化学学报 ›› 2012, Vol. 70 ›› Issue (19): 2037-2044.DOI: 10.6023/A12070451 上一篇 下一篇
研究论文
刁智俊a, 赵跃民b, 陈博c, 段晨龙b
Diao Zhijuna, Zhao Yueminb, Chen Boc, Duan Chenlongb
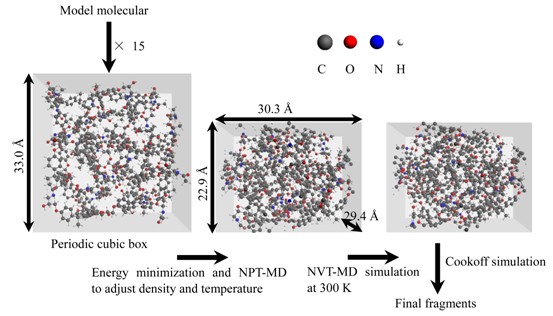
采用ReaxFF动力学方法模拟了非交联固化环氧树脂在不同温度和升温速率下的热解特性. 结果表明, 含N和含O桥键的断裂是热解的引发反应. 观察到H2O的4种主要的生成途径, 而这些反应途径都涉及到含羟基的前驱体. 当反应温度较低时, H2O为热解的主要产物. 而在高温条件下, 热解的主要产物为H2, 它主要为分子内/分子间脱氢反应和氢自由基的夺氢反应的产物; 高温同时促进了含石墨烯结构且分子量较大的碳团簇的形成. 除此之外, 还观察到了CH4, HCN, NH3和CO等小分子产物. 本文用ReaxFF动力学方法模拟所得的气体产物以及含类似石墨烯结构的碳团簇与实际实验结果一致, 说明ReaxFF动力学方法能为从分子水平上研究有机物高温热解反应提供了一种有效的途径.
