化学学报 ›› 2011, Vol. 69 ›› Issue (10): 1167-1172. 上一篇 下一篇
研究论文
杜瑞1,2, 陈玉红*1,2, 张致龙2, 王伟超2, 张材荣1,2, 康龙1, 罗永春1
Du Rui1,2,Chen Yuhong*,1,2,Zhang Zhilong2,Wang Weichao2,Zhang Cairong1,2,Kang Long1,Luo Yongchun1
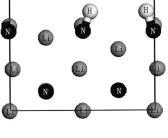
采用第一性原理方法研究了H2分子在两种Li3N(100)晶面的表面吸附情况. 通过研究Li3N(100)/H2体系的吸附位置、吸附能和电子结构, 发现H2分子在Li3N(100)晶面主要是化学吸附, 但也可以发生物理吸附. 在表面终止原子为Li和N的Li3N(100)表面, 吸附的最稳定结构中H2分子被解离, 最终H原子分别趋于两个N原子的顶位, 形成两个NH基, 吸附能为5.157 eV, 属于强化学吸附|此时H2分子与Li3N(100)表面的相互作用主要源于H1s轨道与Li3N表层N原子的2s, 2p轨道重叠杂化的贡献, 且N—H键为共价键. 在表面终止原子为Li的Li3N(100)表面, 吸附的最稳定结构中H2分子也被解离, H原子趋于穴位, 吸附能为2.464 eV, 也属于强化学吸附|此时Li和H之间为较强的离子键相互作用.
中图分类号:
