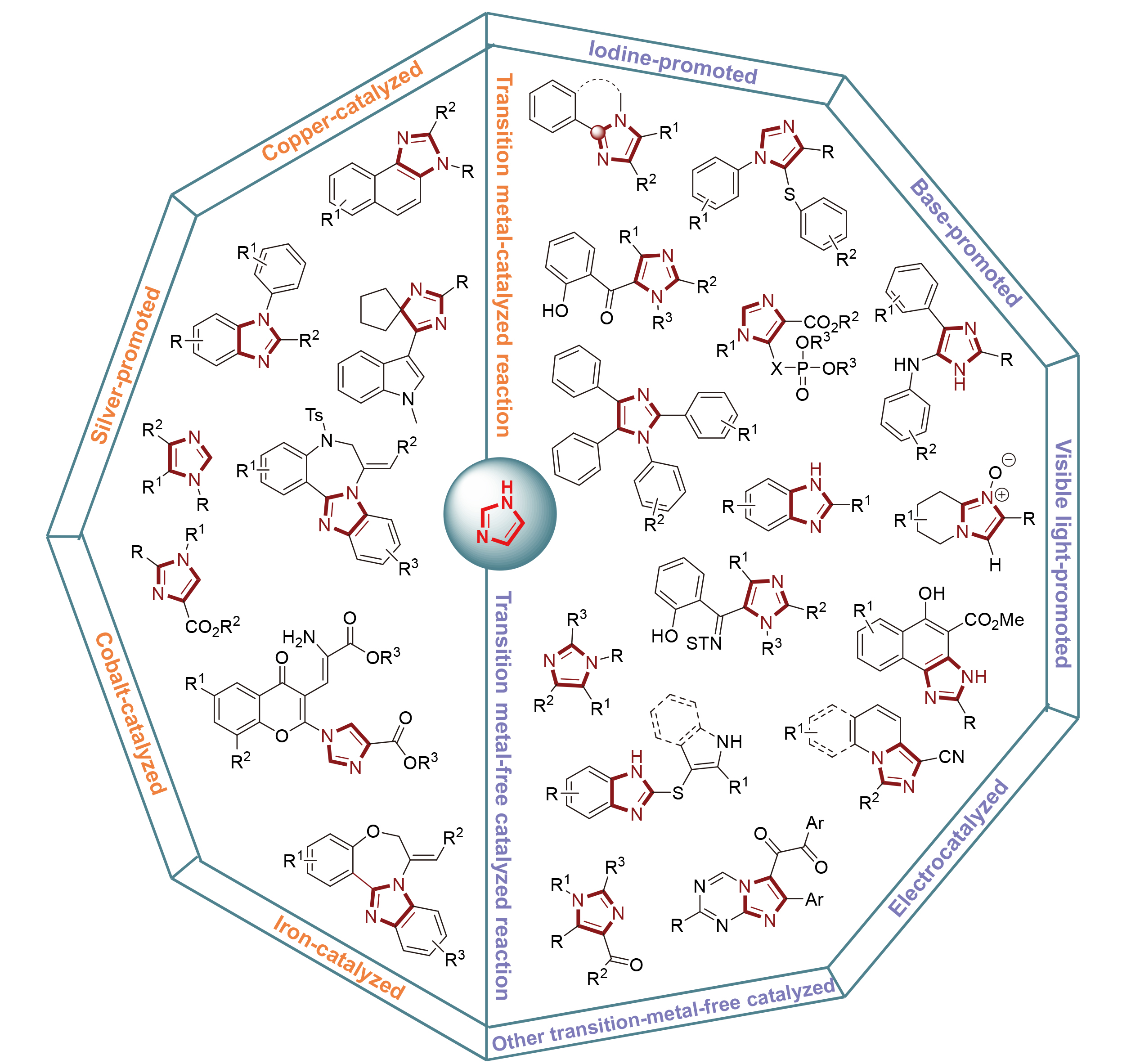
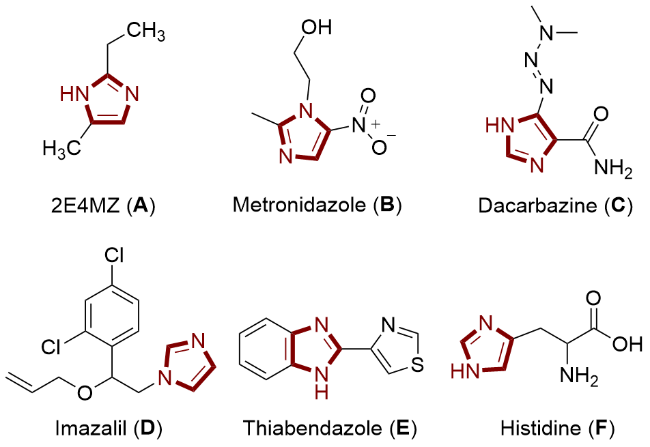
分子结构中含有两个间位氮原子的五元芳杂环化合物——咪唑, 是一种重要的含氮杂环化合物, 其不仅广泛存在于各种天然产物、药物和材料中[1-2], 而且在许多化合物中表现出显著的生物活性特征[3-5]. 此外, 咪唑类化合物还具有良好的光物理性质[6-8], 可作为配体用于均相催化[9]和功能化材料[10-11]. 因此, 咪唑类化合物是化工生产中比较重要的中间体, 被广泛应用到各个领域: (1)在工业与材料领域, 主要用作离子液体和耐高温材料, 例如作为环氧树脂固化剂的2-乙基-4-甲基咪唑(A); (2)在医药领域, 主要是用于药物的合成, 例如治疗厌氧菌感染的甲硝唑(B)以及抗恶性黑色素瘤的甲氮咪胺(C); (3)在农药领域, 主要用于合成杀菌剂, 如抑霉唑(D)和噻菌灵(E); (4)在生物化学领域, 可作为缓冲液, 如作为酶催化活性中心的组氨酸(F)(图1).
1 过渡金属催化合成咪唑类化合物
过渡金属催化在咪唑类化合物的合成中具有广泛应用, 常用的过渡金属催化剂包括钯、铜、银、铁和钴等. 这些催化剂能够通过有效活化底物分子中的化学键, 促进多种反应的进行, 从而高效构建咪唑类化合物的核心骨架.
1.1 铜催化合成咪唑类化合物
2021年, Wang课题组[22]制备了一种稳定的碳化八面体骨架材料Cu-PC@OFM, 并以其为催化剂, 实现了邻苯二胺1与苄醇2的环化反应, 高效地制备了1-苄基- 2-芳基-1H-苯并[d]咪唑衍生物3 (Scheme 1, a). 此方法的优点在于生成物产率较高、催化剂绿色高效且在水中回收性能良好. 值得注意的是, 在反应过程中负载纳米铜的碳基体载体不仅保持了原有的结构, 而且原位生成的纳米铜颗粒显著提高了此反应的催化性能和稳定性.
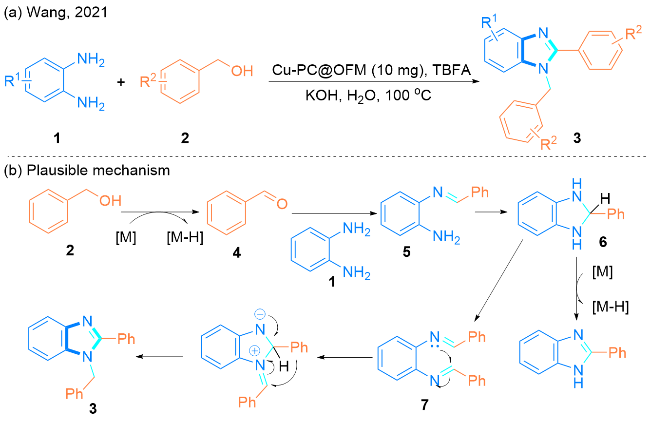
该课题组提出了可能的反应机理(Scheme 1, b): 首先, 苄醇2经过脱氢生成醛4, 然后醛4与邻苯二胺1缩合得到亚胺5, 随后亚胺5通过环化反应得到中间产物6, 中间产物6可以与另一等价物4反应得到中间产物7, 最后, 中间产物7经过分子内环化和重排后, 生成最终产物1-苄基-2-芳基-1H-苯并[d]咪唑3.
2021年, Kadu课题组[23]报道了一种CuI催化芳基醛8、2-羟基-2-苯基苯乙酮9与乙酸铵10发生三组分串联反应合成2,4,5-三取代咪唑11的方法(Scheme 2, a).
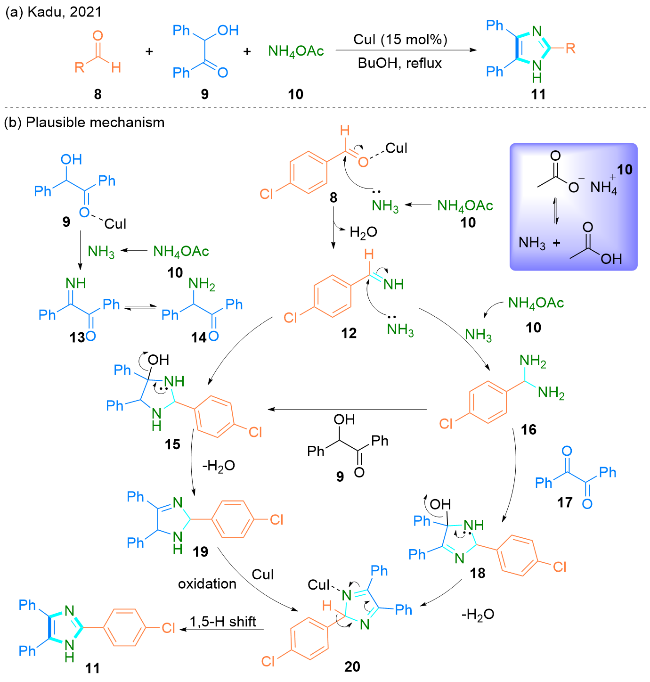
该课题组提出了两种可能的合成路径(Scheme 2, b). 首先, 4-氯苯甲醛8与乙酸铵10发生氨缩合生成中间体12, 同时, 2-羟基-2-苯基苯乙酮9与乙酸铵反应生成中间体13, 中间体13通过互变异构进一步转化为中间体14; 然后, 14与中间体12反应生成中间体15. 另一方面, 中间产物12通过与另一个氨分子反应, 转化为(4-氯苯基)甲二胺16. 然后, (4-氯苯基)甲二胺16分别与2-羟基-2-苯基苯乙酮9和二苯乙二酮17反应生成相应的中间体15和中间体18. 之后, 中间产物15通过失水转化为19, 19在CuI催化剂的作用下氧化得到中间产物20. 中间产物18由于失水而形成相同的中间产物20. 最后, 通过[1,5]-氢迁移得到目标产物2,4,5-三取代咪唑11.
2022年, Wang课题组[24]报道了一种在Cu(I)催化下, 2-甲氧基-2H-氮杂环丙烯21和肟酯22发生[3+2]环加成反应来合成2H-咪唑23的新方法(Scheme 3, a). 在反应过程中, 2H-氮杂环丙烯发生了C—N键选择性裂解.
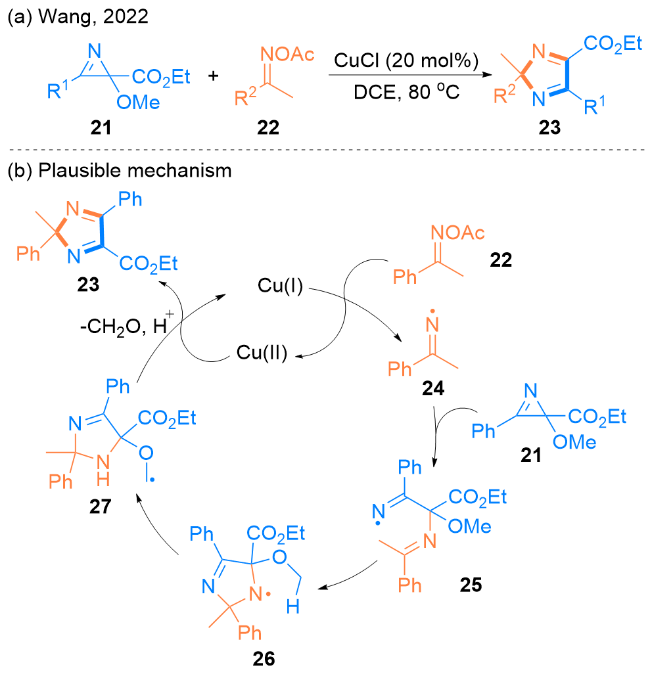
作者提出了一种可能的反应机理(Scheme 3, b). 首先, 该反应是由Cu(I)激活肟酯22开始的, 生成自由基中间体24. 然后, 当二氯乙烷(DCE)被用作溶剂时, 24与氮杂环丙烯21发生自由基加成生成中间体25, 中间体25经过环加成反应得到了咪唑核心26, 在咪唑核心26的氢转移之后, 形成自由基中间体27, 它与Cu(II)进行单电子转移, 并消除一分子醛和一个质子, 最终得到2H-咪唑23.
2022年, Deng课题组[26]报道了一种Cu2O催化下2-(吲哚-3-基)环己酮31和脒32发生环化重排反应得到螺咪唑衍生物33的方法(Scheme 5, a). 该课题组对2-(吲哚-3-基)环己酮31和脒32底物进行拓展, 分别以54%~76%和39%~76%的产率得到了一系列螺咪唑类衍生物.
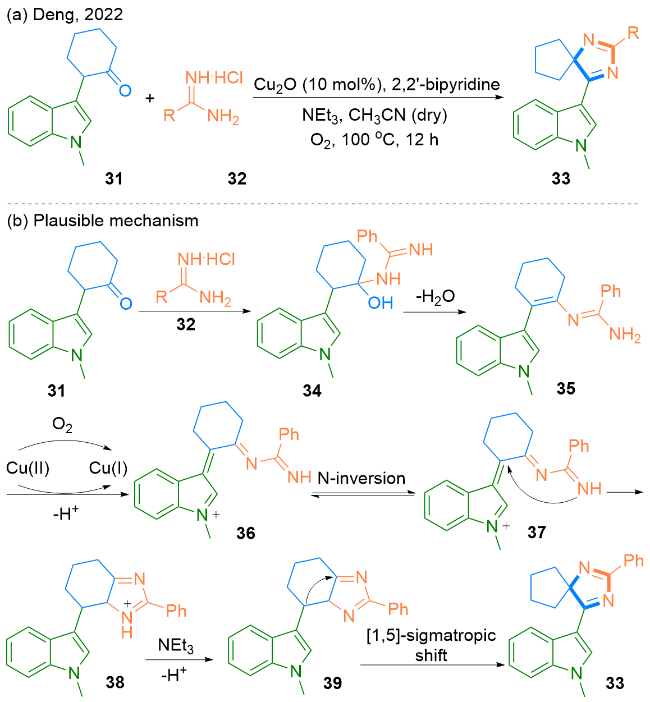
作者提出了一个可能的反应机理(Scheme 5, b). 首先, 2-(吲哚-3-基)环己酮31与盐酸苯甲脒32发生缩合反应, 生成中间体34, 中间体34脱去一分子水伴随互变异构, 生成中间体35. 然后在Cu(II)的催化下, 中间体35被氧化为中间体36. 随后, 中间体36发生N原子反转和分子内亲核加成反应, 生成中间体38. 中间体38通过去质子化进一步转化为中间体39. 最后, 中间体39发生[1,5]-σ迁移反应, 生成目标产物33.
2022年, Sridharan课题组[28]报道了一种高效的微波辅助铜(II)催化的方法. 此方法在温度为100 ℃的条件下, 以K2CO3为碱, N,N-二甲基甲酰胺(DMF)为溶剂, 实现了2-丙炔氨基芳基醛44/2-丙炔氧基芳基醛45和邻苯二胺46的串联环化反应(Scheme 7), 以高达93%的产率合成了苯并[f]咪唑并[1,2-d][1,4]氧氮杂䓬衍生物47和苯并[f]咪唑并[1,2-d][1,4]二氮杂䓬衍生物48 (Scheme 7). 值得注意的是, 此反应能很好地容忍各种取代基(包括给电子取代基和吸电子取代基), 并且能在一次合成操作中产生两个新的杂环(七元环和五元环)和三个新的 C—N键.
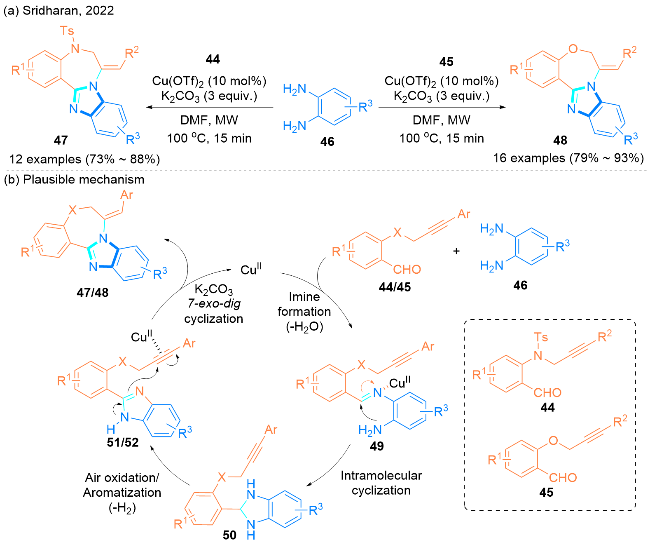
该课题组提出了可能的反应机理(Scheme 7, b). 首先, 醛44/45与邻苯二胺46反应生成中间产物亚胺49, 亚胺49经过分子内5-endo-trig环化及空气氧化/芳构化, 经物种50生成咪唑中间产物51/52. 最后, 在碱存在下, 铜(II)催化剂激活中间产物51/52的内部炔, 引发了7-exo-dig环化, 得到所需产物苯并[f]咪唑并[1,2-d][1,4]氧氮杂䓬47和苯并[f]咪唑并[1,2-d][1,4]二氮杂䓬48.
2022年, Guo课题组[29]报道了通过铜催化α,β-不饱和酮肟53、多聚甲醛54和胺55的三组分反应合成氮杂环化合物的方法. 值得注意的是, 通过控制反应条件能得到相应的咪唑56和二氢咪唑57 (Scheme 8, a).
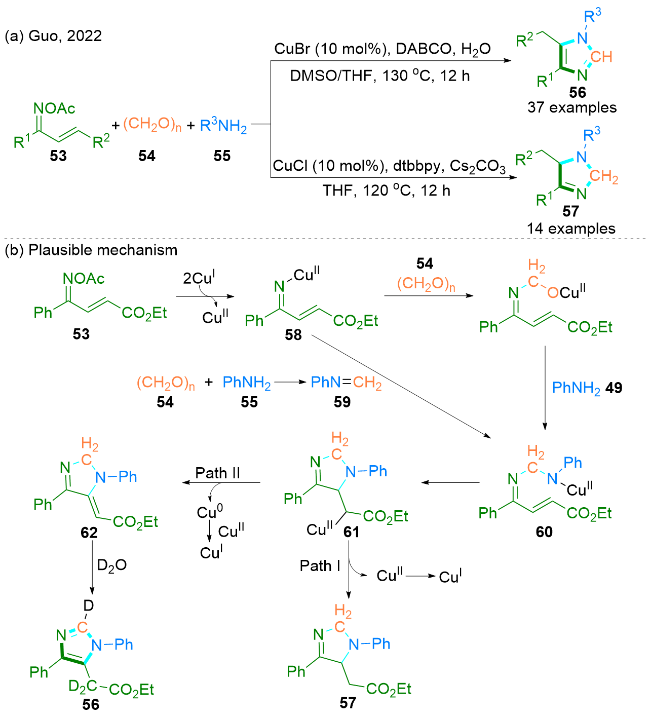
作者提出了一个可能的反应机理(Scheme 8, b): 最初, α,β-不饱和酮肟53与Cu(I)盐反应生成亚氨基Cu(II)中间产物58和Cu(II)物种. 接下来, 中间产物58和原位生成的N-苯基甲酰亚胺59之间发生亲核加成, 得到中间产物60. 另外, 60也可以通过58与多聚甲醛54发生亲核加成, 再与苯胺55偶联, 并与铜催化剂配位形成. 随后, 60的分子内迈克尔加成生成61, 61在不同的反应条件下经过质子去金属化作用(路径I)或β-氢消除作用(路径II), 得到二氢咪唑产物57和62. 最后, 在水的作用下, 通过62的互变异构化得到咪唑产物56.
1.2 银促进合成咪唑类化合物
2021年, Wu课题组[30]报道了一种新颖的银促进亚甲胺叶立德63和三氟乙酰胺酰氯64发生[3+2]环加成反应合成5-(三氟甲基)咪唑衍生物65的方法(Scheme 9, a). 值得注意的是, 该方案成功应用于构建γ-氨基丁酸A型受体(GABAA)的特异性调节剂类似物, 并且银可以通过简单过滤操作进行回收.
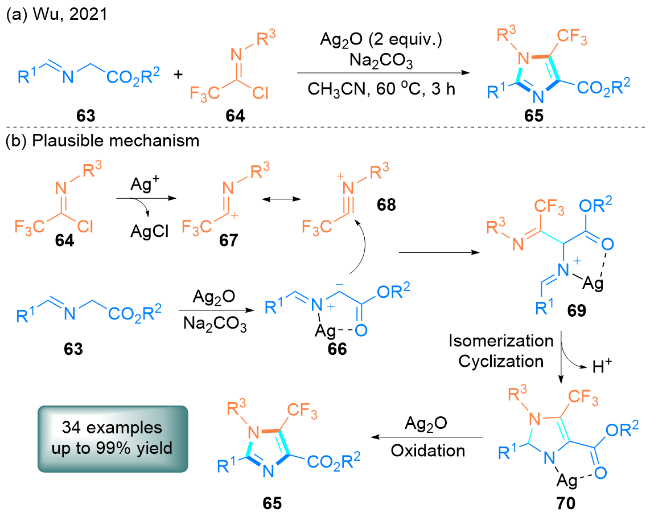
该课题组提出了一个可能的反应机理(Scheme 9, b): 首先, 亚甲胺叶立德63与银和碱的相互作用生成偶极中间体66, 三氟乙酰胺酰氯64与银的反应生成三氟乙酰胺阳离子67, 它可以发生异构化得到络合物68. 然后, 偶极中间体66亲核进攻络合物68生成中间体69, 中间体69经过异构化和环化反应生成二氢咪唑-银配合物70. 最后, 在Ag2O的作用下, 70进一步氧化得到所需产物5-(三氟甲基)咪唑65.
2022年, Yan课题组[31]报道了一种Ag2CO3促进3-氰基色酮71和α-异氰酸酯72发生一锅串联反应来高效合成2-(1H-咪唑-1-基)-4H-色烯-4-酮衍生物73的方法(Scheme 10, a). 该方案不仅可以合成官能团化的咪唑, 而且还可以合成官能团化的烯胺结构.
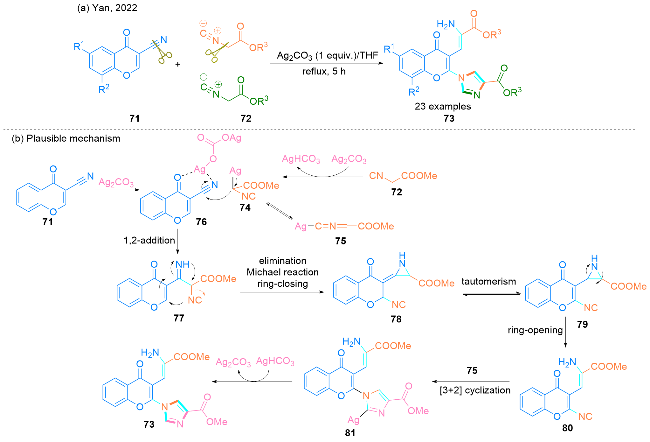
该串联反应的机理为(Scheme 10, b): 首先, α-异氰酸酯72的酸性α-质子被Ag2CO3去质子化, 激活Ag+并产生α-金属化的异氰酸酯74和其互变异构体75. 同时, 3-氰基色酮71与路易斯酸Ag2CO3结合得到中间产物76, 增加了氰基碳的亲核性. 然后, 中间产物74的α-碳攻击中间产物76的氰基, 通过1,2-加成反应形成中间产物77. 中间体77经历一系列的串联反应, 包括消除异氰基、异氰基与77的双键进行迈克尔加成以及闭环反应, 形成关键的中间体78, 然后发生互变异构得到中间体79. 随后, 中间体79通过开环反应转化为中间体80. 接下来, 中间体80与异氰化物75发生[3+2]偶极环加成反应得到中间体81. 最后, 在AgHCO3的帮助下, 中间体81经过Ag+的协同消除, 得到所需的目标化合物2-(1H-咪唑-1-基)-4H-色烯-4-酮73.
1.3 钴催化合成咪唑类化合物
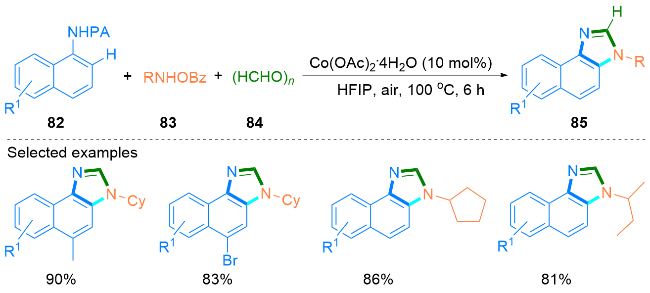
1.4 铁催化合成咪唑类化合物
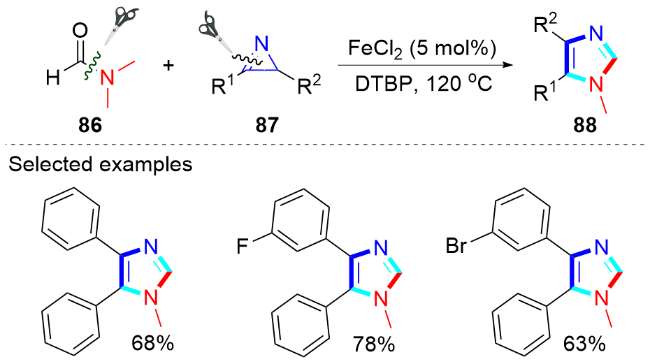
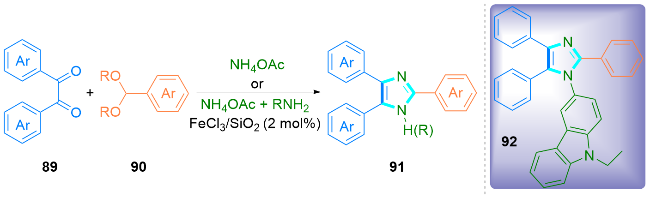
2 无过渡金属催化合成咪唑类化合物
2.1 碘促进合成咪唑类化合物
2021年, Gao课题组[39]报道了一种碘促进下烯胺93与叠氮基三甲基硅烷(TMSN3, 94)发生的氧化[4+1]环化反应, 高效地合成了一系列具有不同取代基的2,5-二取代-1H-咪唑-4-羧基衍生物95 (Scheme 14, a). 此反应适用于克级规模的合成, 且反应过程中无过渡金属参与.
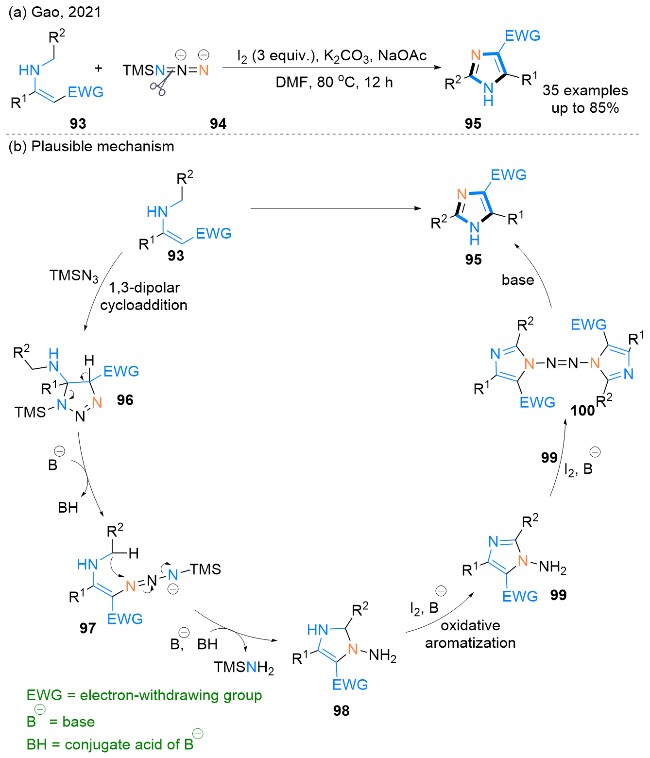
根据实验结果, 该课题组提出了一个可能的反应机理(Scheme 14, b). 最初, 94与烯胺化合物93通过发生1,3-偶极环加成反应生成1,2,3-三唑啉中间体96. 然后, 中间体96开环得到α-叠氮烯胺97. 进一步碱诱导的 N=N双键发生分子内加成得到2,3-二氢咪唑98, 98被碘进一步氧化芳构化生成中间体99. 随后, 99在I2/碱体系中被氧化, 与另一分子99偶联, 得到1,1'-偶氮咪唑100. 最后, 100水解得到所需产物2,5-二取代-1H-咪唑- 4-羧基衍生物95. 在该反应中, 碘的作用是多样的, 除了在互变异构化和生成100的过程中充当氧化剂外, 还可以在1,3-偶极环加成和亲核加成的步骤中充当有用的催化剂.
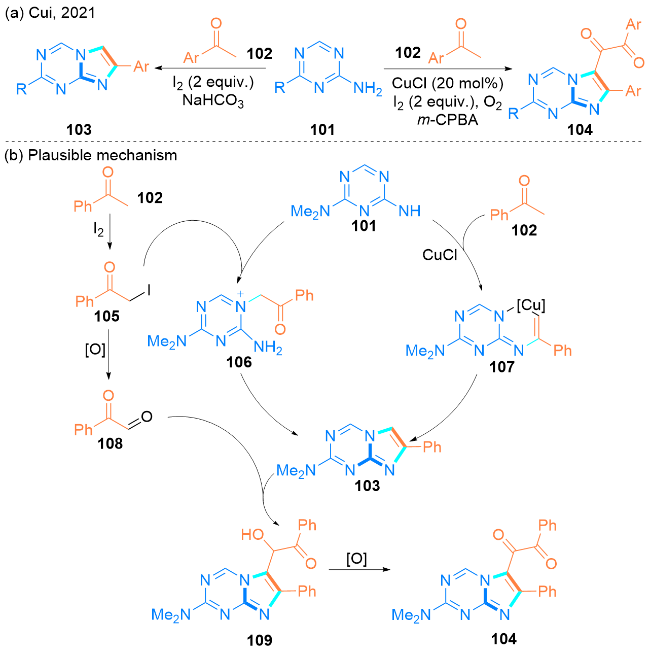
该课题组提出了一个可能的反应机理(Scheme 15, b). 一方面, 苯乙酮102与I2作用, 在反应条件下产生中间产物α-碘苯乙酮105. 然后α-碘苯乙酮105与2-氨基[1,3,5]三嗪101发生分子间的亲核取代反应, 得到中间体106, 随后经分子内环化, 得到所需产物咪唑[1,2-a][1,3,5]三嗪103. 另一方面, 在Cu催化剂和O2的存在下, 2-氨基[1,3,5]三嗪101也可以与苯乙酮102反应, 得到中间产物107, 然后进行还原消除, 形成咪唑[1,2-a][1,3,5]三嗪103. 随后, 103与苯基乙二醛108反应, 并且在路易斯酸I2和铜催化剂的活化下, 直接转化为中间产物109, 然后进一步快速氧化, 得到产物咪唑[1,2-a][1,3,5]三嗪基乙二酮104.
2021年, Feng课题组[41]报道了一种I2O5促进吡啶- 2-甲醛110或2-甲基喹啉111、苄胺112和NH4SCN (113)的串联Strecker/C(sp3)—H胺化反应(Scheme 16, a). 这种多组分反应实现了一步合成具有重要生物学价值的氰基官能团化咪唑并[1,5-a]吡啶衍生物114. 该方法还可以通过一锅两步法合成具有氰基取代的咪唑并[1,5-a]喹啉类化合物. 值得注意的是, 使用安全和易于处理的NH4SCN作为替代氰化剂, 使得该方案具有潜在的应用价值.
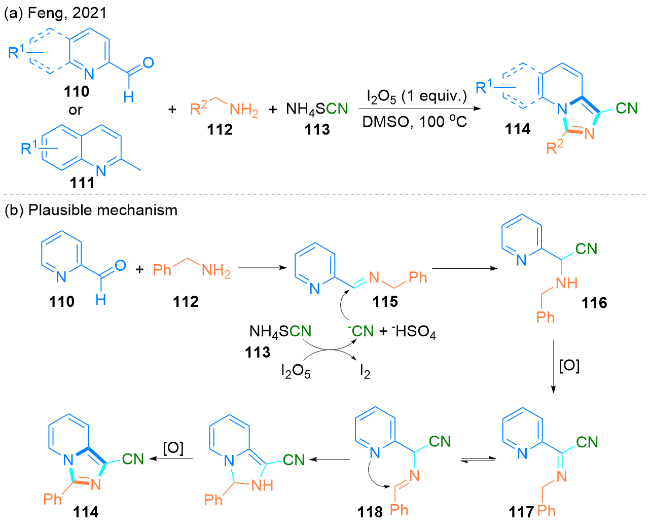
该课题组提出了一种可能的反应机理(Scheme 16, b): 首先, 吡啶-2-甲醛110与苄胺112通过简单缩合, 生成亚胺115, 在I2O5的存在下, 由NH4SCN (113)生成的亲核氰化物进攻亚胺115, 得到中间体α-氨基腈116. 随后, 中间体α-氨基腈116进一步氧化产生中间体117, 它可以通过共振得到中间体118. 最后, 通过分子内亲核加成反应和氧化芳烃化反应得到最终产物氰基官能团化咪唑并[1,5-a]吡啶衍生物114.
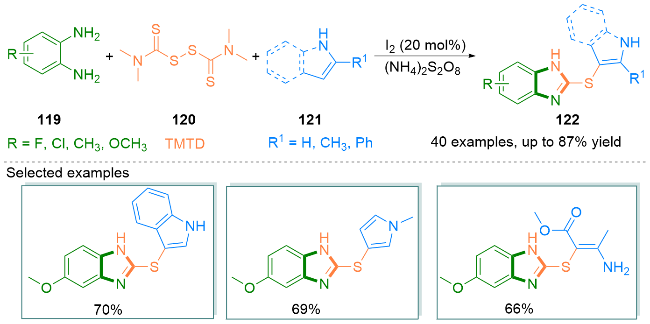
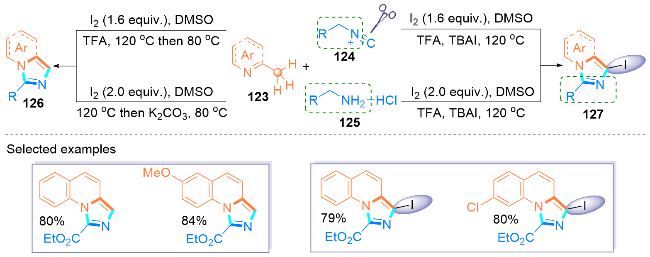
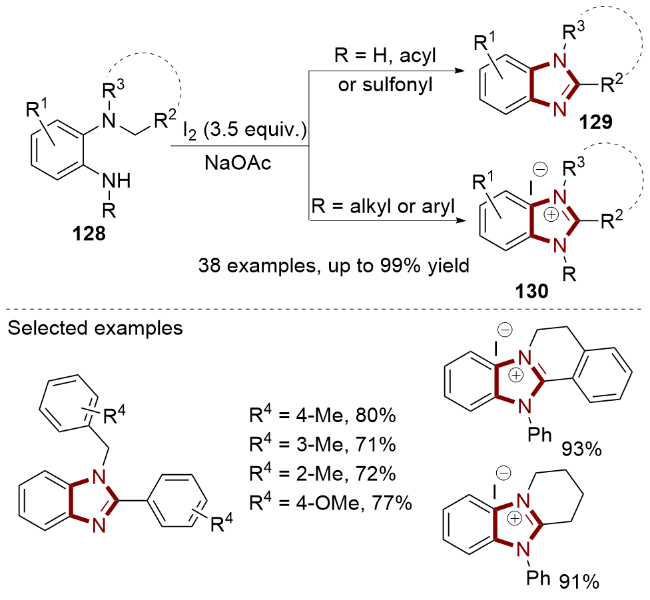
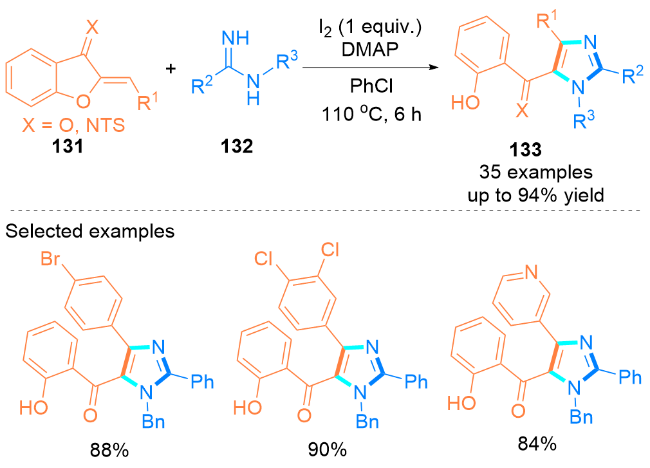
2022年, Gao课题组[46]设计了KI介导下烯胺134和tBuONO (135)的无金属氧化环化反应, 合成了咪唑- 4-羧基衍生物136 (Scheme 21, a). 在这个环化过程中, tBuONO的使用不仅保证了比TMSN3更安全、更少的毒性操作, 而且有效地促进了活性较低的烯烃发生该反应. 值得注意的是, 该反应的底物范围广泛, 并且对未活化的烯胺类化合物也具有良好的官能团耐受性.
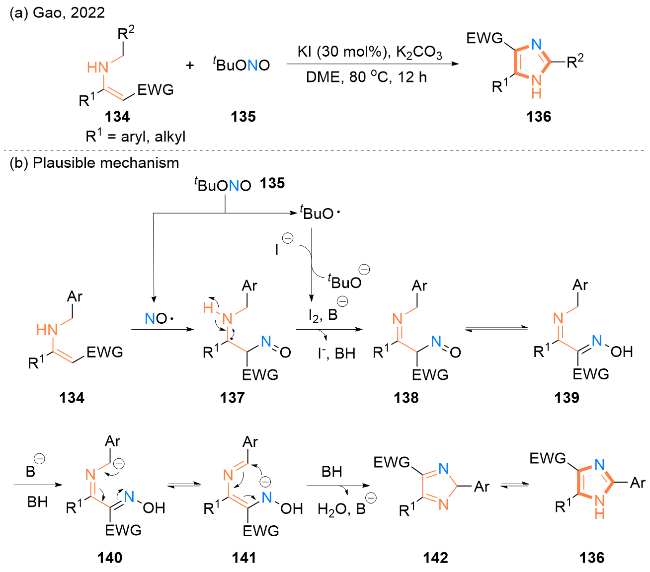
作者提出了一个可能的自由基反应机理(Scheme 21, b): 最初, tBuONO (135)生成的NO•自由基与烯胺134反应, 生成中间产物137. 然后用分子碘进一步氧化中间体137, 得到α-C-亚硝基化中间体138, 中间体138与肟139互变异构. 随后, 由碱促进质子离去生成140, 140发生互变异构产生141. 最后, 141发生分子内亲核环化和OH-消除得到中间体142, 中间体142互变异构得到咪唑-4-羧基衍生物136. 此外, tBuO•自由基将I-氧化为I2, 它是一种重要的路易斯酸催化剂和中间产物形成过程中的氧化剂, 也是部分NO•自由基的引发剂.
2.2 碱促进合成咪唑类化合物
2021年, Xiong课题组[48]报道了由CF3-氢苯并咪唑146合成一系列2-芳基苯并咪唑147的新方法(Scheme 23, a). 值得注意的是, 2-芳基苯并噻唑和2-芳基苯并噁唑也能够以类似的方式被顺利合成. 此外, 反应可以以克级规模进行, 表明该方法具有实用性.
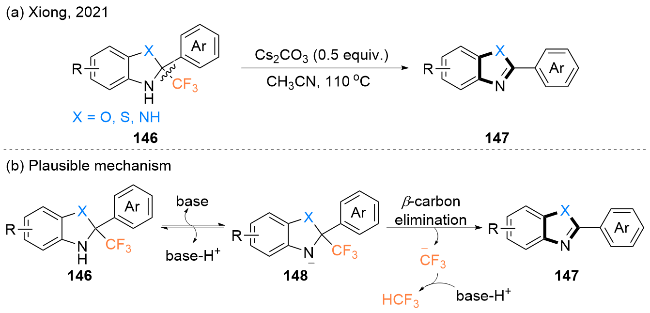
该反应可能的机理是(Scheme 23, b): 首先, CF3-氢苯并咪唑146通过碱去质子化生成中间体148, 这是一个可逆的过程. 然后, 中间体148经过芳构化驱动β-碳消除过程, 生成2-芳基苯并咪唑147和$\mathrm{CF}_{3}^{-}$, 后者进一步质子化得到CF3H.
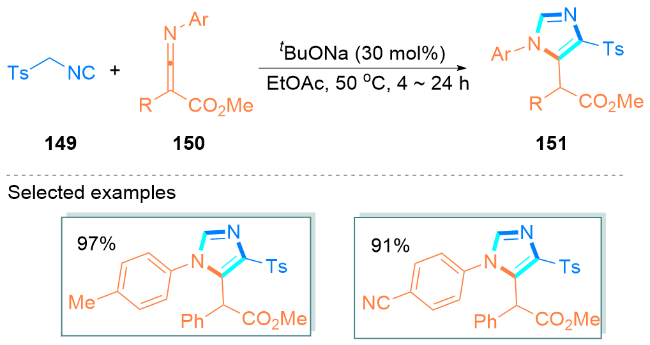
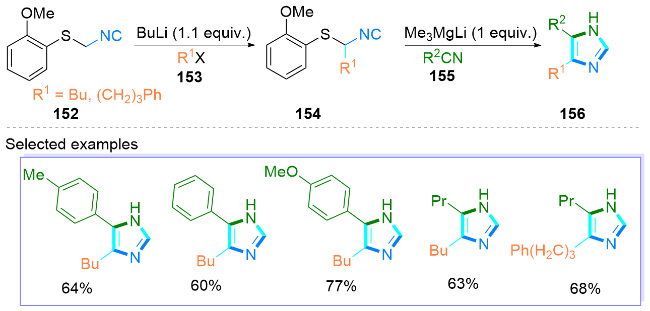
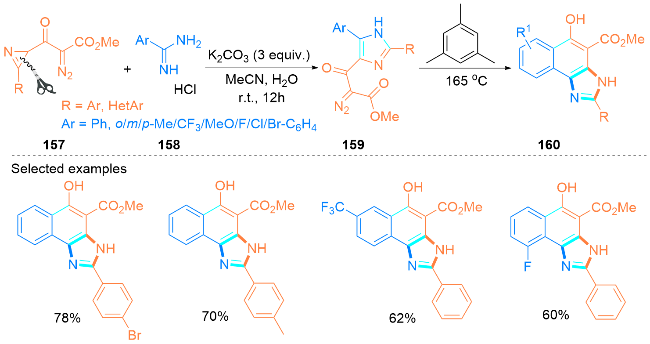
2.3 可见光促进合成咪唑类化合物
2021年, Sharada课题组[52]报道了一种在可见光介导下, 无金属有机染料催化的四氢异喹啉(THIQ)烯胺161发生叠氮化/环胺化反应, 合成高取代的咪唑和二氢异喹啉基咪唑衍生物162的方法(Scheme 27, a). 值得注意的是, 这种环保且温和的一步C—H胺化方法, 避免了化学计量的氧化剂的使用, 并且在一次合成操作中就能够形成两个C—N键. 此外, 该方法能够以64%~75%的良好产率合成CF3取代的咪唑.
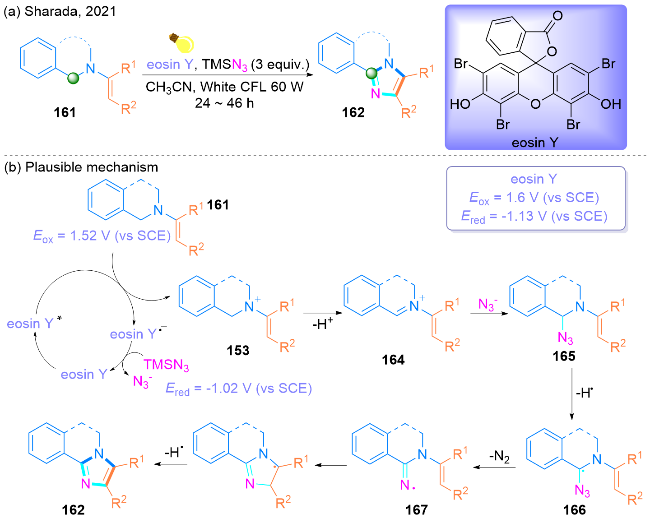
该课题组提出了一种合成2,4,5-三取代咪唑的反应机理(Scheme 27, b). 最初, 在白色紧凑型荧光灯(CFL)光的存在下, 曙红Y (eocin Y)被光激发. 通过还原猝灭循环, THIQ烯胺161通过单电子转移(SET)机理氧化为THIQ烯胺自由基阳离子163. 随后, 阳离子163被氧化成亚氨基离子164, 它被$\mathrm{N}_{3}^{-}$攻击生成165, 然后被氧化得到α-叠氮基166. 随后, 从166中放出N2生成亚胺基自由基157. 最后, 亚胺基自由基167经过氧化生成二氢异喹啉基咪唑衍生物162.
2022年, Zhu课题组[54]报道了一种新颖的光氧化还原/铜协同催化的多米诺环化反应. 该方法使用肟酯172、醛173和胺174作为反应底物, 三乙胺盐酸盐作为添加剂, DMF作为溶剂, 以32%~90%的产率得到了一系列完全取代的1H-咪唑衍生物175 (Scheme 29, a). 此方法可以进行克级规模的合成.
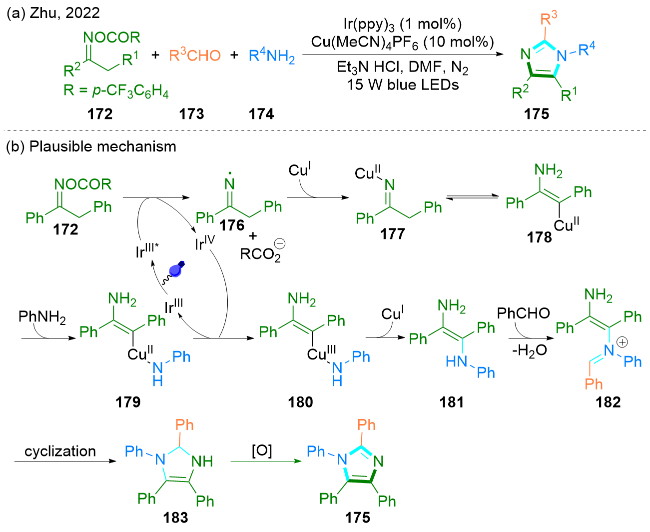
该课题组提出了一个可能的反应机理(Scheme 29, b). 在可见光的照射下, 基态光催化剂IrIII(ppy)3被激发为激发态*IrIII(ppy)3, 然后作为还原剂通过单电子转移过程还原肟酯172, 同时释放亚氨基自由基176. 随后, 自由基176被Cu(I)捕获产生亚氨基Cu(II)络合物177. Cu(II)络合物177发生连续异构化得到中间体178, 中间体178与苯胺配位得到Cu(II)中间体179. 然后, 中间体179被IrIV(ppy)3氧化, 产生Cu(III)络合物180和IrIII(ppy)3. 络合物180经过还原消除, 得到中间体181. 中间体181进一步与苯甲醛反应, 通过脱水缩合得到中间体182. 中间体182经过分子内亲核加成得到中间体183. 最终, 经氧化芳构化生成完全取代的1H-咪唑175.

2.4 电催化合成咪唑类化合物
2021年, Chen课题组[57]报道了一种在无隔膜电解条件下合成1,2,4,5-四取代咪唑194的电化学方法(Scheme 32, a). 该方法的优点在于避免了过渡金属和氧化剂的使用、官能团容忍性好以及为合成GABAA受体类似物提供了一个简单、绿色的途径.
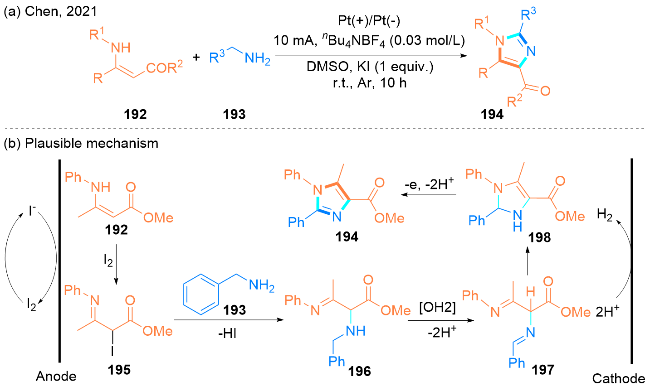
该课题组提出了一个合理的机理(Scheme 32, b): 首先, 分子碘在阳极上原位生成, 然后碘与β-烯胺酯192反应生成碘化中间体195, 中间体195与苄胺193发生亲核取代得到中间体196. 随后, 中间体196经过电化学氧化生成197, 197发生分子内环化形成中间体198. 最后, 中间体198通过去质子作用和电化学氧化生成目标产物1,2,4,5-四取代咪唑194.
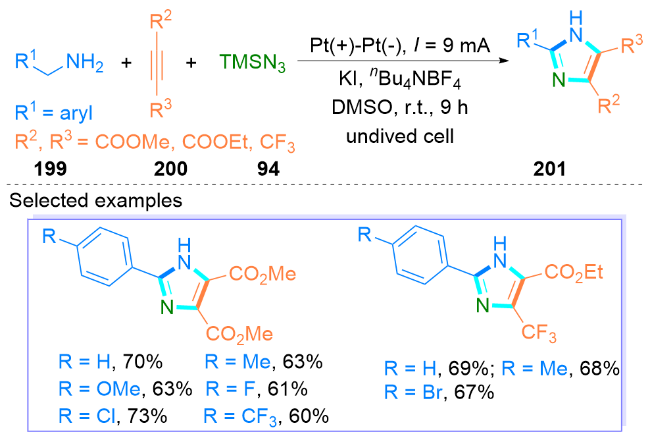
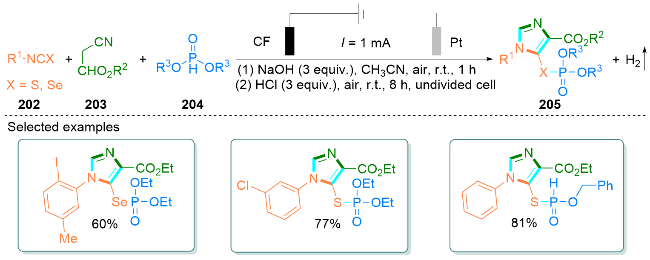
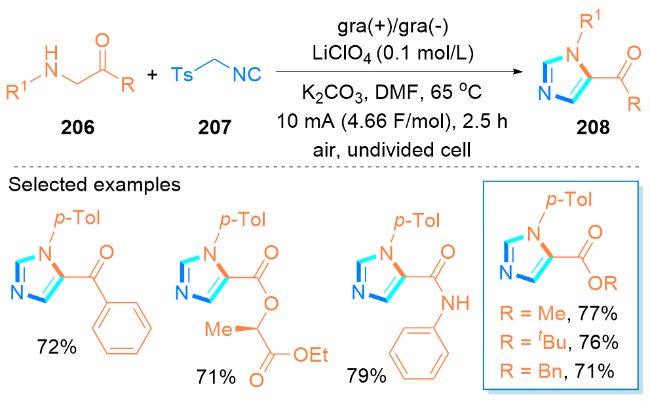
2.5 其他无过渡金属催化合成咪唑类化合物
2022年, Lee课题组[61]报道了一种在甲醇作溶剂的条件下, 乙二醛209、芳香胺210和脒211发生一锅反应合成5-氨基咪唑衍生物212的方法(Scheme 36, a). 值得注意的是, 5-氨基咪唑是多种生物活性和药用化合物的重要结构单元. 此外, 合成的5-氨基咪唑可以与2-溴苯甲酰氯反应, 进一步得到咪唑并[5,1-b]喹唑啉-9(4H)-酮. 此方法的优点在于可以快速合成5-氨基咪唑, 适用于克级规模的合成且不需要色谱纯化.
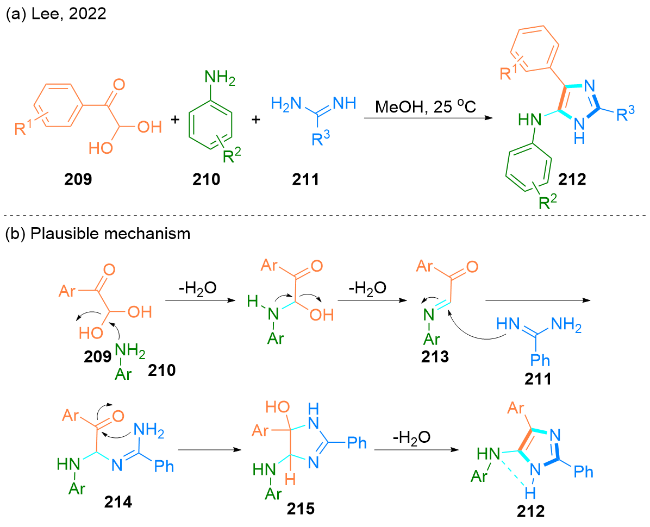
作者提出了一种可能的反应机理(Scheme 36, b): 最初, 芳基乙二醛209和芳基胺210反应生成重要的C-苯甲酰亚胺中间体213, 伴随失去两个水分子. 随后, 苯甲脒211对中间体213的亚胺碳进行亲核进攻, 得到氨基-β-酮胺加合物214. 苯甲酰基的羰基易受脒的氨基的亲核进攻, 并通过4,5-二氢咪唑215中间体, 以5-exo-trig的方式进行分子内环化. 最后, 中间体4,5-二氢咪唑215经过脱水和互变异构反应生成5-氨基咪唑212, 这可能是由于分子内氢键的稳定作用.
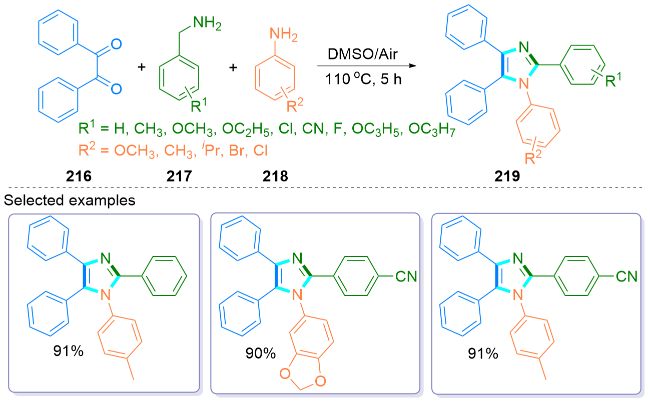
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 总结与展望
对近五年咪唑类化合物的合成方法进行了综述. 从文中的讨论可以看出, 该领域一直是人们关注和研究的热点, 随着化学工作者研究的不断深入, 此类化合物的合成方法丰富多样. 多种类型的过渡金属催化合成咪唑类化合物的反应被发现, 包括铜催化、银催化、铁催化以及一些其他过渡金属催化的反应. 随着绿色环保理念的提出, 化学工作者又迫切寻找一种既要符合工业生产要求, 又要顺应绿色环保理念的方法来合成咪唑类化合物. 因此, 光催化、电催化和无过渡金属催化反应的研究也取得了一定的进展. 这些研究进展不仅丰富了咪唑类化合物的合成方法, 而且在应用方面也提供了新的实践和理论基础. 值得注意的是, 传统咪唑类化合物的工业生产往往伴随着高污染, 随着国家环保力度的不断加大, 高额的环保成本使得咪唑类化合物生产变得更加困难. 因此为了满足工业化的需求, 拓展更多反应底物, 设计合成新的催化方法和优化反应体系, 发展助力更绿色高效的方法来合成咪唑类化合物, 仍是今后研究的主要方向.
(Zhao, C.)