化学学报 ›› 2022, Vol. 80 ›› Issue (7): 888-895.DOI: 10.6023/A22030109 上一篇 下一篇
研究论文
李海茹a,*( ), 张层b, 李思殿c
), 张层b, 李思殿c
投稿日期:2022-03-15
发布日期:2022-05-05
通讯作者:
李海茹
基金资助:
Hairu Lia(), Ceng Zhangb, Sidian Lic
Received:2022-03-15
Published:2022-05-05
Contact:
Hairu Li
Supported by:文章分享
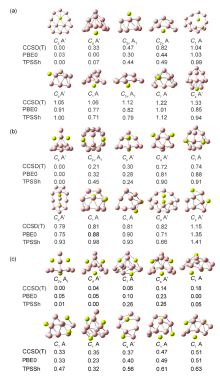
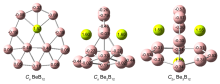
基于第一性原理, 系统地研究了Ben (n=1~3)对B12团簇结构的调控. 结果表明: 团簇BeB12全局极小结构为Cs对称性准平面结构, 而Be2B12和Be3B12最稳定的结构均为笼状结构, 对称性分别为Cs和C2v. 随着Ben (n=1~3)原子数的增加, 团簇B12由准平面结构过渡到笼状结构, 且Be倾向内嵌在B12笼状结构表面的B7或B8单元环中, 通过离子和共价作用形成稳定Be&B7和Be&B8单元, 从而稳定笼状结构. 自然键轨道(NBO)分析表明, 团簇Cs BeB12, Cs Be2B12, C2v Be3B12内部存在电子转移情况, Be原子2s轨道上失去电子, Be—B键主要以离子作用为主, 同时也存在共价作用. 成键分析显示Cs Be2B12和C2v Be3B12的π键遵循球状芳香性2(n+1)2 (n=1)电子计数规则, 表明该团簇具有球状芳香性. 预测了三个结构的红外和拉曼光谱, 为以后的合成实验和数据表征提供了理论基础.
李海茹, 张层, 李思殿. 碱土金属Ben (n=1~3)对B12团簇结构的调控研究[J]. 化学学报, 2022, 80(7): 888-895.
Hairu Li, Ceng Zhang, Sidian Li. Study on the Regulation of Alkali-earth Metal Ben (n=1~3) on the Structure of B12 Clusters[J]. Acta Chimica Sinica, 2022, 80(7): 888-895.


| BeB12 | Be2B12 | Be3B12 | |
|---|---|---|---|
| B—B键长范围 | 0.155~0.183 | 0.153~0.187 | 0.151~0.196 |
| B—Be键长范围 | 0.185~0.206 | 0.191~0.225 | 0.188~0.216 |
| Be—Be键长范围 | — | 0.240 | 0.261~0.269 |
| Be—B WBIs | 0.01~0.17 | 0.01~0.15 | 0.02~0.17 |
| Be—Be WBIs | 0.00 | 0.00~0.01 | 0.00~0.01 |
| B价电子构型 | 2s0.65–0.932p2.05–2.81 | 2s0.60–0.892p2.09–2.86 | 2s0.80–0.872p2.44–2.67 |
| Be价电子构型 | 2s0.212p0.19 | 2s0.222p0.15 | 2s0.16–0.282p0.15–0.16 |
| BeB12 | Be2B12 | Be3B12 | |
|---|---|---|---|
| B—B键长范围 | 0.155~0.183 | 0.153~0.187 | 0.151~0.196 |
| B—Be键长范围 | 0.185~0.206 | 0.191~0.225 | 0.188~0.216 |
| Be—Be键长范围 | — | 0.240 | 0.261~0.269 |
| Be—B WBIs | 0.01~0.17 | 0.01~0.15 | 0.02~0.17 |
| Be—Be WBIs | 0.00 | 0.00~0.01 | 0.00~0.01 |
| B价电子构型 | 2s0.65–0.932p2.05–2.81 | 2s0.60–0.892p2.09–2.86 | 2s0.80–0.872p2.44–2.67 |
| Be价电子构型 | 2s0.212p0.19 | 2s0.222p0.15 | 2s0.16–0.282p0.15–0.16 |
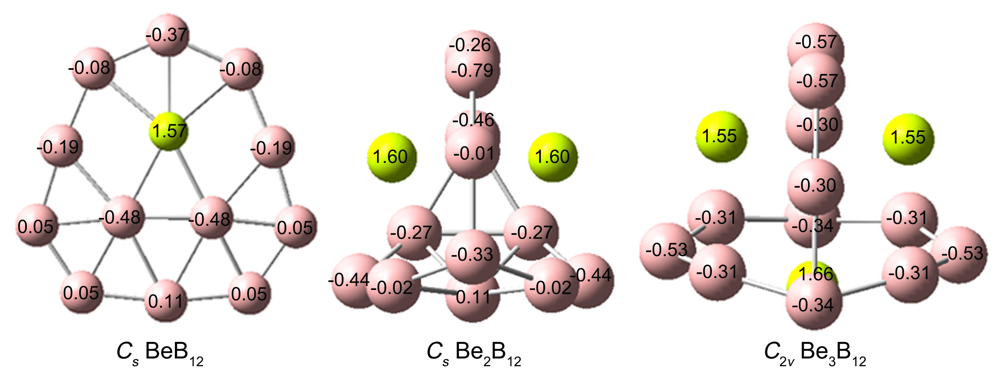
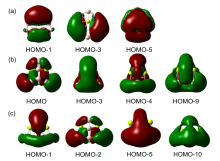




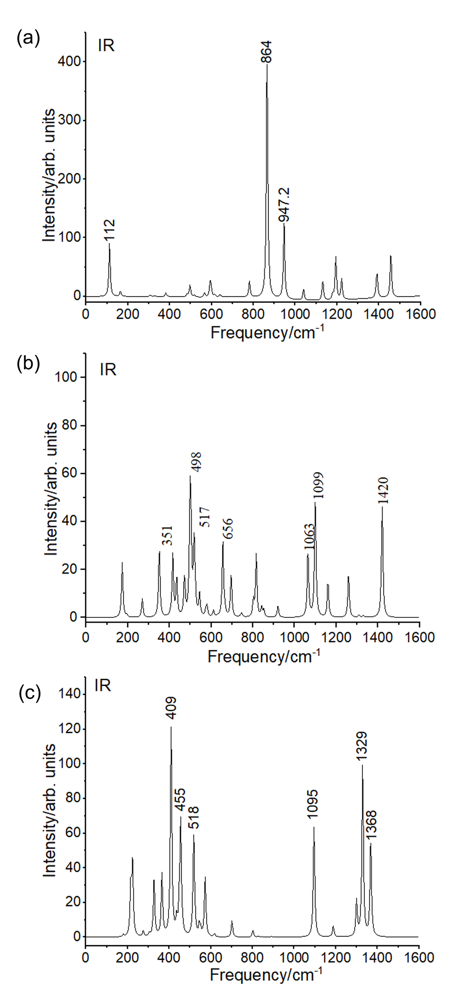
| [1] |
Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162.
doi: 10.1038/318162a0 |
| [2] |
Krätschmer, W.; Lamb, L. D.; Fostiropoulos, K.; Huffman, D. R. Nature 1990, 347, 354.
doi: 10.1038/347354a0 |
| [3] |
Iijima, S. Nature 1991, 354, 56.
doi: 10.1038/354056a0 |
| [4] |
Iijima, S.; Ichihashi, T. Nature 1993, 363, 603.
doi: 10.1038/363603a0 |
| [5] |
Alexandrova, A. N.; Boldyrev, A. I.; Zhai, H. J.; Wang, L. S. Coordin. Chem. Rev. 2006, 250, 2811.
doi: 10.1016/j.ccr.2006.03.032 |
| [6] |
Sergeeva, A. P.; Popov, I. A.; Piazza, Z. A.; Li, W. L.; Romanescu, C.; Wang, L. S.; Boldyrev, Alexander I. Acc. Chem. Res. 2014, 47, 1349.
doi: 10.1021/ar400310g |
| [7] |
Wang, L. S. Int. Rev. Phys. Chem. 2016, 35, 69.
doi: 10.1080/0144235X.2016.1147816 |
| [8] |
Zhai, H. J.; Zhao, Y. F.; Li, W. L.; Chen, Q.; Bai, H.; Hu, H. S.; Piazza, Z. A.; Tian, W. J.; Lu, H. G.; Wu, Y. B.; Mu, Y. W.; Wei, G. F.; Liu, Z. P.; Li, J.; Li, S. D.; Wang, L. S. Nat. Chem. 2014, 6, 727.
doi: 10.1038/nchem.1999 |
| [9] |
Chen, Q.; Li, H. R.; Tian, W. J.; Lu, H. G.; Zhai, H. J.; Li, S. D. Phys. Chem. Chem. Phys. 2016, 18, 14186.
doi: 10.1039/C6CP02369J |
| [10] |
Romanescu, C.; Galeev, T. R.; Li, W. L.; Boldyrev, A. I.; Wang, L. S. Angew. Chem., Int. Ed. 2011, 50, 9334.
doi: 10.1002/anie.201104166 |
| [11] |
Li, W. L.; Romanescu, C.; Galeev, T. R.; Piazza, Z. A.; Boldyrev, A. I.; Wang, L. S. J. Am. Chem. Soc. 2012, 134, 165-168.
doi: 10.1021/ja209808k |
| [12] |
Galeev, T. R.; Romanescu, C.; Li, W. L.; Wang, L. S.; Boldyrev, A. I. Angew. Chem., Int. Ed. 2012, 51, 2101.
doi: 10.1002/anie.201107880 pmid: 22298320 |
| [13] |
Romanescu, C.; Galeev, T. R.; Li, W. L.; Boldyrev, A. I.; Wang, L. S. J. Chem. Phys. 2013, 138, 134315.
doi: 10.1063/1.4798935 |
| [14] |
Li, W. L.; Ivanov, A. S.; Federi, J.; Romanescu, C.; ernušák, I.; Boldyrev, A. I.; Wang, L. S. J. Chem. Phys. 2013, 139, 104312.
doi: 10.1063/1.4820401 |
| [15] |
Romanescu, C.; Galeev, T. R.; Li, W. L.; Boldyrev, A. I.; Wang, L. S. Acc. Chem. Res. 2013, 46, 350.
doi: 10.1021/ar300149a |
| [16] |
Popov, I. A.; Li, W. L.; Piazza, Z. A.; Boldyrev, A. I.; Wang, L. S. J. Phys. Chem. A 2014, 118, 8098.
doi: 10.1021/jp411867q pmid: 24428747 |
| [17] |
Popov, I. A.; Jian, T.; Lopez, G. V.; Boldyrev, A. I.; Wang, L. S. Nat. Commun. 2015, 6, 8654.
doi: 10.1038/ncomms9654 pmid: 26456760 |
| [18] |
Jian, T.; Li, W. L.; Popov, I. A.; Lopez, G. V.; Chen, X.; Boldyrev, A. I.; Li, J.; Wang, L. S. J. Chem. Phys. 2016, 144, 154310.
doi: 10.1063/1.4946796 |
| [19] |
Jian, T.; Li, W. L.; Chen, X.; Chen, T. T.; Lopez, G. V.; Li, J.; Wang, L. S. Chem. Sci. 2016, 7, 7020.
doi: 10.1039/C6SC02623K |
| [20] |
Li, W. L.; Jian, T.; Chen, X.; Chen, T. T.; Lopez, G. V.; Li, J.; Wang, L. S. Angew. Chem., Int. Ed. 2016, 55, 7358.
doi: 10.1002/anie.201601548 |
| [21] |
Li, H. R.; Liu, H.; Tian, X. X.; Zan, W. Y.; Mu, Y. W.; Lu, H. G.; Li, J.; Wang, Y. K.; Li, S. D. Phys. Chem. Chem. Phys. 2017, 19, 27025.
doi: 10.1039/C7CP05179D |
| [22] |
Li, H. R.; Liu, H.; Lu, X. Q.; Zan, W. Y.; Tian, X. X.; Lu, H. G.; Wu, Y. B.; Mu, Y. W.; Li, S. D. Nanoscale 2018, 10, 7451.
doi: 10.1039/C8NR01087K |
| [23] |
Li, H. R. Ph.D. Dissertation, Shanxi University, Taiyuan, 2019. (in Chinese)
|
|
(李海茹, 博士论文, 山西大学, 太原, 2019.)
|
|
| [24] |
Wang, Y. J.; Feng, L. Y.; Guo, J. C.; Zhai, H. J. Chem-Asian J. 2017, 12, 2899.
doi: 10.1002/asia.201701310 |
| [25] |
Li, S. X.; Chen, D. L.; Zhang, Z. L.; Long, Z. W. Acta Phys. Sin. 2020, 69, 193101. (in Chinese)
doi: 10.7498/aps.69.20200756 |
|
(李世雄, 陈德良, 张正良, 隆正文, 物理学报, 2020, 69, 193101.)
|
|
| [26] |
Dong, X.; Jalife, S.; Vásquez-Espinal, A.; Ravell, E.; Pan, S.; Cabellos, J. L.; Liang, W. Y.; Cui, Z. H.; Merino, G. Angew. Chem. 2018, 130, 1.
doi: 10.1002/ange.201712460 |
| [27] |
Feng, L. Y. Ph.D. Dissertation, Shanxi University, Taiyuan, 2021. (in Chinese)
|
|
(冯林雁, 博士论文, 山西大学, 太原, 2021.)
|
|
| [28] |
Cui, Z. H.; Yang, W. S.; Zhao, L. L.; Ding, Y. H.; Frenking, G. Angew. Chem., Int. Ed., 2016, 55, 7841.
doi: 10.1002/anie.201601890 |
| [29] |
Feng, L. Y.; Guo, J. C.; Li, P. F.; Zhai, H. J. Phys. Chem. Chem Phys. 2018, 20, 22719.
doi: 10.1039/C8CP04332A |
| [30] |
Guo, J. C.; Feng, L. Y.; Wang, Y. J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J. L.; Pan, S.; Merino, G.; Zhai, H. J. Angew. Chem., Int. Ed. 2017, 56, 10174.
doi: 10.1002/anie.201703979 |
| [31] |
Feng, L. Y.; Wang, K.; Zhai, H. J. Phys. Chem. Chem. Phys. 2020, 22, 25574.
doi: 10.1039/D0CP05012A |
| [32] |
Feng, L. Y.; Guo, J. C.; Li, P. F.; Zhai, H. J. Chem. Asian J. 2020, 15, 1094.
doi: 10.1002/asia.201901640 |
| [33] |
Zhai, H. J.; Kiran, B.; Li, J.; Wang, L. S. Nat. Mater. 2003, 2, 827.
doi: 10.1038/nmat1012 |
| [34] |
Zhao, Y. F.; Chen, X.; Li, J. Nano Research. 2017, 10, 3407.
doi: 10.1007/s12274-017-1553-z |
| [35] |
Chen, X.; Zhao, Y. F.; Wang, L. S.; Li, J. Comput. Theor. Chem. 2017, 1107, 57.
doi: 10.1016/j.comptc.2016.12.028 |
| [36] |
Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158.
doi: 10.1063/1.478522 |
| [37] |
Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Phys. Rev. Lett. 2003, 91, 146401.
doi: 10.1103/PhysRevLett.91.146401 |
| [38] |
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980. 72, 650.
|
| [39] |
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16 Revision, A. 03, Gaussian Inc., Wallingford, CT, 2016.
|
| [40] |
Zubarev, D. Y.; Boldyrev, A. I. Phys. Chem. Chem. Phys. 2008, 10, 5207.
doi: 10.1039/b804083d pmid: 18728862 |
| [41] |
Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Bohmann, J. A.; Morales, C. M.; Weinhold, F.NBO 5.0/6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001.
|
| [42] |
Vondele, J. V.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Comput. Phys. Commun. 2005, 167, 103.
doi: 10.1016/j.cpc.2004.12.014 |
| [43] |
Pyykkö, P. J. Phys. Chem. A. 2015, 119, 2326.
doi: 10.1021/jp5065819 pmid: 25162610 |
| [44] |
Li, S. D.; Zhai, H. J.; Wang, L. S. J. Am. Chem. Soc. 2008, 130, 2573.
doi: 10.1021/ja0771080 |
| [45] |
Kalemos, A. J. Chem. Phys. 2016, 145, 214302.
doi: 10.1063/1.4967819 |
| [46] |
Wang, G.; Zhou, M.; Goettel, J. T.; Schrobilgen, G. J.; Su, J.; Li, J.; Schlöder, T.; Riedel, S. Nature 2014, 514, 475.
doi: 10.1038/nature13795 |
| [1] | 王海朋, 蔡文生, 邵学广. 抗冻剂抗冻机制的近红外光谱与分子模拟研究★[J]. 化学学报, 2023, 81(9): 1167-1174. |
| [2] | 张凯, 武晓君. 具有室温铁磁性的二维Janus钛硫属化物★[J]. 化学学报, 2023, 81(9): 1142-1147. |
| [3] | 王娟, 肖华敏, 谢丁, 郭元茹, 潘清江. 铜掺杂与氮化碳复合氧化锌材料结构和二氧化氮气体传感性质的密度泛函理论计算[J]. 化学学报, 2023, 81(11): 1493-1499. |
| [4] | 郭瑞, 魏星, 曹末云, 张研, 杨云, 樊继斌, 刘剑, 田野, 赵泽坤, 段理. AlAs/InSe范德华异质结构的光学和可调谐电子特性[J]. 化学学报, 2022, 80(4): 526-534. |
| [5] | 位亚茹, 马晶, 袁婷婷, 姜嘉伟, 段银利, 薛娟琴. 氯化锂插层氮化碳材料的可控制备及吸附性能[J]. 化学学报, 2022, 80(4): 494-502. |
| [6] | 龚雪, 马新国, 万锋达, 段汪洋, 杨小玲, 朱进容. 二维单层MoSi2X4 (X=N, P, As)的电子结构及光学性质研究[J]. 化学学报, 2022, 80(4): 510-516. |
| [7] | 熊昆, 陈伽瑶, 杨娜, 蒋尚坤, 李莉, 魏子栋. 理论探究水溶液条件对TMNxCy催化氮还原性能的增强机制[J]. 化学学报, 2021, 79(9): 1138-1145. |
| [8] | 邱凯, 严铭霞, 赵守旺, 安胜利, 王玮, 贾桂霄. Al掺杂的锂离子电池层状正极材料Li(Li0.17Ni0.17Al0.04Fe0.13Mn0.49)O2结构稳定性及氧离子氧化的理论研究[J]. 化学学报, 2021, 79(9): 1146-1153. |
| [9] | 陆远, 王继芬, 谢华清. LiMn2O4尖晶石氧化物的低指数表面结构优化及表面能的第一性原理研究[J]. 化学学报, 2021, 79(8): 1058-1064. |
| [10] | 杜英喆, 张恒, 苑世领. Al2O3/PDMS复合材料热传导的分子动力学模拟[J]. 化学学报, 2021, 79(6): 787-793. |
| [11] | 付雯雯, 李严, 梁长海. 乙醇在Co(111)表面脱氢反应机理的第一性原理研究[J]. 化学学报, 2019, 77(6): 559-568. |
| [12] | 曹爱华, 吴波, 甘利华. Pc-carbon:一种可能的超硬碳材料[J]. 化学学报, 2019, 77(5): 455-460. |
| [13] | 徐婕, 魏雨晨, 伍智蔚, 易忠胜. 基于酸度诱导的HSA与BDE154的光谱和计算模拟研究[J]. 化学学报, 2018, 76(5): 408-414. |
| [14] | 吴苗苗, 刘世强, 陈浩, 魏雪虎, 李洺阳, 杨志宾, 马向东. 超卤素掺杂立方相卤化物钙钛矿太阳能电池材料第一性原理研究[J]. 化学学报, 2018, 76(1): 49-54. |
| [15] | 郭宇, 姚远, 李慧, 赫兰兰, 朱尊伟, 杨忠志, 宫利东, 刘翠, 赵东霞. 光合释氧机理的ABEEM/MM/MD和BS-DFT理论研究[J]. 化学学报, 2017, 75(9): 903-913. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
