


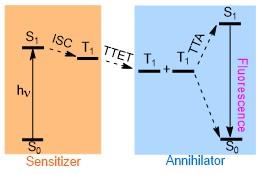
光子上转换是一种实现低能量光子向高能量光子转变的基本光学过程, 目前在超分辨显微镜、生物成像以及光伏材料等领域得到了广泛的应用[1]. 1996年, Auzel及其同事[2]在这一领域做出了开创性的工作, 他们率先提出了在光伏器件底层放置三价镧系元素Ln3+的上转换层的策略, 以此来提高太阳能的利用效率. 除了基于无机Ln3+的上转换系统, 在有机或者混合有机/无机分子体系中, 也可以通过三重态-三重态湮灭上转换(Triplet-triplet annihilation photon upconversion, TTA- UC)的机制来实现[3]. TTA-UC的原理如Scheme 1所示, 经典的TTA-UC体系常常包括两个组分, 即敏化剂(Sensitizer)和湮灭剂(Annihilator). 敏化剂分子首先吸收光能跃迁到单重激发态S1, 随后通过系间窜越(Inter- system crossing, ISC)过程发生电子自旋的翻转, 跃迁至三重激发态T1. 之后, 敏化剂和湮灭剂分子间发生三重态能量转移(Triplet-triplet energy transfer, TTET), 两个三重态的湮灭剂分子可以生成一个具有更高能量的单重激发态S1和一个回到基态的分子. 最后, S1态的分子跃迁回基态, 发出上转换荧光, 通过这一过程来实现光子能量的上转换.
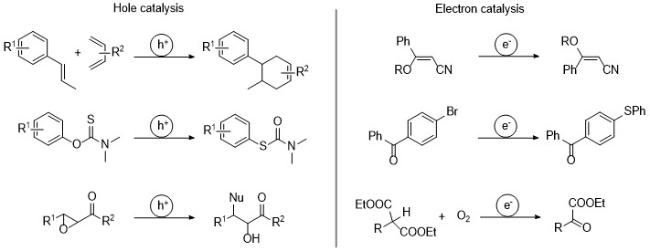
在有机合成领域, 光催化和电催化表现出巨大的应用潜力[4]. 在光氧化还原中, 单电子转移(SET)产生的自由基离子可以引发自由基链式反应[5]; 在电化学催化中, 电极向底物中注入或者移除电子来实现一些氧化还原中性的反应[6]. 由此人们提出真正起催化作用的是在反应中不断再生的电子和空穴, 从而以温和和原子经济的方式进行分子重排[7]、Diels-Alder型环加成[8]和自由基取代反应[9]等(Scheme 2).
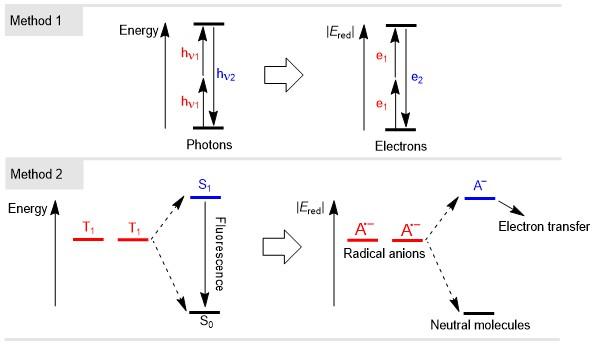
电子和光子都是物质世界的基本粒子, 电子是否可以像光子一样发生上转换过程, 从而变成具有更强供电子能力的还原剂. 2019年, Alabugin指出, 电子上转换可以产生高达20~25 kcal/mol (1 cal=4.1828 J)的能量提升, 使氧化还原电位增加0.9~1.1 V. 在此之前电子/还原剂上转换这一概念还没有被提出, Alabugin等[10]系统地从现有文献中举例介绍了电子上转换的基本原理, 并对其进行了分类, 从分子设计的角度为未来设计合成这种电子上转换还原剂提供了思路.
电子上转换可以与光子上转换的原理进行类比, 例如Scheme 3中的Method 1, 某些物质在强激光的作用下发生双光子吸收, 同时吸收相同或者不同频率的两个光子从低能态跃迁至高能态, 其产生的双光子激发荧光相比于激发光能波长更短, 能量更高, 然而这种方法不适用于电子转移, 因为第二次电子转移不会储存额外的能量, 并且上转换物种的浓度受到平衡常数的影响, 能量较高的物种不稳定, 因此其平衡浓度较小, 这种能量上升的过程在热力学上十分不利. 对于Scheme 3中的Method 2, 基于上述讨论的TTA-UC过程, 不难发现在生成能量较高的物种的同时, 也伴随着一个能量比较低的基态物种的生成. 受此启发, 人们可以通过将上转换过程与一个热力学上很有利的反应(即放能更多、产物更稳定)进行耦合, 利用释放的能量来弥补上转换过程的能量提升, 即在不违背热力学基本定律的前提下实现电子上转换.
1 电子上转换的热力学基础
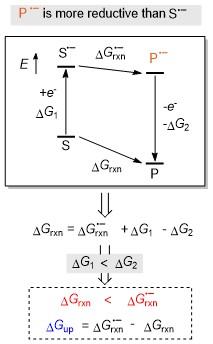
当电子转移与放能化学反应相结合时, 氧化还原上转换在热力学上是有利的. 如Scheme 4所示, 反应底物S首先通过某种方式(如光催化或者电催化等)得到一个电子, 启动整个反应. 底物S得电子过程的能量变化定义为ΔG1, 生成的自由基阴离子S•-可以转换为产物的自由基阴离子P•-, 并通过释放一个电子得到产物P, 该过程的能量变化定义为-ΔG2. 上述逆过程可用来衡量P•-的给电子能力. 通过简单的数学推导可知, 若要使产物P的还原性强于原料S, 需要满足ΔG1<ΔG2, 即 ΔGrxn<ΔGrxn•-, 需要保证相比于中性过程, 自由基阴离子转化过程的放能要更少, 甚至可能需要吸收能量, 并将二者的Gibbs自由能之差定义为上转换的Gibbs自由能变ΔGup, 以此来衡量上转换变化的程度.
在化学反应中, 除了要考虑热力学因素的影响, 动力学分析也是必要的, 这两种过程的动力学可能存在着显著差异. 对于中性反应而言, 反应能垒应该是比较高的, 转化速率较慢, 然而与上转换相结合的自由基阴离子过程应该会很快. 因此, 动力学和热力学的要求实际上是相反的, 即中性反应的放能大而速率慢, 阴离子反应的放能小而速率快, 换句话说, 中性反应物必须在动力学上稳定并储存大量能量, 电子的注入会使反应更快, 但不会使其在热力学上更有利.
以上是对所有还原剂上转换过程通用的热力学分析, 人们也根据具体的反应特征以及分子结构提出了更为实用和直观的判断方式. 总体而言, 上转换过程主要通过两种类型的反应来实现: 解离和缔合. 下面将按类别举例介绍.
2. 电子上转换的主要机制和分子设计
2.1 解离型上转换
如前所述, 基于单分子解离型上转换的热力学仍需要满足阴离子解离过程释放的能量更少, 且速率更快. 其基本原理是, 当一个分子被还原时, 电子会布居到反键轨道上, 导致分子整体能量升高, 且稳定性显著降低, 使得某些不稳定的键发生断裂. 解离型上转换概念的优势是可以分别考虑反应物和解离产物, 利用模块化的方式来调节上转换还原剂的性质, 因此可以将其分为以下三种类型(Scheme 5): 反应物驱动型(Type Ⅰ)、产物驱动型(Type Ⅱ)以及混合型(Type Ⅲ).
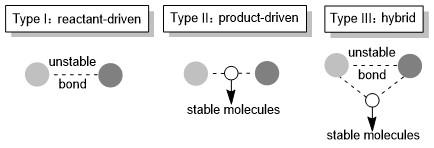
其中, 反应物驱动型上转换的主要特征是分子中存在一个不太稳定的化学键, 如N—O键、O—O键等. 当该分子获得一个电子后, 整个体系的能量得到提升, 当增加的能量与该化学键的解离能相匹配时, 该化学键发生断裂, 解离生成的两个片段分子的还原能力均得到增强. 产物驱动型上转换过程则是伴随有稳定副产物的生成, 这些稳定产物大多为一些中性小分子, 如N2, CO2, SO2,[11] 乙烯分子等, 因此在设计分子结构时, 我们需考虑将这些结构单元引入体系中. 基于以上两种类型, 混合型则是将不稳定化学键的断裂以及稳定副产物的生成相结合来实现还原剂的上转换.
2.1.1 反应物驱动型
N-氧基邻苯二甲酰亚胺(Ered=-1.30 V)是一种稳定且易于获得的化合物(Scheme 6), 当它得到一个电子之后, 会发生N—O键的断裂, 生成邻苯二甲酰亚胺阴离子以及苄氧自由基[12]. 一般来说, 自由基邻位碳上的质子会更活泼, 显示出更强的酸性, 苯甲醛自由基易被邻苯二甲酰亚胺阴离子拔去质子. 由于C—O上的两中心三电子键的形成, 上转换得到苯甲醛自由基阴离子(Ered=-1.82 V), 其给出电子(还原)的能力极大地提高, 可以发生上转换. 由于苯甲醛自由基阴离子和邻苯二甲酰亚胺相距较近, 二者之间可以优先发生电子转移, 生成苯甲醛和邻苯二甲酰亚胺自由基阴离子(Ered=-1.47 V); 接下来邻苯二甲酰亚胺自由基阴离子可以将电子传递给底物, 构成完整的自由基链式循环. 通过理论计算可知, ΔGup可以达到12 kcal/mol[10].
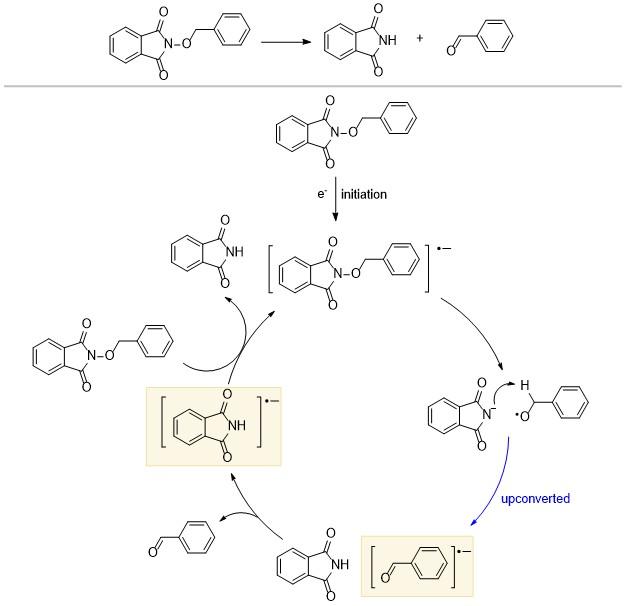
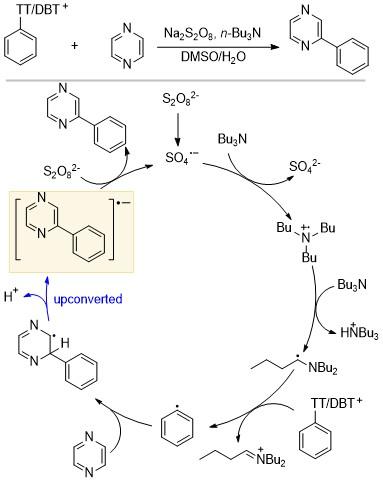
2021年Ritter课题组[13]报道了一种利用α-氨基烷基自由基催化实现杂芳基和芳基偶联的合成策略, 其反应机理与之前报道的基于卤素原子转移(XAT)的机理不同(Scheme 7). 芳基硫鎓盐的新活化模式可以在无过渡金属和没有光照的条件下进行, 具有较好的底物普适性, 可用于多种杂芳烃. 首先过二硫酸根离子分解生成的硫酸根一价阴离子与三叔丁基胺反应; 产生的胺自由基阳离子脱去质子, 上转换生成α-氨基烷基自由基, 其还原电位E=-1.12 V (vs. SCE); 接着α-氨基烷基自由基将电子传递给芳基硫鎓盐Ar-TT+. 尽管E(Ar-TT+/Ar- TT•)=-1.5 V (vs. SCE), 其值小于E($S_{2}O_{8}^{2-}/SO_{4}^{2-}+SO_{4}^{-·}$)=+1.4 V (vs. SCE), 但是α-氨基烷基自由基还是选择克服0.4 eV的能垒, 将电子传递给芳基硫鎓盐Ar-TT+, 这可能是因为芳基硫鎓盐具有比较大的π体系, 二硫酸根离子需要进行结构重组, 以及在这种两相混合体系其溶解度较低. 最后自由基α位的质子被碱拔除, 从而生成产物的自由基阴离子来实现上转换. 产物的自由基阴离子将电子传递给$S_{2}O_{8}^{2-}$后, 开启下一轮的催化.
2.1.2 产物驱动型
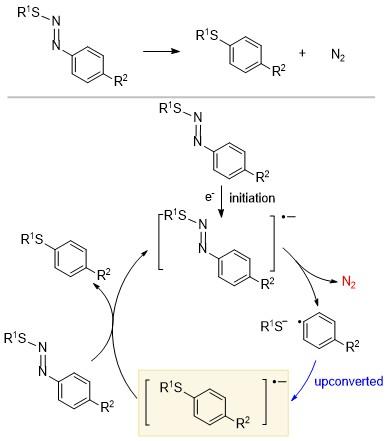
某些具有特定结构的分子在得到一个电子之后, 由于该电子布居到反键轨道上会发生不稳定化学键的断裂, 同时产生稳定的产物, 这使得反应体系的总能量下降, 从而在一定程度上弥补了上转换所造成的能量提升, 在不违背热力学定律的前提下实现还原剂的上转换. 一个比较经典的例子是偶氮硫苯化合物的裂解和重组(Scheme 8)[14]. 早在1993年, 人们便研究了在溶剂笼中, 芳偶氮硫化物的自由基通过芳香亲核取代(SRN1)与亲核试剂的快速重组. 研究发现, 该反应涉及到C—N键和S—N键的协同裂解, 释放一分子的氮气; 然后芳基自由基和烷基/芳基硫负离子发生结合, 生成上转换的中间体. 电化学分析表明, 当R1为苯基、R2为氰基时, 底物的还原电位为-1.02 V, 而产物的还原电位为-1.98 V, 其增加的还原电位高达0.96 V. 该结果得到了理论计算的支持(ΔGup=22.1 kcal/mol)[10]. 除了生成氮气外, 某些过氧化物可以通过O—O键的断裂生成稳定的副产物CO2来实现上转换[15].
2.1.3 混合型
混合型上转化过程同时涉及不稳定化学键的断裂以及稳定产物的生成. 代表性的例子是环过氧化物的上转换过程(Scheme 9)[16]. 环状骨架内的过氧键广泛存在于生物活性化合物中, 如青蒿素和软骨素等. 人们对该类环状过氧化物的电子转移过程进行了研究, 发现底物得到电子之后发生O—O的断裂, 生成两个氧上分别带有自由基和阴离子的物种. 该物种通过β裂解产生相应的酮和烯烃, 同时上转换生成酮自由基阴离子, 引发后续的自由基阴离子链式反应. 电化学测试表明, 这种二苯基酮的标准电势比底物低30 mV, 而当苯基上带有甲氧基时, 这种电势差可以增大到60 mV. 由此, 这一上转换中间体与底物之间的电子转移过程是热力学有利的, 是推动电子循环的关键步骤.
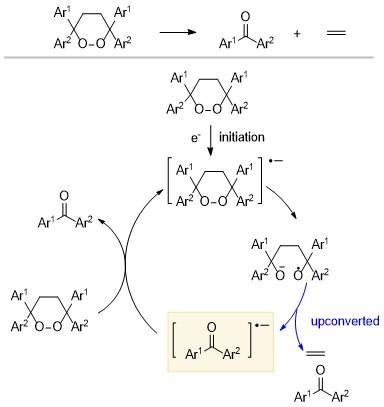
Studer和Pratt课题组[17]发现一些碱性物质(如正丁基锂和1,4-二氮杂双环[2.2.2]辛烷DABCO等)可以使高烯丙醇发生Cα—Cβ的断裂, 即高烯丙醇可以通过强碱脱质子或者与弱碱的氢键相互作用预活化Cα—Cβ, 最终使得全氟烷基碘化物以及烷基吡啶鎓盐发生烯丙基化反应(Scheme 10). 其反应机理为, 在光照条件下R—X键断裂生成烷基自由基, 烷基自由基与高烯丙醇发生双键加成; 在碱的存在下, 进一步发生C—C键的断裂, 生成烯丙基化产物, 同时上转换生成酮自由基阴离子. 这类具有更强还原能力的酮自由基将电子传递给底物, 引发下一个催化循环. 理论计算结果表明, 碱的存在降低了上转换过程的能垒ΔG‡(高达15 kcal/mol), 并在一定程度上降低了酮自由基阴离子的能量.
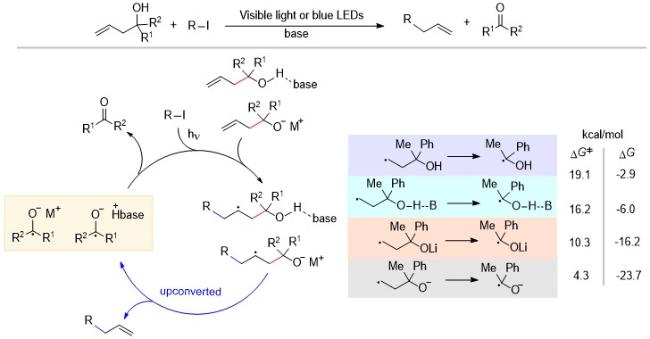
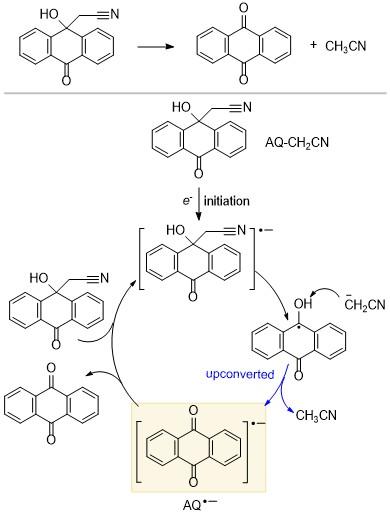
Yuasa课题组[18]发现可持续的电催化链式反应可以成为分子识别的单电子开关, 即催化量的电子就可以控制分子之间的电荷转移(Charge transfer, CT)相互作用, 并且电子的注入可以引发这种催化反应. 如Scheme 11所示, 该过程先发生C—C键断裂, 然后再释放乙腈分子上转换得到蒽醌自由基阴离子(AQ•-), 其氧化还原电位增加了大约0.15 V (3.5 kcal/mol). AQ•-再将电子传递给底物, 生成蒽醌(AQ), 由于客体分子AQ为平面结构, 可以更好地与主体分子发生CT相互作用, 即实现主客体识别功能. 同时, 萘钠作为还原剂可以将电子分别传递给AQ-CH2CN和AQ. 研究人员使用电子自旋共振(ESR)法, 在两个体系中都观察到了AQ•-物种, 没有检测出AQ-CH2CN•-, 这说明AQ-CH2CN•-的上转换过程很快, 证明产生了上转换物种AQ•-.
2.2 缔合型上转换
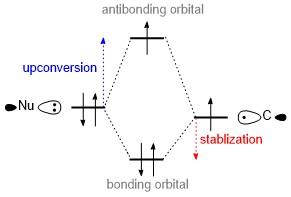
除上述通过化学键断裂或稳定产物生成实现还原剂上转换的策略外, 还存在一种通过形成化学键提升还原剂给电子能力的方法, 即下文要讨论的两中心三电子(2c, 3e)键的形成. 如Scheme 12所示的轨道相互作用的分析表明, 一个亲核试剂和一个自由基物种结合可以形成2c, 3e键, 往往可以导致上转换的发生. 众所周知, 两个原子轨道的重叠可以形成化学键, 产生两个新的分子轨道, 分别为成键轨道和反键轨道. 成键轨道中的电子能量降低, 这成为了稳定化学键的热力学基础. 然而, 由于Pauli反对称性的要求, 成键轨道只能容纳两个电子, 第三个电子必须填充到能量更高的反键轨道中, 即这个电子的能量得到提升, 从而实现了还原剂的上转换. 由于2c, 3e键的形成比经典的2c, 2e键的形成释放的能量更少, 满足Scheme 4所阐述的热力学要求.
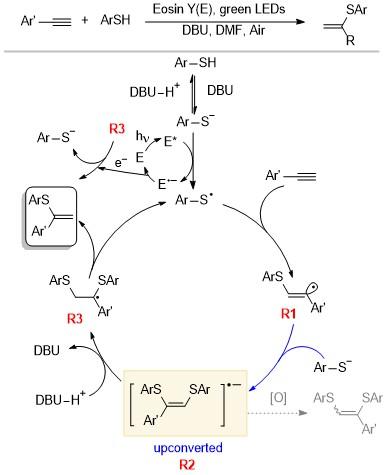
缔合型电子上转换在巯基和炔烃点击反应的选择性中起到了关键作用, 可以生成四种类型的乙烯基硫化物, 从而实现无过渡金属条件下的巯基对炔基的加成[19]. 研究人员可以通过控制反应条件, 如光照、光催化剂、碱、空气或者氩气等条件, 控制不同产物的生成. 研究人员在ESI-MS装置中添加绿色激光器, 利用质谱直接监测光化学过程, 通过实验和密度泛函理论(DFT)计算得到了该反应可能的机理(Scheme 13). 在碱性条件和光催化剂的存在下, 硫酚转换为巯基自由基, 对炔基进行加成得到中间体R1. R1再与硫酚负离子结合, 生成2c, 3e键, 从而实现上转换, 生成具有更强还原能力的中间体R2. 若此时存在氧气, R2则可以将电子传递给氧气从而失活, 生成三取代的烯烃产物. 这种电子转移过程依赖于中间体R2的结构. 通过分析不同R2的HOMO能量可知, 芳香族取代基比脂肪族取代基更能稳定中间体, 例如, 当取代基为苯基时, HOMO的能量为-3.40 eV; 当取代基为正己基时, HOMO的能量为-3.00 eV. 说明在烷基取代的情况下, 中间体R2具有更强的给电子能力, 更容易被氧化进而失活.
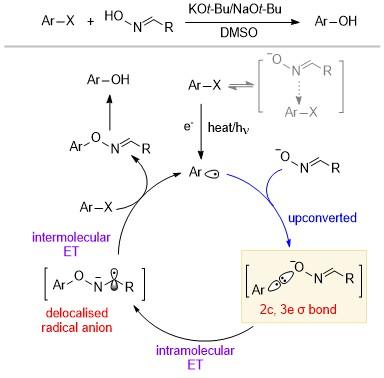
该反应机理涉及到芳基自由基与肟阴离子形成2c, 3e键的过程, 并由此引发还原剂的上转换. 首先, 芳基溴化物与肟阴离子以1∶1的比例生成复合物, 然后该复合物在光或者热化学的条件下生成芳基自由基, 接下来该自由基与肟阴离子发生自由基-阴离子偶联生成上转换产物. DFT计算表明该过程是放能的(ΔG=-17.2 kcal/mol), 并具有较小的活化能垒(ΔG‡=15 kcal/mol), 因此该过程发生的速率很快, 几乎是一个熵控的反应(ΔH‡=0.4 kcal/mol), 甚至这种含有2c, 3e键的物种可以在气相中微弱形成. 计算出的上转换的自由基阴离子的氧化还原电位是-2.14 V (vs. SCE), 而芳基溴化物的氧化还原电位是-1.89 V (vs. SCE), 说明从自由基阴离子到底物芳基溴化物的分子间电子转移过程是热力学有利的, 从而实现芳基自由基的再生, 然后所得芳基肟可以在碱性反应条件下裂解, 得到苯酚产物.
2.3 超级电子供体
基于分子的裂解或者缔合方法引发电子上转换的体系一般涉及到自由基阴离子中间体, 此外人们在研究中也发现一些中性的有机小分子表现出很强的还原能力, 被称为超级电子供体(Super electron donors, SED, Scheme 15). 基于对有机小分子S1和S2的研究[21], 人们发展合成了一系列具有更强还原能力的电子给体S3~S6[22], 这些分子中均含有杂原子取代在乙烯的结构片段. 以S3为例, 这种超强给电子特性来源于当其被氧化后, π共轭体系引发的共振使得这种自由基中间体具有芳香性. 正是这种稳定化作用使该自由基阳离子寿命较长, 容易捕获底物生成的自由基而失活, 限制了其在有机催化领域的合成应用.
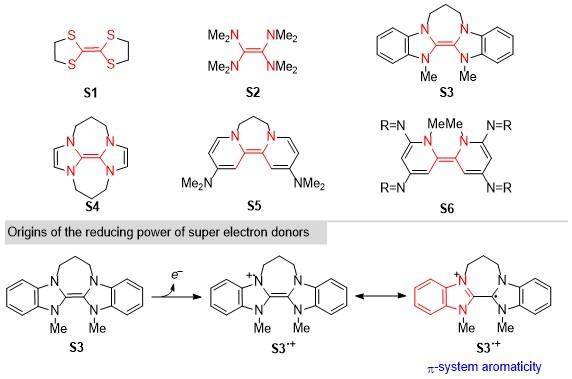
Tuttle和Murphy课题组[23]从概念上提出了一种解决这一问题的新方法, 从而发展了第一例利用中性有机超电子供体催化有机反应的体系(Scheme 16). 从苯并咪唑鎓盐出发, 在加入NaBH4的条件下, 反应以较高的产率(83%)获得SED前体(pre-SED). 以一个自由基环化反应为例, 作者对反应条件进行了优化, 发现在空气条件下, 加入pre-SED和0.2 equiv.的十二烷硫醇作为极性反转催化剂, 以87%的收率得到环化产物. 其可能的反应机理如Scheme 16所示, 首先二氢苯并咪唑为催化剂前体(pre-SED), 产物自由基物种R2•通过攫取氢原子得到氢化产物, 同时生成SED物种. 随后, 该还原剂与卤代芳烃发生电子转移, 产生芳基自由基, 再通过与较为温和的氢化试剂(如硼氢化钠)反应再生二氢苯并咪唑. 由于生成的SED物种无法通过芳香体系来稳定自由基, 因此其寿命较短, 浓度较低, 无法有效地与底物自由基相结合, 从而抑制SED物种被猝灭. 同时, 根据文献报道, SED的氧化电位可以达到-2.1 V, 展现出较强的给电子能力. 研究人员将分子S3和基于苯并咪唑结构的SED进行了反应活性的比较, 发现新开发的这种反应体系具有较好的底物普适性, 环化反应均得到比较满意的收率, 对于各种官能团的兼容性较好.

近些年来发展的SED体系不可避免地使用一些牺牲性电子给体(如溶剂或者金属氢化物). 它们是将电子送入催化循环所必不可少的物质, 但是反应过程会产生相应的氧化副产物, 如上述例子中产生的金属卤化物以及硼烷. 由于Ar—I的还原电位小于-2 V, 在非光照条件下需要使用较强的还原剂才能将其还原. 针对上述问题Martin和Broggi课题组[24]利用氮杂环卡宾(NHC)与醛相结合生成Breslow中间体, 上转换得到了一种新型的超级电子供体, 并将其应用于取代烯烃的氢芳基化反应中. 该反应的氧化副产物(酯类化合物)具有重要的应用和经济价值. 其催化机理如Scheme 17所示, G-作为SED发生了两次电子转移过程. 第一次电子转移活化芳基碘, 第二次电子转移和后续质子化生成烯烃氢化产物, 最后在醇和碱的作用下再生NHC和生成相应的醛. 然而, 对于苯乙烯化合物来说, 其还原电位小于G•, 因此会生成苄基位酰基化的产物.

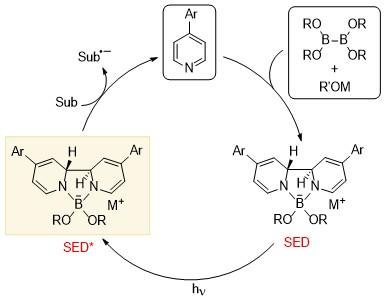
单电子转移是有机化学中的一个重要过程, 其中单电子还原剂(电子供体)是关键组分. 焦雷课题组发展了基于吡啶基硼配合物的SED体系[25], 发现由二硼化合物、甲醇盐和吡啶衍生物组成的反应体系可以在原位顺利产生超级电子供体, 观察并表征了两种基于硼基吡啶的物质. 该体系中主要的SED是反式-2H,20H-[20-联吡啶]-1,10-硼酸盐络合物(SED-1), 在甲氧基频哪醇硼酸酯的作用下进一步生成吡啶基阴离子-硼酸盐络合物(SED-2) (Scheme 18). 研究人员得到了SED-1的单晶结构. 电化学测试表明, SED-1的THF溶液在-1.11 V (vs. Fc+/0)处表现出不可逆的氧化峰. 该氧化电位与之前报道的许多SED体系相似. 其可以将电子传递到多种底物上, 引发还原反应. 研究人员通过DFT理论计算阐明了SED-1的生成机理, 并得到了过程中所涉及到的热力学数据. 与之前报道的氰基吡啶不同, 苯基吡啶通过 B—B键异裂的方式生成硼阳离子和阴离子, 利用苯基吡啶来稳定硼阴离子. DFT计算表明, 异裂过程所释放的能量为12.4 kcal/mol, 活化势垒为27.9 kcal/mol. 这说明异裂途径在热力学和动力学上都是可行的. 同时考虑到另一种可能的途径, 即[3,3]-σ重排, 计算得到其能垒为30.0 kcal/mol, 在动力学上稍有不利. 接下来另一分子苯基吡啶与硼配位, 并与之前的吡啶形成C—C键, 最终形成SED-1. 该过程的反应势垒为23.1 kcal/mol, 释放2.1 kcal/mol的能量.
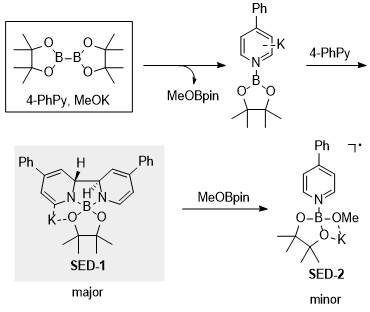
基于上述SED分子体系, 焦雷课题组进一步发展了基于光诱导电子转移(Photo-induced electron transfer, PET)的吡啶硼盐催化体系[26], 通过PET进一步提升其还原能力, 首先, 4取代的芳基吡啶与双硼酸酯、醇负离子的作用下生成SED, 然后在光照的条件下, 基态的SED跃迁至激发态SED*, 并引入模块化的概念, 通过改变吡啶硼盐上的取代基来更好地调节和修饰分子结构, 以此得到氧化还原能力适中的还原剂(Scheme 19). 这种模块化光氧化还原系统的特点是原位形成光氧化还原活性物种, 能够实现各种具有挑战性的单电子还原反应, 包括还原性C—O/C—N/C—F键断裂、甲苯磺酰胺的还原性脱磺酰化和Birch型还原等反应, 其底物的还原电位可以低至-3.3 V.
3 电子上转换在有机催化反应中的应用
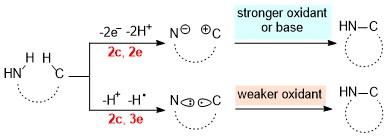
通过C(sp3)—H键和N—H键偶联直接构建C—N键是有机化学中具有挑战性的反应, 尤其是当使用非保护的伯胺和惰性的C—H键来进行偶联时, 在大部分情况下依赖于过渡金属催化. 从总的结果来看(Scheme 20), 该反应需要脱去两个质子和两个电子, 即脱去一分子的H2, 涉及到去质子化、氧化和氢原子的攫取和氢负离子的攫取等基元过程. 其反应路径可分为两大类: 一是阳离子与亲核试剂的结合(形成2e键), 二是自由基与亲核试剂的结合(形成3e键). 对于第一种途径来说, 该反应不可避免地要使用较强的氧化剂或者碱. 而人们发现含有奇数个电子的化学键的生成往往会导致电子上转换[27], 这一概念对于电子催化的化学反应具有重要的意义. 这种上转换的中间体可以与温和的或者较弱的氧化剂发生电子交换从而得到产物, 并引发下一个循环过程, 即完成电子催化剂的再生. 这为利用电子上转换的原理来实现C—N键构筑奠定了基础.
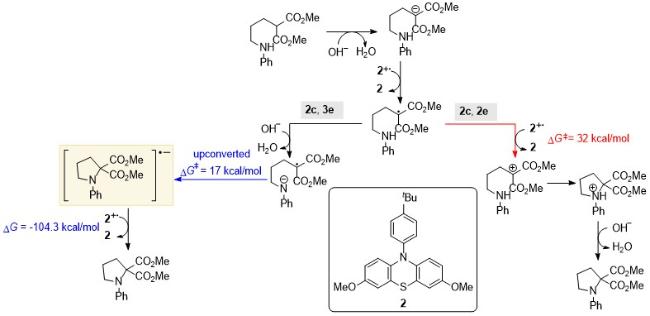
2018年徐海超课题组[28]报道了一种实现C—N键构筑的环化反应(Scheme 21). 在电化学条件下, 作者以酚噻嗪2作为电子转移受体, 氢氧化钠作为碱, 实现了C—N键的偶联反应. Alabugin和Opatz等[29]针对以上两种可能的反应路径进行了理论计算分析, 并得到了相关的反应能垒和热力学数据. 其中两种路径的前两步都经过了碱脱质子以及酚噻嗪衍生物2作为电子受体得到一个电子的过程. 然而对于2c, 2e机理, 反应路径为阳离子与亲核试剂结合来进行环化, 该过程具有较高的反应能垒(32 kcal/mol), 说明这一途径的速率较慢, 在动力学上较为不利. 对于2c, 3e机理, 自由基和亲核试剂结合可以生成上转换物种, 这一过程的过渡态能垒有所降低(17 kcal/mol). 该中间体可以与比较温和的氧化剂(如氧气)发生电子转移; 并且与较弱的氧化剂发生反应时, 由于反应过程的放能比较小, 电子转移势能面从动力学上不利的Marcus反转区转移到正常的Marcus区. 例如, Alabugin课题组[30]基于还原剂上转换的基本原理, 在温和条件下使用t-BuOK、分子氧和N,N-二甲基甲酰胺(DMF), 实现了C(sp3)—H键的亚胺化. 该反应需要弱酸性的N—H键和较为活泼的C(sp3)—H键(BDE<85 kcal/mol). 这一反应特性源于碱性、自由基和氧化物种之间的协同作用, 它们共同促进脱质子化及后续的H原子转移和氧化过程, 形成新的C—N键. 这种2c, 3e键的形成使得反应能垒下降25 kcal/mol, 并释放出20 kcal/mol的能量.
除了上述提到的利用自由基和阴离子结合导致的缔合型还原剂上转换外, 解离型上转换也在相应的有机催化体系中得到应用. 一般来说, 对于X—Y化学键的断裂, 还原性裂解通常引发电子上转换, 需要反键轨道σ*X—Y的能量较低; 而氧化性裂解通常引发空穴上转换, 需要成键轨道σX—Y的能量较高. 含有较大电负性元素的分子一般会发生还原性裂解(电子上转换), 如N-苄氧基邻苯二甲酰亚胺中的N—O键(Scheme 6), 偶氮硫自由基阴离子中的S—N键(Scheme 8), 环状过氧化物中的O—O键(Scheme 9). 反之, 能发生氧化性裂解(空穴上转换)的体系通常含有电负性较小的元素. 同时一些键能较小、更易断裂的化学键既能发生还原性裂解, 也能发生氧化性裂解. 例如对于Si—Si键来说, 它的电负性适中, 既能进行电子催化也能进行空穴催化. 主要是因为其成键和反键轨道的分离程度较小, 既是很好的电子给体也是很好的电子受体. 在氧化和还原过程中Si—Si键都很容易断裂.
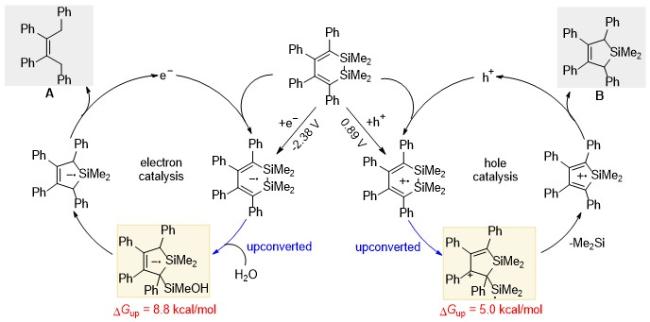
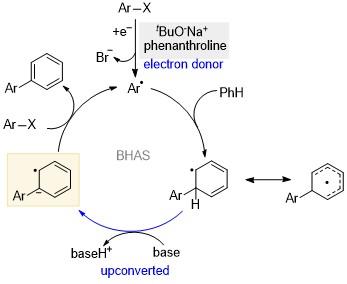
Alabugin课题组[31]使用电化学或者光氧化还原的方法实现二硅杂环己二烯的催化反应, 发现空穴或者电子催化会导向不同的产物, 说明二者经历了不同的反应路径(Scheme 22). 前线轨道分析也证明了这一点, 最高占据分子轨道(HOMO)主要分布在Si—Si σ键上, 而最低空分子轨道(LUMO)主要分布在双键的π体系上, 说明空穴上转换更有可能发生Si—Si键的断裂. 电化学分析表明该反应体系只能发生空穴上转换(ΔE=0.26 V, 相当于5 kcal/mol的能量). 二硅杂环己二烯得到电子后会形成一个较为稳定的自由基, 只有在淬灭剂水的存在下, 该自由基才能生成比较稳定的Si—O化合物和开链烯烃的产物, 从而完成催化循环. 在芳烃偶联反应中, 碱促进的均裂芳香取代反应(Base-promoted homolytic aromatic substitution, BHAS)以其操作简单、价格低廉等优势, 正在逐步取代过渡金属催化[32]. 2008年, Itami课题组[33]意外发现, 在微波条件下等物质的量的叔丁醇钾可以催化碘苯和吡嗪的直接偶联. 目前已被广泛认可的机理如Scheme 23所示[34]. 首先双胺配体、杂环卡宾和杂环芳烃(如邻菲罗啉)协同叔丁醇钾的复合物作为电子给体, 引发该反应的启动. 卤代芳烃经过脱卤素阴离子生成芳基自由基, 然后该自由基与芳烃发生偶联. 如前所述, 自由基的α-H酸性变强, 易被碱攫取质子, 发生上转换生成相应的自由基阴离子. 该上转换中间体具有很强的给电子能力, 可以作为下一轮催化循环的电子引发剂.
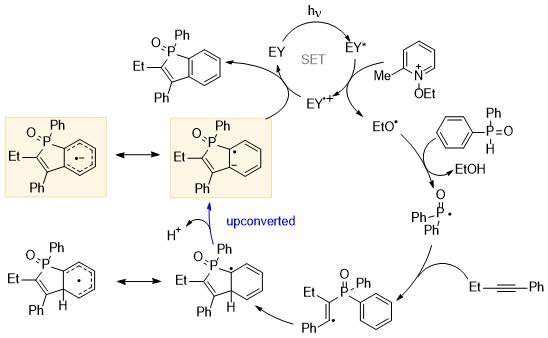
除了上述基态的反应外, 光氧化还原体系通过单电子转移过程也可以诱导产生活性自由基中间体, 发生BHAS反应. 曙红Y (Eosin Y, EY)、有机金属配合物(钌、铱等配合物)、酚噻嗪衍生物等光催化剂在光激发的条件下, 其氧化还原能力得到提升, 通过向体系中注入或者移除电子来引发反应. 目前广泛应用于C—H键活化与官能团化、交叉偶联反应和自由基加成环化等有机反应体系中. 在光氧化还原催化领域, 烷氧自由基也有着广泛的应用, 可以发生分子内和分子间的氢原子转移、不饱和键的加成, 以及单分子的β裂解. Ravelli课题组[35]利用曙红Y、3-磷酸喹诺酮衍生物和三(联吡啶)铱配合物[IrⅢ(ppy)3]作为有机光催化剂, 借助N-烷氧基氮鎓和唑鎓盐与EY*之间的电子转移过程, 成功产生烷氧自由基. 烷氧自由基可以通过对不饱和键(如碳碳双键和碳碳叁键等)进行加成, 从而实现链传递, 再通过碱攫取质子生成上转换中间体. 该中间体可以再将电子传递给EY自由基阳离子, 实现光氧化还原催化循环(Scheme 24).
金属试剂或电化学催化被认为是实现芳基卤化物脱卤氢化的有效策略, 然而这些策略在促进自由基反应方面存在局限性. 由于反应底物可以在金属表面迅速发生连续的双电子还原, 瞬态自由基会过度还原为相应的阴离子, 极大地限制了其在催化合成中的应用. 2021年Nathan课题组[36]报道了一种高效的自由基还原的方法(Scheme 25): 利用CO2自由基阴离子作为强大的单电子还原剂, 其中CO2自由基阴离子的产生是整个催化循环的关键[37]. 一般来说自由基链引发可以通过光化学或热化学进行, 反应初始是在蓝光条件下, 硫醇与光催化剂(Photocatalyst, PC)相互作用生成巯基自由基, 引发自由基链式反应. 巯基自由基与甲酸根离子发生HAT, 上转换生成CO2自由基阴离子. 该物种也可以通过CO2的电化学还原得到, 其还原电位可以达到-2.2 V (vs SCE). 本文中所用到的芳基氯化物(R为CO2Me)的还原电位为-2.1 V (vs SCE), 说明二者之间的SET过程是热力学允许的. 研究结果表明, 反应物的还原电位与反应结果是相关的. 当反应底物的还原电势大于-2.2 V (vs SCE)时, 即在CO2自由基阴离子还原能力范围内, 反应物经历SET机理; 而当反应底物的还原电势小于-2.2 V (vs SCE)时, 则会通过Giese自由基加成机制生成氢羧基化产物. 作者利用这种超强电子给体实现了很多具有挑战性的反应底物的还原, 如未活化烯烃与芳基氯/溴化物的分子间氢芳基化、芳基铵盐的自由基脱氨基、脂肪酮自由基的形成及磺酰胺裂解等反应.

基于对电子上转换体系的讨论, 对于在催化循环中涉及到的中间体的电位进行了简单的汇总与比较(Scheme 26). 对于不同的反应类型或者电子上转换的类型(如解离性和缔合型), 电子上转换的程度以及电位的提升都稍有不同, 并且可以观察到, 在发生电子上转换的体系中, 产物的还原电位小于反应物的还原电位, 这也满足了上述提出的电子上转换的热力学基础, 从而促进整个催化循环的进行.

4 总结与展望
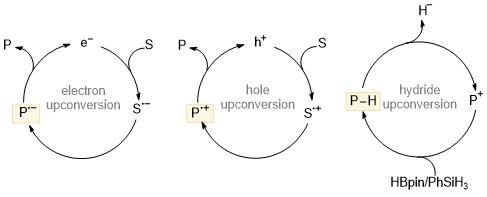
电子转移是促进化学反应的有利工具. 氧化还原介导的化学反应一般是电中性的, 其中反应物和产物处于相同的氧化态. 在这个过程中, 如果产物具有比反应物更高的氧化或者还原电位, 即在电子或者空穴上转换的情况下, 那么电子和空穴就可以成为化学反应真正的催化剂, 从而引发自由基链式反应, 使反应物可以源源不断地转化为产物(Scheme 27). 电子上转换的热力学基础是, 自由基物种相互转换的放能要比中性物种小, 同时反应速率要更快. 为了更好地理解, 讨论了一些能够实现电子上转换的体系, 如基于化学键的解离以及稳定产物生成的策略、两中心三电子键的形成及近些年来发现的具有SED特性的分子结构. 人们利用电子上转换可以实现各种有机催化反应, 如C—N/C—O/C—S键的构筑、卤代芳烃的官能团化以及DNA光损伤的还原修复等.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
电子上转换可以得到具有更强还原能力的电子供体, 与之相类似的还有空穴上转换. 通过从体系中移除一个电子实现氧化剂的上转换, 即提升氧化剂得电子的能力, 例如前文所述的空穴上转换促进的Si—Si键的活化转化. 2023年Alabugin课题组[38]报道了利用空穴来催化Diels-Alder反应, 并发现空穴上转换的发生难易和反应程度主要取决于取代基的位置和性质, 说明人们可以通过调节取代基的类型和位置来调控上转换物种的氧化或者还原电位. 此外, 程津培课题组[39]发现一些常见的负氢源(如频哪醇硼烷HBpin、苯硅烷PhSiH3等)可以与膦阳离子发生反应, 上转换为具有超强的负氢还原能力的氮杂环磷氢. 其作为负氢给体可以应用于碳卤键和碳氧键的催化转化中(Scheme 27). 目前研究主要集中在利用“电子上转换”概念设计催化有机反应, 针对衡量上转换程度的热力学参数的研究则相对较少, 如自由基阴阳离子的氧化还原电位和上转换过程的Gibbs自由能变ΔGup等. 现有数据无法支撑我们合理地设计氧化还原催化循环过程. 此外, 鉴于自由基具有高反应活性, 如何调控其反应活性以实现反应的可控进行, 是该领域极具挑战性的难题.
(Lu, Y.)