Acta Chimica Sinica ›› 2021, Vol. 79 ›› Issue (7): 948-952.DOI: 10.6023/A21040172 Previous Articles
Article
满清敏a,b, 付尊蕴b, 刘甜甜b, 郑明月b,*( ), 蒋华良a,b,*()
), 蒋华良a,b,*()
投稿日期:2021-04-22
发布日期:2021-06-02
通讯作者:
郑明月, 蒋华良
基金资助:
Qingmin Mana,b, Zunyun Fub, Tiantian Liub, Mingyue Zhengb(), Hualiang Jianga,b()
Received:2021-04-22
Published:2021-06-02
Contact:
Mingyue Zheng, Hualiang Jiang
Supported by:Share
Qingmin Man, Zunyun Fu, Tiantian Liu, Mingyue Zheng, Hualiang Jiang. DFT Mechanism of Cu Catalyzed Coupling Reaction to Alkyl Aryl Ethers[J]. Acta Chimica Sinica, 2021, 79(7): 948-952.
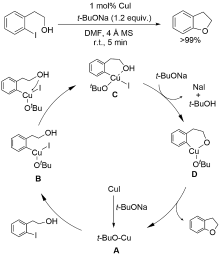

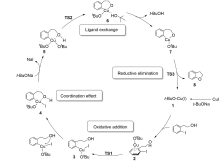
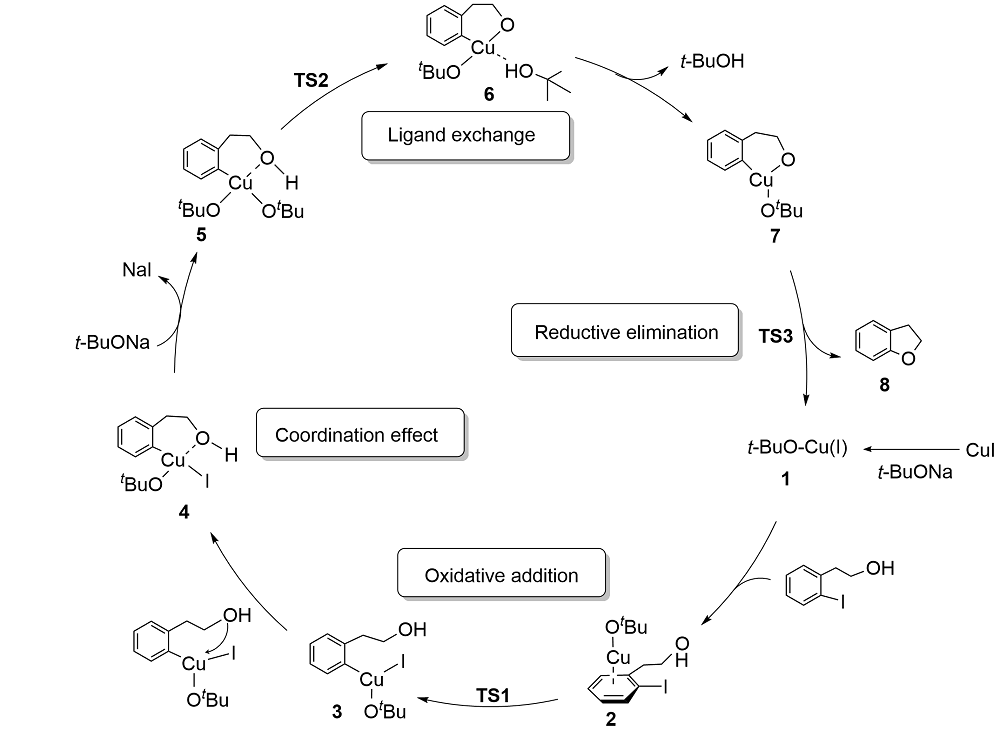
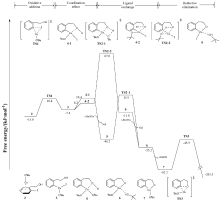

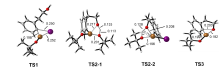
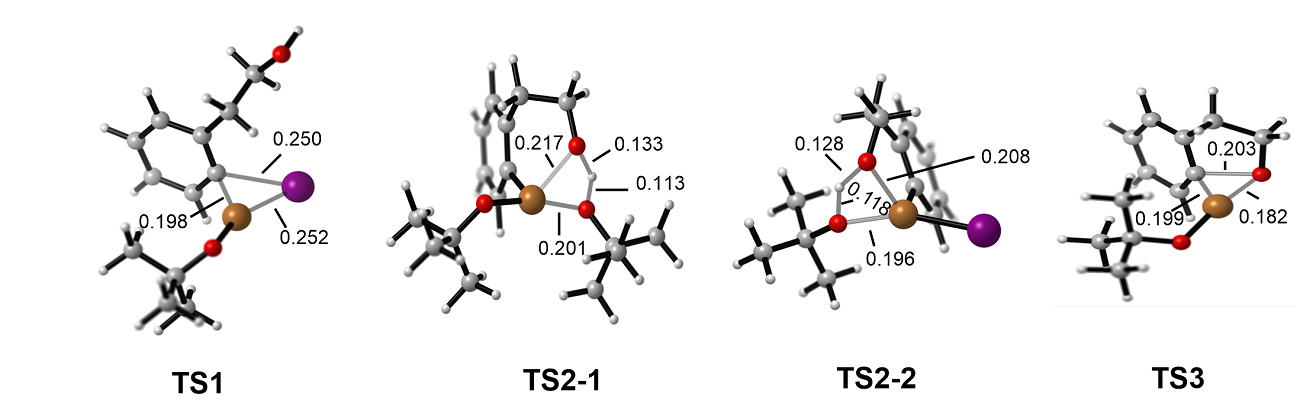
| [1] |
(a) Evano, G.; Wang, J. J.; Nitelet, A. Org. Chem. Front. 2017,2480.
|
|
(b) Ding, H.-W.; Li, J.; Guo, Q.-H.; Xiao, Y. Chinese J. Org. Chem. 2017, 37, 3112. (in Chinese)
doi: 10.6023/cjoc201706006 |
|
|
(丁怀伟, 李娟, 郭庆辉, 肖琰, 有机化学, 2017, 37, 3112.)
doi: 10.6023/cjoc201706006 |
|
| [2] |
(a) Liu, Y. J.; Zhang, D. M.; Xiao, S. H.; Qi, Y.; Liu, S. F. Asian J. Org. Chem. 2019, 8, 858.
doi: 10.1002/ajoc.v8.6 |
|
(b) Merritt, J. M.; Andiappan, M.; Pietz, M. A.; Richey, R. N.; Sullivan, K. A.; Kjell, D. P. Org. Process Res. Dev. 2016, 20, 178.
doi: 10.1021/acs.oprd.5b00324 |
|
|
(c) Monnier, F.; Taillefer, M. Angew. Chem. Int. Ed. 2009, 48, 6954.
doi: 10.1002/anie.v48:38 |
|
|
(d) Cho, G. Y.; Remy, P.; Jansson, J.; Moessner, C.; Bolm, C. Org. Lett. 2004, 6, 3293.
doi: 10.1021/ol048806h |
|
| [3] |
Yang, J.-P.; Zhang, L.; Jin, X.-P.; Gao, H.-Q.; Fang, J.-H.; Li, R.-F.; Fang, Y.-W. Chinese J. Org. Chem. 2013, 33, 1647. (in Chinese)
doi: 10.6023/cjoc201301016 |
|
(杨建平, 张莉, 金小平, 高浩其, 房江华, 李瑞丰, 方烨汶, 有机化学, 2013, 33, 1647.)
doi: 10.6023/cjoc201301016 |
|
| [4] |
(a) Ouali, A.; Taillefer, M.; Spindler, J. F.; Jutand, A. Organometallics 2007, 26, 65.
doi: 10.1021/om060706n |
|
(b) Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525.
doi: 10.1039/C3CS60289C |
|
| [5] |
Weingarten, H. J. Org. Chem. 1964, 29, 3624.
doi: 10.1021/jo01035a046 |
| [6] |
Paine, A. J. J. Am. Chem. Soc. 1987, 109, 1496.
doi: 10.1021/ja00239a032 |
| [7] |
Marcoux, J. F.; Doye, S.; Buchwald, S. L. J. Am. Chem. Soc. 1997, 119, 10539.
doi: 10.1021/ja971901j |
| [8] |
Mayoral, J. A.; Rodriguez-Rodriguez, S.; Salvatella, L. Chem. Eur. J. 2008, 14, 9274.
doi: 10.1002/chem.v14:30 |
| [9] |
Tye, J. W.; Weng, Z.; Johns, A. M.; Incarvito, C. D.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 9971.
doi: 10.1021/ja076668w |
| [10] |
Yu, H. Z.; Jiang, Y. Y.; Fu, Y.; Liu, L. J. Am. Chem. Soc. 2010, 132, 18078.
doi: 10.1021/ja104264v |
| [11] |
Chen, Z. X.; Jiang, Y. W.; Zhang, L.; Guo, Y. L.; Ma, D. W. J. Am. Chem. Soc. 2019, 141, 3541.
doi: 10.1021/jacs.8b12142 |
| [12] |
Legon, A. C. Angew. Chem, Int. Ed. 1999, 38, 2687.
|
| [13] |
Lefevre, G.; Franc, G.; Adamo, C.; Jutand, A.; Ciofini, I. Organometallics 2012, 31, 914.
doi: 10.1021/om200952v |
| [14] |
(a) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
pmid: 9944570 |
|
(b) Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200.
doi: 10.1016/0009-2614(89)87234-3 pmid: 9944570 |
|
|
(c) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
doi: 10.1063/1.464913 pmid: 9944570 |
|
|
(d) Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623.
doi: 10.1021/j100096a001 pmid: 9944570 |
|
| [15] |
(a) Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257.
doi: 10.1063/1.1677527 |
|
(b) Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724.
doi: 10.1063/1.1674902 |
|
| [16] |
Bergner, A.; Dolg, M.; Kuchle, W.; Stoll, H.; Preuss, H. Mol. Phys. 1993, 80, 1431.
doi: 10.1080/00268979300103121 |
| [17] |
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104.
doi: 10.1063/1.3382344 |
| [18] |
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378.
doi: 10.1021/jp810292n |
| [19] |
(a) Jover, J. Phys. Chem. Chem. Phys. 2017, 19, 29344.
doi: 10.1039/C7CP05709A |
|
(b) Jover, J. J. Chem. 2015, 2015, 430358.
|
|
|
(c) Jover, J.; Spuhler, P.; Zhao, L. G.; McArdle, C.; Maseras, F. Catal. Sci. Technol. 2014, 4, 4200.
doi: 10.1039/C4CY00322E |
|
|
(d) Jover, J.; Maseras, F. J. Org. Chem. 2014, 79, 11981.
doi: 10.1021/jo501837p |
|
|
(e) Jover, J. ACS Catal. 2014, 4, 4389.
doi: 10.1021/cs500872m |
|
| [20] |
(a) Fukui, K. J. Phys. Chem. 1970, 74, 4161.
doi: 10.1021/j100717a029 |
|
(b) Fukui, K. Acc. Chem. Res. 1981, 14, 363.
doi: 10.1021/ar00072a001 |
| [1] | Guanglong Huang, Xiao-Song Xue. Computational Study on the Mechanism of Chen’s Reagent as Trifluoromethyl Source [J]. Acta Chimica Sinica, 2024, 82(2): 132-137. |
| [2] | Xuefeng Liang, Jian Jing, Xin Feng, Yongze Zhao, Xinyuan Tang, Yan He, Lisheng Zhang, Huifang Li. Electronic Structure of Covalent Organic Frameworks COF66 and COF366: from Monomers to Two-Dimensional Framework [J]. Acta Chimica Sinica, 2023, 81(7): 717-724. |
| [3] | Lei Yang, Jiaoyang Ge, Fangli Wang, Wangyang Wu, Zongxiang Zheng, Hongtao Cao, Zhou Wang, Xueqin Ran, Linhai Xie. A Theoretical Study on the Effective Reduction of Internal Reorganization Energy Based on the Macrocyclic Structure of Fluorene [J]. Acta Chimica Sinica, 2023, 81(6): 613-619. |
| [4] | Jie Yang, Lin Ling, Yuxue Li, Long Lu. Density Functional Theory Study on Thermal Decomposition Mechanisms of Ammonium Perchlorate [J]. Acta Chimica Sinica, 2023, 81(4): 328-337. |
| [5] | Shaoqin Zhang, Meiqing Li, Zhongjun Zhou, Zexing Qu. Theoretical Study on the Multiple Resonance Thermally Activated Delayed Fluorescence Process [J]. Acta Chimica Sinica, 2023, 81(2): 124-130. |
| [6] | Jinjing Liu, Na Yang, Li Li, Zidong Wei. Theoretical Study on the Regulation of Oxygen Reduction Mechanism by Modulating the Spatial Structure of Active Sites on Platinum★ [J]. Acta Chimica Sinica, 2023, 81(11): 1478-1485. |
| [7] | Wenchao Bi, Linfeng Zhang, Jian Chen, Ruixue Tian, Hao Huang, Man Yao. Lithiation Mechanism and Performance of Monoclinic ZnP2 Anode Materials [J]. Acta Chimica Sinica, 2022, 80(6): 756-764. |
| [8] | Xuefei Luan, Congzhi Wang, Liangshu Xia, Weiqun Shi. Theoretical Studies on the Interaction of Uranyl with Carboxylic Acids and Oxime Ligands [J]. Acta Chimica Sinica, 2022, 80(6): 708-713. |
| [9] | Luocong Wang, Zhewei Li, Caiwei Yue, Peihuan Zhang, Ming Lei, Min Pu. Theoretical Study on the Isomerization Mechanism of Azobenzene Derivatives under Electric Field [J]. Acta Chimica Sinica, 2022, 80(6): 781-787. |
| [10] | Yinghui Wang, Simin Wei, Jinwei Duan, Kang Wang. Mechanism of Silyl Enol Ethers Hydrogenation Catalysed by Frustrated Lewis Pairs: A Theoretical Study [J]. Acta Chimica Sinica, 2021, 79(9): 1164-1172. |
| [11] | Yan Wang, Yingqi Tian, Zhong Jin, Bingbing Suo. Hartree-Fock and Density Functional Calculations on Graphics Processing Unit [J]. Acta Chimica Sinica, 2021, 79(5): 653-657. |
| [12] | Yu Mohan, Cheng Yuanyuan, Liu Yajun. Mechanistic Study of Oxygenation Reaction in Firefly Bioluminescence [J]. Acta Chimica Sinica, 2020, 78(9): 989-993. |
| [13] | Lu Xiaoqing, Cao Shoufu, Wei Xiaofei, Li Shaoren, Wei Shuxian. Investigation on Oxygen Reduction Reaction Mechanism on S Doped Fe-NC lsolated Single Atoms Catalyst [J]. Acta Chimica Sinica, 2020, 78(9): 1001-1006. |
| [14] | Yang Zhice, Tian Jianan, Cai Hongxue, Li Li, Pan Qingjiang. Theoretical Probe for Tris(aryloxide)arene Complexed Low-valent Actinide Ions and Their Structural/Redox Properties [J]. Acta Chimica Sinica, 2020, 78(10): 1096-1101. |
| [15] | Bo Yifan, Liu Yuyu, Chang Yongzheng, Li Yinxiang, Zhang Xiaofei, Song Chunyuan, Xu Weifeng, Cao Hongtao, Huang Wei. Theoretical and Experimental Studies on Raman Spectroscopy of Cyclic Fluorene-Based Strained Semiconductors [J]. Acta Chim. Sinica, 2019, 77(5): 442-446. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
