

1 Introduction
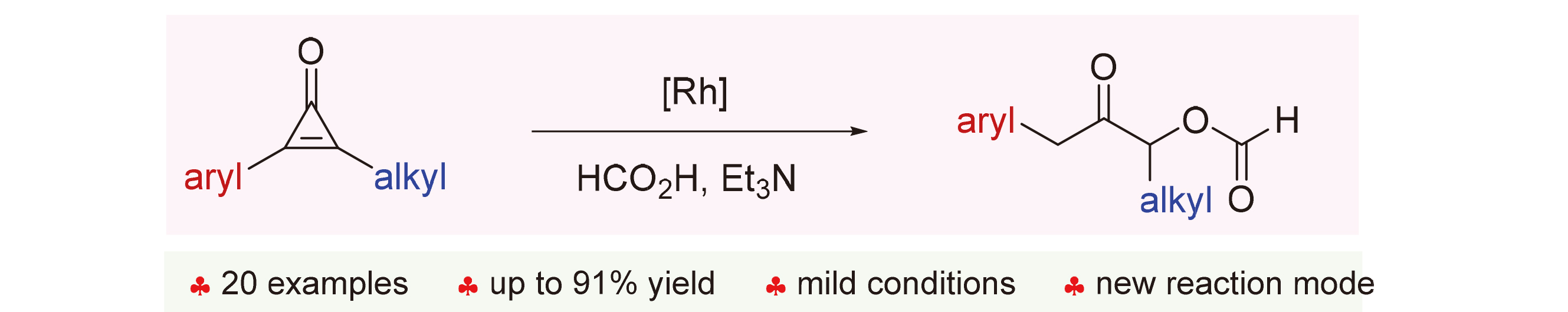
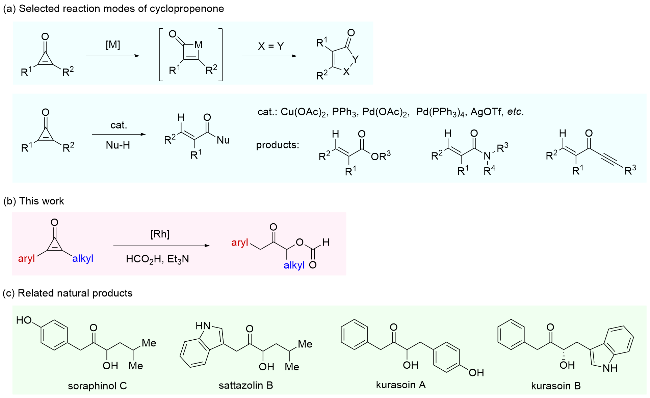
Developing new reaction modes mediated by widely used and readily available starting materials plays a vital role in organic synthesis. For instance, cyclopropenone is a class of highly reactive reagents that has been extensively studied.[1] Known reaction modes include transition-metal-catalyzed annulations that afford various cyclic products[2] and ring-opening reactions that produce unsaturated esters,[3] amides,[4] and ynones,[5] which are catalyzed by either transition metals or organocatalysts (Scheme 1, a). Considering the great importance of cyclopropenones, new reaction modes that generate valuable compounds are still highly desirable. We have been interested in chemical transformations using small ring molecules[6] and herein we report the rhodium-catalyzed ring-opening of aryl-alkyl- substituted cyclopropenones under transfer hydrogenation conditions, and the products are acyloin derivatives bearing benzyl and alkyl substituents (Scheme 1, b), which are ubiquitous in natural products that show various promising activities (Scheme 1, c).[7]
2 Results and discussion
As shown in Table 1, we commenced examining the reaction of 1a under the catalysis of a series of half-sandwich Rh or Ru catalysts[8] that have been widely used in Noyori- Ikariya transfer hydrogenation reactions.[9-10] No reaction happened when A was tested (Table 1, Entry 1), and catalyst B could catalyze the reaction to deliver acyloin 2a in moderate yield, together with a small amount of 3a (Table 1, Entry 2). Ruthenium catalysts C, D, and E were found to be ineffective in catalyzing the reaction (Table 1, Entries 3~5). Then, using catalyst B, a range of bases such as TMEDA, DABCO, tBuOK, Cs2CO3 was tested, but no better results were observed (Table 1, Entries 6~9). Increasing the dosage of HCO2H/Et3N resulted in a lower yield of 2a (Table 1, Entry 10), but decreasing the amount of HCO2H/ Et3N produced a higher yield of 2a (Table 1, Entry 11). Adding solvents such as tetrahydrofuran (THF) and MeOH impeded the reaction (Table 1, Entries 12, 13), and the reaction in MeCN afforded 2a in 70% yield with 4∶1 regioselectivity (Table 1, Entry 14). To our delight, using CH2Cl2 as solvent, 2a was obtained in 72% yield with 6∶1 rr (Table 1, Entry 15), but CHCl3 and CH2ClCH2Cl gave inferior results (Table 1, Entries 16, 17). Acetone was proven not to be a suitable solvent (Table 1, Entry 18), and toluene did not provide better outcomes (Table 1, Entry 19). We also attempted to add Lewis acid as additive, such as Zn(OTf)2, Mg(OTf)2 and MgBr2•Et2O, but the yield of 2a wasn’t improved (Table 1, Entries 20~22). Furthermore, we found that the catalyst was necessary (Table 1, Entry 23).
Table 1 Optimization of reaction conditions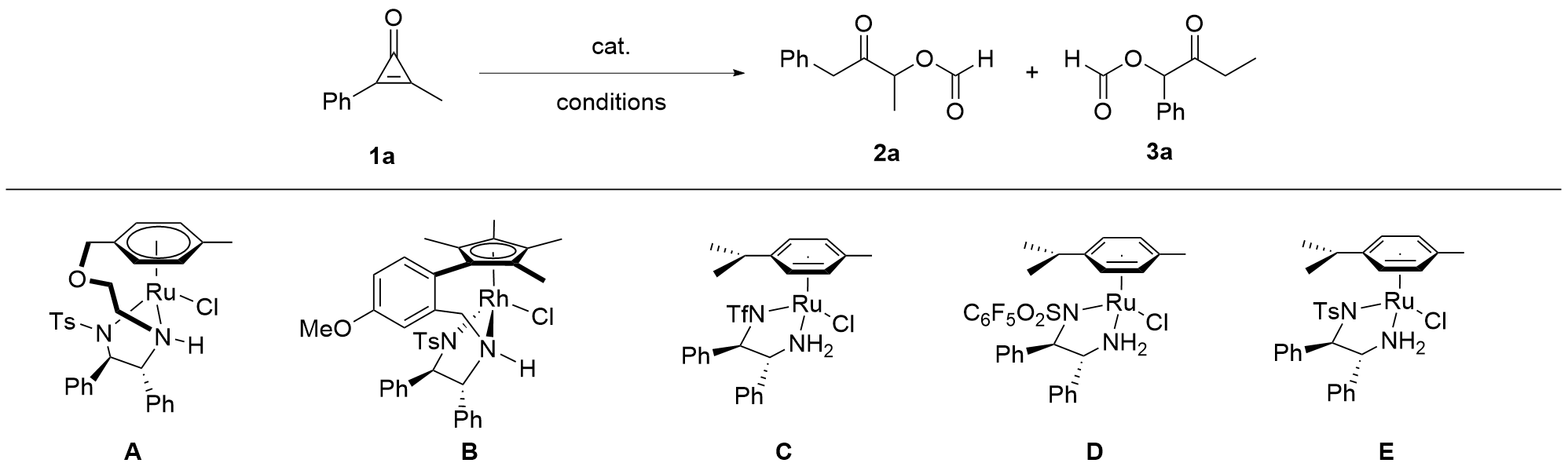 |
| Entry | Cat. | Hydrogen donor/base | HCO2H/equiv. | Solven | Additive | Temp./℃ | Time/h | Yieldb/% of 2a | rrc (2a∶3a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | HCO2H/Et3N | 60 | — | — | 20 | 9.5 | Trace | — | |||||||||
| 2 | B | HCO2H/Et3N | 60 | — | — | 20 | 19 | 51 | 6∶1 | |||||||||
| 3 | C | HCO2H/Et3N | 60 | — | — | 30 | 11 | Trace | — | |||||||||
| 4 | D | HCO2H/Et3N | 60 | — | — | 30 | 11 | Trace | — | |||||||||
| 5 | E | HCO2H/Et3N | 60 | — | — | 30 | 11 | Trace | — | |||||||||
| 6 | B | HCO2H/TMEDA | 60 | — | — | 30 | 12 | 11 | 3∶1 | |||||||||
| 7 | B | HCO2H/DABCO | 60 | — | — | 30 | 13 | Trace | — | |||||||||
| 8 | B | HCO2H/tBuOK | 60 | — | — | 30 | 12 | Trace | — | |||||||||
| 9 | B | HCO2H/Cs2CO3 | 60 | — | — | 30 | 12 | Trace | — | |||||||||
| 10 | B | HCO2H/Et3N | 120 | — | — | 30 | 24 | 32 | 5∶1 | |||||||||
| 11 | B | HCO2H/Et3N | 30 | — | — | 30 | 24 | 66 | 6∶1 | |||||||||
| 12 | B | HCO2H/Et3N | 30 | THF | — | 30 | 24 | 29 | 1∶1 | |||||||||
| 13 | B | HCO2H/Et3N | 30 | MeOH | — | 30 | 24 | Trace | — | |||||||||
| 14 | B | HCO2H/Et3N | 30 | MeCN | — | 30 | 24 | 70 | 4∶1 | |||||||||
| 15 | B | HCO2H/Et3N | 30 | CH2Cl2 | — | 30 | 24 | 72 | 6∶1 | |||||||||
| 16 | B | HCO2H/Et3N | 30 | CHCl3 | — | 30 | 24 | 52 | 4∶1 | |||||||||
| 17 | B | HCO2H/Et3N | 30 | CH2ClCH2Cl | — | 30 | 24 | 70 | 5∶1 | |||||||||
| 18 | B | HCO2H/Et3N | 30 | Acetone | — | 30 | 24 | Trace | — | |||||||||
| 19 | B | HCO2H/Et3N | 30 | Toluene | — | 30 | 24 | 71 | 4∶1 | |||||||||
| 20 | B | HCO2H/Et3N | 30 | CH2Cl2 | Zn(OTf)2 | 30 | 24 | 45 | 5∶1 | |||||||||
| 21 | B | HCO2H/Et3N | 30 | CH2Cl2 | Mg(OTf)2 | 30 | 24 | 52 | 5∶1 | |||||||||
| 22 | B | HCO2H/Et3N | 30 | CH2Cl2 | MgBr2•Et2O | 30 | 24 | 54 | 8∶1 | |||||||||
| 23 | — | HCO2H/Et3N | 30 | CH2Cl2 | — | 30 | 24 | Trace | — | |||||||||
a Reaction conditions: 1a (0.2 mmol), cat. (1 mol%), HCOOH/base (V∶V=1∶1), solvent (0.5 mL), additive (20 mol%), under argon protection, 30 ℃. DIPEA=N,N,-diisopropylethylamine, TMEDA=N,N,N',N'-tetramethylethylenediamine, DBU=1,8-diazabicyclo[5.4.0]undec-7-ene, DABCO=1,4-diazabicyclo[2.2.2]- octane, MTBE=methyl tert-butyl ether. b Isolated yields based on 1a. c Determined by crude 1H NMR. |
Having established the optimal conditions for the acyloin formation, we then started to survey the scope of the reaction (Table 2). It was found that the aryl rings bearing electron-donating groups retard the reaction, and both 2b and 2c were obtained in low yield with low rr values. To our delight, electron-deficient aromatic rings worked well, delivering 2d, 2e and 2f in good yields with good regioselectivities. The substrate having 4-PhC6H4 group was also compatible with the reaction conditions, affording 2g in 63% yield with 4∶1 rr. The substrate having heterocyclic thienyl group released 2h in moderate yield with 2∶1 rr, and 3-Me-4-ClC6H3 group was suitable for the reaction (Table 2, 2i). To our delight, the substrate having electron-deficient 3,5-Cl2C6H3 worked well to deliver 2j in 91% yield with>20∶1 rr. Next, a range of starting materials having butyl groups were tested. It was found that phenyl, 1-naphthyl, and 3,5-Cl2C6H3 were able to produce the corresponding products in moderate to high yields (Table 2, 2k~2m). Similarly, chloropropyl group did not affect the reaction, affording 2n and 2o in good yields. Subsequently, a series of alkyls bearing different functional groups, such as phenyl, CH3OCH2O (OMOM), OBn, and acetal, were surveyed, and in all cases, the corresponding products were obtained smoothly (Table 2, 2p~2s). Finally, the secondary cyclohexyl group was proven to generate 2t in moderate yield with 3∶1 rr. Dialkyl and diphenyl-substituted substrates 1u and 1v have also been tested, but the reactions were ineffective in providing the desired products (Table 2). The results indicate that both the electronic and steric properties of the substrates affect the reaction.
Table 2 Substrate scope of 2a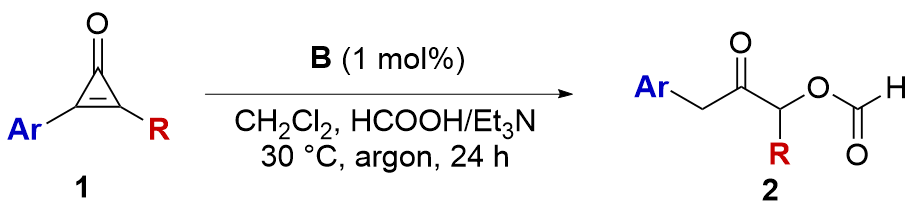 |
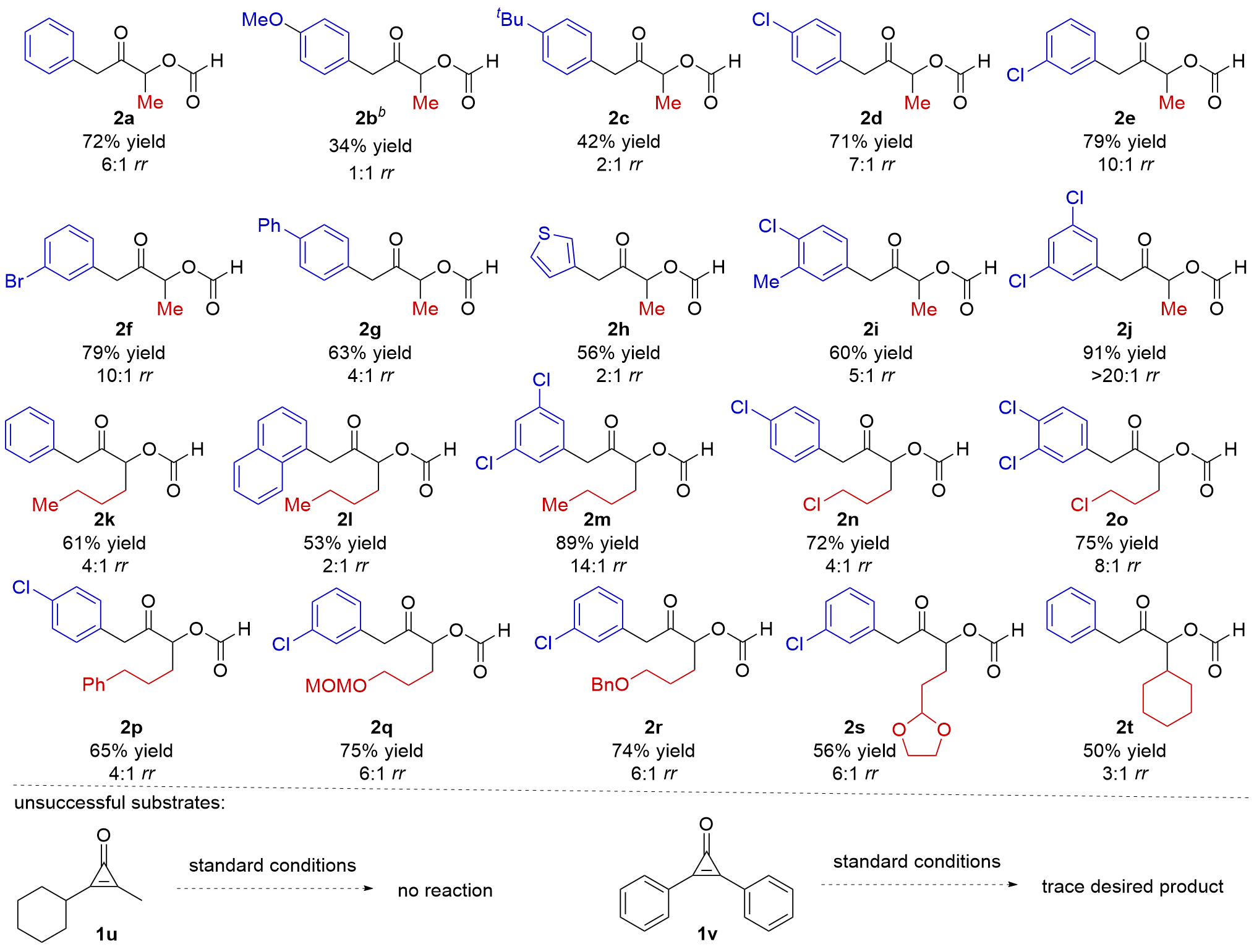 |
aReaction conditions: 1 (0.2 mmol), cat. B (1 mol%), HCOOH/Et3N (V∶V=1∶1, 30 equiv.), CH2Cl2 (0.5 mL), 30 ℃, 24 h; 99% HCOOH was used; all yields were of isolated products; rr values were determined via 1H NMR analysis of the reaction mixtures. b Compound 2b was synthesized using Et2O as the solvent. |
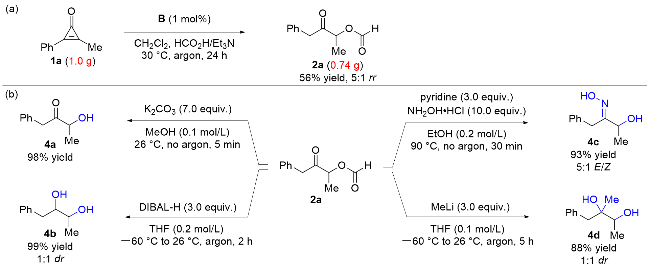
The gram-scale reaction using 1.0 g of 1a proceeded smoothly to provide 2a in 56% yield with 5∶1 rr (Scheme 2, a). Furthermore, the product could undergo a series of further reactions (Scheme 2, b). For instance, the hydrolysis of 2a afforded acyloin 4a in 98% yield, and the reduction of 2a using DIBAL-H led to the formation of diol 4b, the reaction of 2a with hydroxyamine generated imine 4c, and the addition of MeLi allowed access to diol 4d.
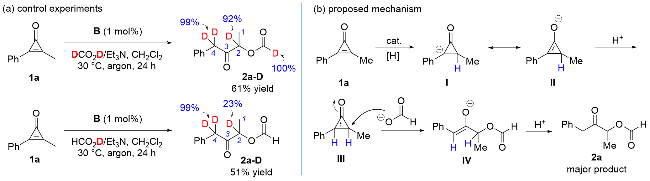
To get more information about the reaction mechanism, we conducted related control experiments (Scheme 3, a). The reaction using DCO2D delivered 2a-D with 92% D at the 2-position and 99% D at the 4-position. For comparison, the reaction using HCO2D released 2a-D with only 23% D at the 2-position. Based on the above information, we postulated the following mechanism (Scheme 3, b). The rhodium-catalyzed transfer hydrogenation of 1a afforded intermediate I, which has a resonance structure with II; After protonation, cyclopropenone III is formed. Then, formate anion attacks III to give ring opening intermediate IV, and after the second protonation, 2a is obtained. From the comparison of the regioselectivity of 2a, 2k, and 2t, formate anion attacks preferentially from the less sterically hindered position. Moreover, various methods have been employed to synthesize the key intermediate III, but all failed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 Conclusions
In summary, we have developed for the first time the rhodium catalyzed synthesis of benzyl-alkyl substituted acyloin derivatives through the ring-opening of cyclopropenones. The reaction represents a new reaction mode of cyclopropenone, and the acyloin products are highly valuable synthon in organic synthesis and are key moiety in numerous natural products.
4 Experimental section
4.1 General experimental information
Commercially available chemicals were directly used without further purification, unless otherwise mentioned all experiments and manipulations involving air- or moisture-sensitive compounds were performed using the standard Schlenk technique. All solvents were purified and dried using typical procedures. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker AVANCE III HD400 (400 MHz) and ECZ600S (600 MHz) spectrometers using tetramethylsilane (δ 0.00) as internal standard. Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on Bruker AVANCE III HD400 (101 MHz) and ECZ600S (151 MHz) spectrometers. High resolution mass spectral analysis (HRMS) was performed on a Thermo Fisher Scientific Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer.
4.2 General procedure for the synthesis of cyclopropenones (method A, 1a~1i, 1k, 1l, 1n, 1p~1t)
To a dry two-necked 250 mL flask were added iodobenzene (4.1 g, 20 mmol), CuI (152 mg, 0.8 mmol) and PdCl2(PPh3)2 (140 mg, 0.2 mmol) under an argon atmosphere. Then Et3N (67 mL) and propyne (24 mL, 1.0 mol/L in THF, 24 mmol) were added to the system. The reaction mixture was stirred at 25 ℃ for 6 h. After completion of the reaction as indicated by thin layer chromatography (TLC), it was quenched with saturated aqueous NH4Cl, extracted with EtOAc (5 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by chromatography on silica gel (petroleum ether) to afford prop-1-yn-1-ylbenzene (2.0 g, 85% yield) as colorless oil.
To a solution of prop-1-yn-1-ylbenzene (2.0 g, 17.2 mmol) and dry CHCl3 (2.8 mL, 34.4 mmol) in dry THF (17 mL) at -78 ℃ was added dropwise nBuLi (10.4 mL, 2.5 mol/L in hexane, 25.8 mmol) for 1 h under an argon atmosphere. The reaction mixture was then stirred for 4 h at room temperature. Then the mixture was quenched with HCl (ω=37%, 10 mL), extracted with CH2Cl2 (10 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by chromatography (petroleum ether/ethyl acetate, V∶V=1∶1) to afford 1a (248 mg, 10% yield) as brown solid. Compounds 1b~1i, 1k, 1l, 1n, 1p~1t were prepared using the same method.
2-(4-(tert-Butyl)phenyl)-3-methylcycloprop-2-en-1-one (1c): Crystalline brown solid, 482 mg, 14% yield. m.p. 91~94 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.66 (d, J=8.4 Hz, 2H), 7.48 (d, J=8.4 Hz, 2H), 2.41 (s, 3H), 1.28 (s, 9H); 13C NMR (151 MHz, CDCl3) δ: 156.7, 156.3, 154.5, 150.2, 130.9, 126.2, 121.0, 35.2, 31.0, 11.5; IR (KBr) ν: 1848, 1628, 1362, 1328, 1106, 846, 827, 682, 567 cm-1; HRMS (ESI) calcd for C14H16ONa [M+Na]+ 223.1093, found 223.1094.
2-(4-Chlorophenyl)-3-methylcycloprop-2-en-1-one (1d): Crystalline brown solid, 399 mg, 13% yield. m.p. 99~103 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.67 (d, J=8.2 Hz, 2H), 7.46 (d, J=8.2 Hz, 2H), 2.45 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 156.4, 153.7, 152.1, 138.9, 132.3, 129.8, 122.1, 11.7; IR (KBr) ν: 1847, 1630, 1587, 1400, 1089, 1011, 825, 680, 550, 459 cm-1; HRMS (ESI) calcd for C10H7OClNa [M+Na]+ 201.0078, found 201.0076.
2-(3-Chlorophenyl)-3-methylcycloprop-2-en-1-one (1e): Crystalline brown solid, 369 mg, 12% yield. m.p. 85~87 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.67 (s, 1H), 7.64~7.57 (m, 1H), 7.50~7.36 (m, 2H), 2.45 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 156.2, 153.6, 153.5, 135.2, 132.5, 130.63, 130.59, 129.1, 125.0, 11.8; IR (KBr) ν: 1863, 1831, 1639, 1467, 1426, 1313, 1158, 1067, 872, 813, 749, 690, 580 cm-1; HRMS (ESI) calcd for C10H7OClNa [M+Na]+ 201.0078, found 201.0072.
2-(3-Bromophenyl)-3-methylcycloprop-2-en-1-one (1f): Crystalline brown solid, 230 mg, 6% yield. m.p. 85~88 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.84 (s, 1H), 7.65 (dd, J=14.2, 7.6 Hz, 2H), 7.36 (t, J=7.9 Hz, 1H), 2.47 (s, 3H);13C NMR (101 MHz, CDCl3) δ: 156.3, 153.6, 153.5, 135.4, 133.6, 130.9, 129.6, 125.3, 123.2, 11.8; IR (KBr) ν: 1813, 1631, 1584, 1555, 1410, 1324, 1068, 877, 788, 730, 686, 571 cm-1; HRMS (ESI) calcd for C10H7OBrNa [M+Na]+ 244.9572, found 244.9570.
2-([1'-Biphenyl]-4-yl)-3-methylcycloprop-2-en-1-one (1g): Crystalline brown solid, 379 mg, 10% yield. m.p. 142~145 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.80 (d, J=8.1 Hz, 2H), 7.70 (d, J=8.1 Hz, 2H), 7.59 (d, J=7.4 Hz, 2H), 7.44 (t, J=7.4 Hz, 2H), 7.38 (t, J=7.4 Hz, 1H), 2.46 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 156.7, 154.3, 151.2, 145.1, 139.5, 131.5, 129.0, 128.4, 127.8, 127.2, 122.4, 11.6; IR (KBr) ν: 1843, 1626, 1484, 1397, 1329, 1004, 842, 773, 730, 698, 565, 465 cm-1; HRMS (ESI) calcd for C16H12ONa [M+Na]+ 243.0780, found 243.0782.
2-Methyl-3-(thiophen-3-yl)cycloprop-2-en-1-one (1h): Crystalline brown solid, 129 mg, 5% yield. m.p. 38~40 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.02~7.96 (m, 1H), 7.44~7.31 (m, 2H), 2.34 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 155.4, 148.6, 147.4, 134.1, 127.7, 127.4, 124.5, 11.4; IR (KBr) ν: 3426, 3086, 1845, 1605, 1504, 1415, 1308, 1212, 860, 804, 605 cm-1; HRMS (ESI) calcd for C8H6OSNa M+Na]+ 173.0032, found 173.0030.
2-(4-Chloro-3-methylphenyl)-3-methylcycloprop-2-en-1-one (1i): Crystalline white solid, 497 mg, 15% yield. m.p. 111~114 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.57 (s, 1H), 7.48~7.43 (m, 1H), 7.43~7.38 (m, 1H), 2.42 (s, 3H), 2.35 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 156.5, 153.8, 151.7, 139.0, 137.5, 133.4, 130.0, 129.4, 122.0, 20.0, 11.7; IR (KBr) ν: 1845, 1623, 1473, 1399, 1316, 1278, 1136, 1044, 906, 819, 711, 579 cm-1; HRMS (ESI) calcd for C11H9O- ClNa [M+Na]+ 215.0234, found 215.0230.
2-Butyl-3-(naphthalen-1-yl)cycloprop-2-en-1-one (1l): Crystalline brown solid, 569 mg, 14% yield. m.p. 69~72 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.64 (d, J=8.4 Hz, 1H), 8.03 (d, J=8.2 Hz, 1H), 7.95~7.85 (m, 2H), 7.72 (t, J=7.6 Hz, 1H), 7.63~7.53 (m, 2H), 2.93 (t, J=7.3 Hz, 2H), 1.93~1.78 (m, 2H), 1.59~1.46 (m, 2H), 0.98 (t, J=7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 157.2, 154.4, 152.9, 133.7, 133.5, 132.7, 130.1, 128.5, 128.3, 127.2, 126.2, 125.3, 121.8, 28.7, 26.5, 22.6, 13.8; IR (KBr) ν: 2956, 1828, 1612, 1504, 1409, 1345, 1243, 804, 783, 732, 663, 617, 479 cm-1; HRMS (ESI) calcd for C17H16ONa [M+Na]+ 259.1093, found 259.1094.
2-(4-Chlorophenyl)-3-(3-chloropropyl)cycloprop-2-en-1-one (1n): Crystalline brown solid, 290 mg, 7% yield. m.p. 56~59 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.74 (d, J=8.3 Hz, 2H), 7.52 (d, J=8.3 Hz, 2H), 3.76 (t, J=6.2 Hz, 2H), 3.07 (t, J=7.1 Hz, 2H), 2.39~2.25 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 156.0, 154.0, 153.4, 138.8, 132.2, 129.5, 121.6, 43.6, 28.9, 23.8; IR (KBr) ν: 2944, 1842, 1624, 1585, 1482, 1402, 1291, 1088, 826, 720, 535 cm-1; HRMS (ESI) calcd for C12H10OCl2Na [M+Na]+ 263.0001, found 262.9999.
2-(4-Chlorophenyl)-3-(3-phenylpropyl)cycloprop-2-en- 1-one (1p): Crystalline brown solid, 632 mg, 13% yield. m.p. 42~45 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.54 (d, J=8.3 Hz, 2H), 7.34 (d, J=8.3 Hz, 2H), 7.21~7.13 (m, 2H), 7.11~7.04 (m, 3H), 2.72~2.62 (m, 4H), 2.06~1.97 (m, 2H); 13C NMR (151 MHz, CDCl3) δ: 156.1, 155.3, 152.7, 140.2, 138.3, 132.0, 129.3, 128.2, 128.1, 125.9, 121.6, 34.7, 27.7, 25.5; IR (KBr) ν: 2928, 1837, 1623, 1480, 1400, 1289, 1084, 1009, 829, 745, 696, 534 cm-1; HRMS (ESI) calcd for C18H15OClNa [M+Na]+ 305.0704, found 305.0705.
2-(3-Chlorophenyl)-3-(3-(methoxymethoxy)propyl)cyclo-prop-2-en-1-one (1q): Brown liquid, 367 mg, 8% yield. 1H NMR (400 MHz, CDCl3) δ: 7.71 (s, 1H), 7.67~7.60 (m, 1H), 7.50~7.46 (m, 1H), 7.46~7.39 (m, 1H), 4.58 (s, 2H), 3.63 (t, J=5.9 Hz, 2H), 3.30 (s, 3H), 2.93 (t, J=7.3 Hz, 2H), 2.13~2.03 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 156.8, 156.6, 153.3, 135.3, 132.5, 130.8, 130.7, 129.3, 125.0, 96.5, 66.2, 55.4, 26.8, 23.8; IR (KBr) ν: 3446, 2932, 1839, 1622, 1587, 1148, 1107, 1033, 916, 793, 690 cm-1; HRMS (ESI) calcd for C14H15O3ClNa [M+Na]+ 289.0602, found 289.0601.
2-(3-(Benzyloxy)propyl)-3-(3-chlorophenyl)cycloprop-2-en-1-one (1r): Brown liquid, 538 mg, 10% yield. 1H NMR (400 MHz, CDCl3) δ: 7.74 (t, J=1.6 Hz, 1H), 7.65 (t, J=7.6 Hz, 1H), 7.54~7.49 (m, 1H), 7.46~7.41 (m, 1H), 7.34~7.25 (m, 5H), 4.51 (s, 2H), 3.62 (t, J=5.9 Hz, 2H), 2.97 (t, J=7.2 Hz, 2H), 2.17~2.07 (m, 2H); 13C NMR (151 MHz, CDCl3) δ: 156.9, 156.6, 153.2, 138.0, 135.2, 132.4, 130.7, 130.6, 129.2, 128.4, 127.6, 125.0, 73.1, 68.5, 26.7, 23.7; IR (KBr) ν: 2952, 1842, 1714, 1587, 1272, 1071, 788, 711 cm-1; HRMS (ESI) calcd for C19H17O2ClNa [M+Na]+ 335.0809, found 335.0805.
2-(2-(1,3-Dioxolan-2-yl)ethyl)-3-(3-chlorophenyl)cyclo-prop-2-en-1-one (1s): Brown liquid, 137 mg, 3% yield. 1H NMR (400 MHz, CDCl3) δ: 7.76 (s, 1H), 7.67 (d, J=7.4 Hz, 1H), 7.55~7.49 (m, 1H), 7.49~7.43 (m, 1H), 5.01 (t, J=4.0 Hz, 1H), 4.03~3.93 (m, 2H), 3.93~3.83 (m, 2H), 2.96 (t, J=7.3 Hz, 2H), 2.27~2.14 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 156.8, 156.6, 153.1, 135.4, 132.6, 131.0, 130.7, 129.4, 125.1, 102.7, 65.3, 30.4, 20.9; IR (KBr) ν: 3398, 1841, 1622, 1471, 1264, 1148, 1108, 1033, 916, 793, 732, 690 cm-1; HRMS (ESI) calcd for C14H13O3ClNa [M+Na]+ 287.0445, found 287.0440.
2-Cyclohexyl-3-phenylcycloprop-2-en-1-one (1t): Crystalline white solid, 402 mg, 11% yield. m.p. 49~52 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.82~7.73 (m, 2H), 7.58~7.45 (m, 3H), 3.04~2.91 (m, 1H), 2.09~2.00 (m, 2H), 1.83~1.58 (m, 5H), 1.53~1.32 (m, 3H); 13C NMR (151 MHz, CDCl3) δ: 158.1, 157.0, 152.5, 131.9, 130.8, 128.7, 123.1, 35.9, 29.6, 25.1, 24.4; IR (KBr) ν: 2930, 2849, 1838, 1622, 1444, 1290, 1257, 1161, 1018, 897, 768, 690, 600, 466 cm-1; HRMS (ESI) calcd for C15H16ONa [M+Na]+ 235.1093, found 235.1095.
4.3 General procedure for the synthesis of cyclopropenones (method B, 1j, 1m, 1o)
To a dry two-necked 250 mL flask were added 1,3- dichloro-5-iodobenzene (5.5 g, 20 mmol), CuI (152 mg, 0.8 mmol), and PdCl2(PPh3)2 (140 mg, 0.2 mmol) under an argon atmosphere. Then Et3N (67 mL) and propyne (24 mL, 1.0 mol/L in THF, 24 mmol) were added to the system. The reaction mixture was stirred at 25 ℃ for 6 h. After completion of the reaction as indicated by TLC, it was quenched with saturated aqueous NH4Cl, extracted with EtOAc (5 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by chromatography on silica gel (petroleum ether) to afford 1,3-dichloro-5-(prop-1-yn-1-yl)benzene (3.1 g, 85% yield) as a colorless oil.
To a sealed tube were added 1,3-dichloro-5-(prop-1-yn- 1-yl)benzene (3.1 g, 16.8 mmol), NaI (755 mg, 5.1 mmol) and TMSCF3 (7.4 mL, 50.4 mmol) and dry THF (34 mL) under an argon atmosphere. The reaction mixture was stirred at 120 ℃ for 48 h. Then the reaction mixture was quenched with HCl (ω=37%, 10 mL), extracted with CH2Cl2 (10 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by chromatography (petroleum ether/ethyl acetate, V∶V=1∶1) to afford 1j (322 mg, 9% yield) as a crystalline white solid. Compounds 1m and 1o were prepared using the same method.
2-(3,5-Dichlorophenyl)-3-methylcycloprop-2-en-1-one (1j): Crystalline white solid, 322 mg, 9% yield. m.p. 142~145 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.62 (d, J=1.3 Hz, 2H), 7.51 (s, 1H), 2.51 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 155.9, 155.3, 152.8, 136.2, 132.3, 129.0, 125.9, 12.0; IR (KBr) ν: 1838, 1629, 1581, 1560, 1413, 1388, 1317, 1161, 1099, 861, 796, 746, 646 cm-1; HRMS (ESI) calcd for C10H6OCl2Na [M+Na]+ 234.9688, found 234.9683.
2-Butyl-3-(3,5-dichlorophenyl)cycloprop-2-en-1-one (1m): Crystalline white solid, 515 mg, 12% yield. m.p. 60~63 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.57 (s, 2H), 7.45 (s, 1H), 2.81 (t, J=7.2 Hz, 2H), 1.83~1.67 (m, 2H), 1.50~1.37 (m, 2H), 1.00~0.85 (m, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.1, 156.3, 152.0, 136.1, 132.1, 129.0, 125.8, 28.5, 26.6, 22.4, 13.6; IR (KBr) ν: 3049, 2954, 2873, 1830, 1627, 1578, 1558, 1411, 1292, 1104, 865, 800, 761, 648 cm-1; HRMS (ESI) calcd for C13H12OCl2Na [M+Na]+ 277.0157, found 277.0155.
2-(3-Chloropropyl)-3-(3,4-dichlorophenyl)cycloprop-2-en-1-one (1o): Crystalline white solid, 695 mg, 15% yield. m.p. 63~67 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.85 (s, 1H), 7.61 (s, 2H), 3.72 (t, J=6.0 Hz, 2H), 3.05 (t, J=7.1 Hz, 2H), 2.33~2.24 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 155.9, 155.8, 153.0, 137.3, 133.9, 132.6, 131.6, 130.0, 123.0, 43.7, 29.0, 24.1; IR (KBr) ν: 3075, 1841, 1627, 1458, 1385, 1318, 1255, 1115, 1028, 847, 821, 704, 661, 580 cm-1; HRMS (ESI) calcd for C12H9OCl3Na [M+Na]+ 296.9611, found 296.9610.
4.4 Typical procedure for the transfer hydrogenation
A mixture of 1a (29 mg, 0.2 mmol) and the Rh catalyst B (1.5 mg, 0.002 mmol) in CH2Cl2 (0.5 mL) was added to the mixture of formic acid (99%)/triethylamine (0.5 mL, V∶ V=1∶1, 30 equiv.). Then the mixture was stirred at 30 ℃ under an argon atmosphere. After completion of the reaction as indicated by TLC, it was quenched with brine, extracted with CH2Cl2 (10 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by chromatography (petroleum ether/ ethyl acetate, V∶V=5∶1) to afford 2a (28 mg, 72% yield, 6∶1 rr) as colorless oil. Compounds 2b~2t were prepared using the same method.
3-Oxo-4-phenylbutan-2-yl formate (2a): Colorless oil, 28 mg, 72% yield and 6∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.06 (s, 1H), 7.37~7.29 (m, 2H), 7.29~7.23 (m, 1H), 7.23~7.15 (m, 2H), 5.29 (q, J=7.0 Hz, 1H), 3.89~3.73 (m, 2H), 1.40 (d, J=7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 204.2, 160.0, 132.9, 129.6, 128.7, 127.3, 73.8, 45.5, 16.2; IR (KBr) ν: 1716, 1495, 1454, 1164, 1031, 699 cm-1; HRMS (ESI) calcd for C11H12O3Na [M+Na]+ 215.0679, found 215.0676.
4-(4-Methoxyphenyl)-3-oxobutan-2-yl formate (2b): Colorless oil, 15 mg, 34% yield and 1∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.09 (s, 1H), 7.12 (d, J=7.6 Hz, 2H), 6.87 (d, J=7.5 Hz, 2H), 5.30 (q, J=7.1 Hz, 1H), 3.79 (s, 3H), 3.76 (s, 2H), 1.41 (d, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 204.7, 160.1, 159.0, 130.8, 124.9, 114.3, 73.9, 55.4, 44.9, 16.5; IR (KBr) ν: 3358, 1717, 1613, 1512, 1456, 1247, 1166, 1032, 909, 737, 518 cm-1; HRMS (ESI) calcd for C12H14O4Na [M+Na]+ 245.0784, found 245.0785.
4-(4-(tert-Butyl)phenyl)-3-oxobutan-2-yl formate (2c): Colorless oil, 21 mg, 42% yield and 2∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.10 (s, 1H), 7.36 (d, J=8.2 Hz, 2H), 7.14 (d, J=8.1 Hz, 2H), 5.32 (q, J=7.0 Hz, 1H), 3.86~3.74 (m, 2H), 1.43 (d, J=7.0 Hz, 3H), 1.31 (s, 9H); 13C NMR (101 MHz, CDCl3) δ: 204.5, 160.1, 150.3, 129.8, 129.4, 125.8, 74.0, 45.2, 34.6, 31.4, 16.4; IR (KBr) ν: 2959, 1718, 1364, 1268, 1164, 1023, 810, 750, 554 cm-1; HRMS (ESI) calcd for C15H20O3Na [M+Na]+ 271.1305, found 271.1304.
4-(4-Chlorophenyl)-3-oxobutan-2-yl formate (2d): Colorless oil, 32 mg, 71% yield and 7∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.10 (s, 1H), 7.31 (d, J=8.3 Hz, 2H), 7.13 (d, J=8.3 Hz, 2H), 5.29 (q, J=7.0 Hz, 1H), 3.87~3.73 (m, 2H), 1.44 (d, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 203.9, 160.1, 133.4, 131.4, 131.1, 129.0, 74.1, 44.7, 16.4; IR (KBr) ν: 1717, 1492, 1164, 1090, 1016, 802, 751, 495 cm-1; HRMS (ESI) calcd for C11H11O3ClNa [M+Na]+ 249.0289, found 249.0285.
4-(3-Chlorophenyl)-3-oxobutan-2-yl formate (2e): Colorless oil, 36 mg, 79% yield and 10∶1 rr. 1H NMR (600 MHz, CDCl3) δ: 8.10 (s, 1H), 7.26~7.23 (m, 2H), 7.18 (s, 1H), 7.10~7.05 (m, 1H), 5.29 (q, J=7.0 Hz, 1H), 3.88~3.73 (m, 2H), 1.44 (d, J=7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 203.6, 160.1, 134.8, 134.6, 130.0, 129.9, 128.0, 127.7, 74.1, 44.9, 16.4; IR (KBr) ν: 1716, 1574, 1477, 1160, 1029, 870, 766, 683 cm-1; HRMS (ESI) calcd for C11H11- O3ClNa [M+Na]+ 249.0289, found 249.0281.
4-(3-Bromophenyl)-3-oxobutan-2-yl formate (2f): Colorless oil, 43 mg, 79% yield and 10∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.11 (s, 1H), 7.41 (d, J=7.9 Hz, 1H), 7.35 (s, 1H), 7.21 (t, J=7.8 Hz, 1H), 7.12 (d, J=7.6 Hz, 1H), 5.30 (q, J=7.0 Hz, 1H), 3.87~3.72 (m, 2H), 1.45 (d, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 203.6, 160.1, 135.1, 132.8, 130.6, 130.3, 128.4, 122.8, 74.1, 44.9, 16.4; IR (KBr) ν: 1716, 1569, 1475, 1161, 1028, 765; HRMS (ESI) calcd for C11H11O3BrNa [M+Na]+ 292.9784, found: 292.9781.
4-([1'-Biphenyl]-4-yl)-3-oxobutan-2-yl formate (2g): Colorless oil, 34 mg, 63% yield and 4∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.13 (s, 1H), 7.61~7.55 (m, 4H), 7.47~7.41 (m, 2H), 7.37~7.32 (m, 1H), 7.30~7.26 (m, 2H), 5.35 (q, J=6.9 Hz, 1H), 3.93~3.82 (m, 2H), 1.46 (d, J=7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 204.3, 160.1, 140.8, 140.4, 131.9, 130.2, 128.9, 127.6, 127.5, 127.2, 74.1, 45.3, 16.4; IR (KBr) ν: 1718, 1487, 1167, 1031, 758, 698 cm-1; HRMS (ESI) calcd for C17H16O3Na [M+Na]+ 291.0992, found 291.0990.
3-Oxo-4-(thiophen-3-yl)butan-2-yl formate (2h): Colorless oil, 22 mg, 56% yield and 2∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.10 (s, 1H), 7.31 (dd, J=5.0, 3.0 Hz, 1H), 7.16~7.10 (m, 1H), 6.97 (dd, J=5.0, 1.2 Hz, 1H), 5.36~5.26 (m, 1H), 3.92~3.79 (m, 2H), 1.43 (d, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 203.9, 160.1, 132.5, 128.7, 126.2, 123.6, 73.9, 40.1, 16.4; IR (KBr) ν: 1715, 1377, 1163, 1028, 763 cm-1; HRMS (ESI) calcd for C9H10O3SNa [M+Na]+ 221.0243, found 221.0242.
4-(4-Chloro-3-methylphenyl)-3-oxobutan-2-yl formate (2i): Colorless oil, 29 mg, 60% yield and 5∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.10 (s, 1H), 7.29 (d, J=8.1 Hz, 1H), 7.05 (s, 1H), 6.95 (d, J=8.1 Hz, 1H), 5.29 (q, J=7.0 Hz, 1H), 3.84~3.68 (m, 2H), 2.35 (s, 3H), 1.44 (d, J=7.0 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 204.1, 160.1, 136.5, 133.5, 132.3, 131.3, 129.4, 128.4, 74.0, 44.7, 20.2, 16.4; IR (KBr) ν: 1717, 1481, 1161, 1032, 806 cm-1; HRMS (ESI) calcd for C12H13O3ClNa [M+Na]+ 263.0045, found 263.0041.
4-(3,5-Dichlorophenyl)-3-oxobutan-2-yl formate (2j): Colorless oil, 48 mg, 91% yield and 14∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.14 (s, 1H), 7.30 (s, 1H), 7.11 (s, 2H), 5.31 (q, J=7.0 Hz, 1H), 3.92~3.72 (m, 2H), 1.49 (d, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 203.0, 160.0, 136.0, 135.1, 128.3, 127.6, 74.1, 44.3, 16.3; IR (KBr) ν: 3318, 1719, 1569, 1433, 1163, 1031, 907, 858, 796, 728 cm-1; HRMS (ESI) calcd for C11H10O3Cl2Na [M+Na]+ 282.9899, found 282.9892.
2-Oxo-1-phenylheptan-3-yl formate (2k): Colorless oil, 29 mg, 61% yield and 4∶1 rr. 1H NMR (400 MHz, CDCl3 ) δ: 8.12 (s, 1H), 7.39~7.31 (m, 2H), 7.31~7.26 (m, 1H), 7.24~7.16 (m, 2H), 5.23 (dd, J=7.3, 5.0 Hz, 1H), 3.89~3.73 (m, 2H), 1.85~1.68 (m, 2H), 1.37~1.23 (m, 4H), 0.93~0.82 (m, 3H); 13C NMR (151 MHz, CDCl3) 204.0, 160.4, 133.0, 129.8, 128.9, 127.4, 77.7, 46.1, 30.2, 27.3, 22.3, 13.9; IR (KBr) ν: 2957, 1719, 1455, 1167, 750, 701 cm-1; HRMS (ESI) calcd for C14H18O3Na [M+Na]+ 257.1148, found 257.1147.
1-(Naphthalen-1-yl)-2-oxoheptan-3-yl formate (2l): Colorless oil, 30 mg, 53% yield and 2∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.13 (s, 1H), 7.91~7.79 (m, 3H), 7.56~7.47 (m, 2H), 7.45 (t, J=7.6 Hz, 1H), 7.40~7.34 (m, 1H), 5.27 (t, J=6.2 Hz, 1H), 4.35~4.19 (m, 2H), 1.79~1.69 (m, 2H), 1.37~1.16 (m, 4H), 0.83 (t, J=7.1 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 204.1, 160.5, 134.0, 132.4, 129.6, 128.9, 128.52, 128.47, 126.6, 126.1, 125.5, 123.9, 77.6, 44.4, 30.4, 27.2, 22.2, 13.8; IR (KBr) ν: 2956, 1718, 1168, 781 cm-1; HRMS (ESI) calcd for C18H20O3Na [M+Na]+ 307.1305, found 307.1306.
1-(3,5-Dichlorophenyl)-2-oxoheptan-3-yl formate (2m): Colorless oil, 54 mg, 89% yield and 14∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.14 (s, 1H), 7.28 (s, 1H), 7.07 (s, 2H), 5.19 (dd, J=7.3, 5.3 Hz, 1H), 3.86~3.67 (m, 2H), 1.90~1.73 (m, 2H), 1.43~1.27 (m, 4H), 0.95~0.85 (m, 3H); 13C NMR (151 MHz, CDCl3) δ: 202.7, 160.3, 136.1, 135.2, 128.4, 127.7, 77.9, 44.7, 30.2, 27.2, 22.4, 13.9; IR (KBr) ν: 2957, 1719, 1568, 1433, 1162, 857, 796, 738 cm-1; HRMS (ESI) calcd for C14H16O3Cl2Na [M+Na]+ 325.0369, found 325.0364.
6-Chloro-1-(4-chlorophenyl)-2-oxohexan-3-yl formate (2n): Colorless oil, 42 mg, 72% yield and 4∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.13 (s, 1H), 7.31 (d, J=8.3 Hz, 2H), 7.13 (d, J=8.3 Hz, 2H), 5.29~5.21 (m, 1H), 3.88~3.71 (m, 2H), 3.54 (t, J=6.2 Hz, 2H), 2.10~1.99 (m, 1H), 1.99~1.89 (m, 1H), 1.89~1.80 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 202.9, 160.1, 133.5, 131.12, 131.09, 129.0, 76.8, 45.1, 44.2, 27.9, 27.7; IR (KBr) ν: 1717, 1492, 1162, 1091, 1016, 751, 498, 425 cm-1; HRMS (ESI) calcd for C13H14O3Cl2Na [M+Na]+ 311.0212, found 311.0211.
6-Chloro-1-(3,4-dichlorophenyl)-2-oxohexan-3-yl formate (2o): Colorless oil, 49 mg, 75% yield and 8∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.14 (s, 1H), 7.41 (d, J=8.2 Hz, 1H), 7.29 (s, 1H), 7.03 (d, J=8.2 Hz, 1H), 5.25 (dd, J=7.6, 4.2 Hz, 1H), 3.86~3.71 (m, 2H), 3.56 (t, J=6.0 Hz, 2H), 2.13~2.02 (m, 1H), 2.02~1.92 (m, 1H), 1.92~1.83 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 202.4, 160.1, 132.9, 132.7, 131.8, 131.7, 130.7, 129.2, 76.8, 44.6, 44.1, 27.9, 27.6; IR (KBr) ν: 2933, 1721, 1472, 1165, 1032, 750, 440 cm-1; HRMS (ESI) calcd for C13H13O3Cl3Na [M+ Na]+ 344.9822, found 344.9820.
1-(4-Chlorophenyl)-2-oxo-6-phenylhexan-3-yl formate (2p): Colorless oil, 43 mg, 65% yield and 4∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.13 (s, 1H), 7.34~7.26 (m, 4H), 7.25~7.19 (m, 1H), 7.15 (d, J=7.4 Hz, 2H), 7.07 (d, J=8.3 Hz, 2H), 5.22 (dd, J=7.3, 4.7 Hz, 1H), 3.80~3.65 (m, 2H), 2.70~2.55 (m, 2H), 1.89~1.76 (m, 2H), 1.76~1.66 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 203.3, 160.3, 141.2, 133.4, 131.3, 131.0, 128.9, 128.6, 128.5, 126.2, 77.5, 45.0, 35.2, 29.8, 26.7; IR (KBr) ν: 2933, 1717, 1492, 1454, 1163, 1090, 1015, 802, 747, 699, 492 cm-1; HRMS (ESI) calcd for C19H19O3ClNa [M+Na]+ 353.0915, found 353.0918.
1-(3-Chlorophenyl)-6-(methoxymethoxy)-2-oxohexan-3- yl formate (2q): Colorless oil, 47 mg, 75% yield and 6∶1 rr. 1H NMR (600 MHz, CDCl3) δ: 8.14 (s, 1H), 7.29~7.26 (m, 2H), 7.20 (s, 1H), 7.11~7.05 (m, 1H), 5.27 (dd, J=8.5, 4.1 Hz, 1H), 4.60 (s, 2H), 3.85~3.75 (m, 2H), 3.56~3.51 (m, 2H), 3.35 (s, 3H), 2.02~1.94 (m, 1H), 1.93~1.84 (m, 1H), 1.72~1.66 (m, 2H); 13C NMR (151 MHz, CDCl3) δ: 203.0, 160.3, 134.8, 134.6, 130.0, 129.9, 128.0, 127.6, 96.6, 77.5, 66.7, 55.4, 45.3, 27.5, 25.4; IR (KBr) ν: 3421, 2934, 1717, 1476, 1150, 1108, 1035, 917, 765, 683 cm-1; HRMS (ESI) calcd for C15H19O5ClNa [M+Na]+ 337.0813, found: 337.0811.
6-(Benzyloxy)-1-(3-chlorophenyl)-2-oxohexan-3-yl for- mate (2r): Colorless oil, 53 mg, 74% yield and 6∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.12 (s, 1H), 7.39~7.28 (m, 5H), 7.27~7.24 (m, 2H), 7.19 (s, 1H), 7.09~7.03 (m, 1H), 5.26 (dd, J=8.0, 3.8 Hz, 1H), 4.50 (s, 2H), 3.84~3.71 (m, 2H), 3.48 (t, J=5.9 Hz, 2H), 2.05~1.95 (m, 1H), 1.94~1.83 (m, 1H), 1.75~1.67 (m, 2H); 13C NMR (151 MHz, CDCl3) δ: 203.0, 160.2, 138.3, 134.8, 134.5, 130.0, 129.9, 128.6, 128.0, 127.81, 127.78, 127.6, 77.5, 73.1, 69.1, 45.2, 27.4, 25.4; IR (KBr) ν: 3358, 1717, 1477, 1453, 1361, 1163, 1069, 747, 698 cm-1; HRMS (ESI) calcd for C20H21O4ClNa [M+Na]+ 383.1021, found 383.1020.
1-(3-Chlorophenyl)-5-(1,3-dioxolan-2-yl)-2-oxopentan-3-yl formate (2s): Colorless oil, 35 mg, 56% yield and 6∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.13 (s, 1H), 7.29~7.26 (m, 2H), 7.20 (s, 1H), 7.11~7.05 (m, 1H), 5.29 (dd, J=8.2, 4.2 Hz, 1H), 4.88 (t, J=4.2 Hz, 1H), 4.00~3.91 (m, 2H), 3.91~3.84 (m, 2H), 3.84~3.74 (m, 2H), 2.07~1.97 (m, 1H), 1.96~1.86 (m, 1H), 1.82~1.73 (m, 2H); 13C NMR (101 MHz, CDCl3) δ: 202.9, 160.2, 134.8, 134.5, 130.0, 129.9, 128.0, 127.6, 103.4, 77.3, 65.2, 65.1, 45.3, 29.0, 24.5; IR (KBr) ν: 2886, 1717, 1574, 1476, 1141, 1028, 941, 765, 683 cm-1; HRMS (ESI) calcd for C15H17O5ClNa [M+Na]+ 335.0657, found 335.0654.
1-Cyclohexyl-2-oxo-3-phenylpropyl formate (2t): Colorless oil, 26 mg, 50% yield and 3∶1 rr. 1H NMR (400 MHz, CDCl3) δ: 8.14 (s, 1H), 7.37~7.30 (m, 2H), 7.30~7.24 (m, 1H), 7.24~7.14 (m, 2H), 5.11 (d, J=4.3 Hz, 1H), 3.90~3.69 (m, 2H), 2.00~1.89 (m, 1H), 1.81~1.70 (m, 2H), 1.69~1.53 (m, 3H), 1.32~1.08 (m, 5H); 13C NMR (101 MHz, CDCl3) δ: 203.9, 160.5, 133.0, 129.8, 128.8, 127.3, 81.8, 46.7, 39.3, 29.5, 27.3, 26.2, 26.0, 25.9; IR (KBr) ν: 2928, 2855, 1719, 1452, 1168, 701 cm-1; HRMS (ESI) calcd for C16H20O3Na [M+Na]+ 283.1305, found 283.1304.
4.5 Gram scale reaction of 2a
A mixture of 1a (1.0 g, 6.9 mmol) and the Rh catalyst B (51 mg, 0.069 mmol) in CH2Cl2 (17.3 mL) was added to the mixture of formic acid (99%)/triethylamine (17.4 mL, V∶ V=1∶1, 30 equiv.). Then the mixture was stirred at 30 ℃ under an argon atmosphere. After completion of the reaction as indicated by TLC, it was quenched with brine, extracted with CH2Cl2 (20 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by chromatography (petroleum ether/ ethyl acetate, V∶V=5∶1) to afford 2a (743 mg, 56% yield, 5∶1 rr) as a colorless oil.
4.6 Procedures for the derivatizations of product
2a (19 mg, 0.1 mmol) and K2CO3 (97 mg, 0.7 mmol) were dissolved in MeOH (1 mL). The reaction mixture was stirred at 26 ℃ for 5 min. After completion of the reaction as indicated by TLC, it was quenched with saturated aqueous NH4Cl, extracted with EtOAc (5 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate, V∶V=3∶1) to afford 3-hydroxy-1-phenylbutan-2-one (4a) (16 mg, 98% yield) as colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.31~7.24 (m, 2H), 7.24~7.19 (m, 1H), 7.14 (d, J=7.1 Hz, 2H), 4.34~4.23 (m, 1H), 3.83~3.66 (m, 2H), 3.38 (d, J=4.8 Hz, 1H), 1.36 (d, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 210.2, 133.1, 129.5, 128.9, 127.5, 72.4, 44.7, 19.9; IR (KBr) ν: 3388, 1714, 1496, 1454, 1050, 909, 730, 698 cm-1; HRMS calcd for C10H12O2Na (ESI) [M+Na]+ 187.0730, found 187.0735.
To a solution of 2a (19 mg, 0.1 mmol) in dry THF (1 mL) was added DIBAL-H (0.32 mL, 1.0 mol/L in THF, 0.32 mmol) at -60 ℃. After 30 min, the reaction warmed to room temperature and stirred for another 1.5 h. After completion of the reaction as indicated by TLC, it was quenched with saturated aqueous NH4Cl, extracted with EtOAc (5 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate, V∶V=2∶1) to afford 1-phenylbutane-2,3-diol (4b) (16 mg, 99% yield, 1∶1 dr) colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.38~7.28 (m, 4H), 7.28~7.21 (m, 6H), 3.91~3.78 (m, 2H), 3.73~3.64 (m, 1H), 3.63~3.56 (m, 1H), 2.94~2.86 (m, 1H), 2.86~2.79 (m, 1H), 2.74~2.63 (m, 2H), 2.01 (br, 4H), 1.32~1.21 (m, 6H); 13C NMR (101 MHz, CDCl3) δ: 138.4, 138.1, 129.6, 129.5, 128.82, 128.79, 126.73, 126.72, 76.8, 76.0, 70.1, 40.1, 38.2, 19.6, 17.3; IR (KBr) ν: 3352, 1642, 1495, 1454, 1053, 988, 731, 698 cm-1; HRMS (ESI) calcd for C10H14O2Na [M+Na]+ 189.0886, found 189.0882.
2a (19 mg, 0.1 mmol) and NH2OH•HCl (70 mg, 1 mmol) were dissolved in EtOH (0.5 mL), followed by the slow addition of pyridine (24 μL, 0.3 mmol). The reaction mixture was stirred at 90 ℃ for 30 min. After completion of the reaction as indicated by TLC, it was quenched with HCl (1 mol/L), extracted with EtOAc (5 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, V∶V=2∶1) to afford 3-hydroxy-1-phenylbutan-2-one oxime (4c) (17 mg, 93% yield, 5∶1 E/Z) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.38~7.15 (m, 6H), 4.84 (q, J=6.7 Hz, 0.2H), 4.38 (q, J=6.4 Hz, 1H), 3.92 (d, J=14.4 Hz, 1H), 3.66 (d, J=14.4 Hz, 1H), 3.60 (s, 0.4H), 1.30 (d, J=6.5 Hz, 3H), 1.26 (d, J=6.6 Hz, 0.6H); 13C NMR (101 MHz, CDCl3) δ: 163.5, 161.5, 136.6, 136.3, 129.3, 129.0, 128.8, 127.0, 126.7, 68.5, 65.7, 37.2, 31.1, 21.4, 20.3; IR (KBr) ν: 3260, 1602, 1494, 1452, 1071, 962, 889, 728, 697 cm-1; HRMS (ESI) calcd for C10H13O2NNa [M+Na]+ 202.0838, found 202.0833.
To a solution of 2a (19 mg, 0.1 mmol) in THF (1 mL) was added MeLi (0.11 mL, 2.7 mol/L in THF, 0.3 mmol) at -60 ℃. After 1 h, the reaction mixture was warmed to room temperature and stirred for another 4 h. After completion of the reaction as indicated by TLC, it was quenched with saturated aqueous NH4Cl, extracted with EtOAc (5 mL×3), washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate, V∶V=2∶1) to afford 2-methyl-1-phenylbutane-2,3-diol (4d) (16 mg, 88% yield, 1∶1 dr) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.35~7.28 (m, 4H), 7.26~7.19 (m, 6H), 3.72 (q, J=6.4 Hz, 1H), 3.65 (q, J=6.4 Hz, 1H), 2.98 (d, J=13.4 Hz, 1H), 2.82~2.72 (m, 2H), 2.59 (d, J=13.3 Hz, 1H), 1.24 (d, J=6.4 Hz, 3H), 1.20 (d, J=6.4 Hz, 3H), 1.07 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 137.1, 137.0, 130.9, 130.7, 128.5, 128.4, 126.8, 126.7, 75.0, 74.7, 74.0, 72.4, 45.1, 41.1, 23.6, 21.1, 17.5, 17.3; IR (KBr) ν: 3376, 2978, 1495, 1453, 1375, 1085, 910, 700 cm-1; HRMS (ESI) calcd for C11H16O2Na [M+Na]+ 203.1043, found 203.1040.
Supporting Information 1H NMR and 13C NMR spectra of compounds 1c~1j, 1l~1t, 2a~2t, and 4a~4d. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Zhao, C.)