Acta Chimica Sinica ›› 2021, Vol. 79 ›› Issue (5): 653-657.DOI: 10.6023/A21020044 Previous Articles Next Articles
Communication
王岩a, 田英齐b,c, 金钟b,*( ), 索兵兵a,*()
), 索兵兵a,*()
投稿日期:2021-02-03
发布日期:2021-03-30
通讯作者:
金钟, 索兵兵
基金资助:
Yan Wanga, Yingqi Tianb,c, Zhong Jinb,*(), Bingbing Suoa,*()
Received:2021-02-03
Published:2021-03-30
Contact:
Zhong Jin, Bingbing Suo
About author:Supported by:Share
Yan Wang, Yingqi Tian, Zhong Jin, Bingbing Suo. Hartree-Fock and Density Functional Calculations on Graphics Processing Unit[J]. Acta Chimica Sinica, 2021, 79(5): 653-657.
| Molecule | Natoms/NBF | Wall time (Second) and speedup | ||
|---|---|---|---|---|
| 1CPUa | 1GPUb,c | 2GPUb,c | ||
| Becke partition | ||||
| (DNA)1 | 62/625 | 16 | 0.5(32.0) | 0.3(53.3) |
| (DNA)2 | 128/1332 | 138 | 4(34.5) | 2(69.0) |
| (DNA)4 | 260/2746 | 1166 | 33(35.3) | 17(68.6) |
| (DNA)8 | 524/5574 | 9582 | 269(35.6) | 136(70.5) |
| SSF partition | ||||
| (DNA)1 | 62/625 | 6 | 0.6(10.0) | 0.4(15.0) |
| (DNA)2 | 128/1332 | 55 | 4(13.8) | 2(27.5) |
| (DNA)4 | 260/2746 | 517 | 37(14.0) | 18(28.7) |
| (DNA)8 | 524/5574 | 4401 | 287(15.3) | 145(30.4) |
| Molecule | Natoms/NBF | Wall time (Second) and speedup | ||
|---|---|---|---|---|
| 1CPUa | 1GPUb,c | 2GPUb,c | ||
| Becke partition | ||||
| (DNA)1 | 62/625 | 16 | 0.5(32.0) | 0.3(53.3) |
| (DNA)2 | 128/1332 | 138 | 4(34.5) | 2(69.0) |
| (DNA)4 | 260/2746 | 1166 | 33(35.3) | 17(68.6) |
| (DNA)8 | 524/5574 | 9582 | 269(35.6) | 136(70.5) |
| SSF partition | ||||
| (DNA)1 | 62/625 | 6 | 0.6(10.0) | 0.4(15.0) |
| (DNA)2 | 128/1332 | 55 | 4(13.8) | 2(27.5) |
| (DNA)4 | 260/2746 | 517 | 37(14.0) | 18(28.7) |
| (DNA)8 | 524/5574 | 4401 | 287(15.3) | 145(30.4) |
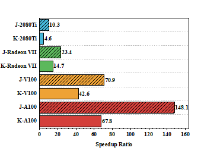
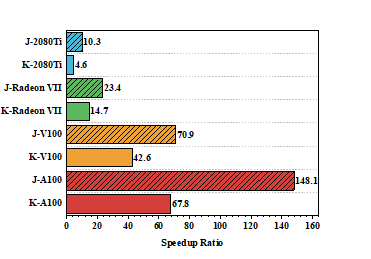
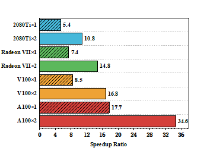

| Molecule | Natoms/NBF | Intel Core i9-9900Kb | AMD Radeon VII | NVIDIA A100 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| One SCF iteration | SCF time | One SCF iteration | SCF time(speedup) | One SCF iteration | SCF time(speedup) | ||||||||
| J | K | Vxc | J | K | Vxc | J | K | Vxc | |||||
| cocaine | 43/240 | 47 | 48 | 12 | 1569 | 3(15.7) c | 5(9.6) | 2(6.0) | 136(11.5) | 0.4(117.5) | 1(48.0) | 0.9(13.3) | 34(46.1) |
| (DNA)1 | 62/369 | 82 | 84 | 19 | 2347 | 4(20.5) | 7(12.0) | 5(3.8) | 191(12.3) | 0.6(136.7) | 2(42.0) | 1(19.0) | 45(52.2) |
| taxol | 110/647 | 718 | 731 | 69 | 22589 | 33(21.8) | 57(12.8) | 14(4.9) | 1524(14.8) | 5(143.6) | 12(60.9) | 4(17.3) | 323(69.9) |
| (DNA)2 | 128/796 | 1333 | 1355 | 113 | 41678 | 57(23.4) | 92(14.7) | 16(7.1) | 2439(17.1) | 9(148.1) | 20(67.8) | 7(16.1) | 542(76.9) |
| valinomycin | 168/882 | 1993 | 2030 | 146 | 61849 | 85(23.4) | 153(13.3) | 21(7.0) | 3855(16.0) | 14(142.4) | 30(67.7) | 9(16.2) | 803(77.0) |
| (DNA)4 | 260/1650 | 9347 | 9492 | 472 | 287831 | 367(25.5) | 572(16.6) | 68(6.9) | 14815(19.4) | 67(139.5) | 122(77.8) | 28(16.9) | 3329(86.5) |
| Molecule | Natoms/NBF | Intel Core i9-9900Kb | AMD Radeon VII | NVIDIA A100 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| One SCF iteration | SCF time | One SCF iteration | SCF time(speedup) | One SCF iteration | SCF time(speedup) | ||||||||
| J | K | Vxc | J | K | Vxc | J | K | Vxc | |||||
| cocaine | 43/240 | 47 | 48 | 12 | 1569 | 3(15.7) c | 5(9.6) | 2(6.0) | 136(11.5) | 0.4(117.5) | 1(48.0) | 0.9(13.3) | 34(46.1) |
| (DNA)1 | 62/369 | 82 | 84 | 19 | 2347 | 4(20.5) | 7(12.0) | 5(3.8) | 191(12.3) | 0.6(136.7) | 2(42.0) | 1(19.0) | 45(52.2) |
| taxol | 110/647 | 718 | 731 | 69 | 22589 | 33(21.8) | 57(12.8) | 14(4.9) | 1524(14.8) | 5(143.6) | 12(60.9) | 4(17.3) | 323(69.9) |
| (DNA)2 | 128/796 | 1333 | 1355 | 113 | 41678 | 57(23.4) | 92(14.7) | 16(7.1) | 2439(17.1) | 9(148.1) | 20(67.8) | 7(16.1) | 542(76.9) |
| valinomycin | 168/882 | 1993 | 2030 | 146 | 61849 | 85(23.4) | 153(13.3) | 21(7.0) | 3855(16.0) | 14(142.4) | 30(67.7) | 9(16.2) | 803(77.0) |
| (DNA)4 | 260/1650 | 9347 | 9492 | 472 | 287831 | 367(25.5) | 572(16.6) | 68(6.9) | 14815(19.4) | 67(139.5) | 122(77.8) | 28(16.9) | 3329(86.5) |
| [1] |
(a) Yasuda, K. J. Comput. Chem. 2008, 29,334.
pmid: 32379450 |
|
(b) Yasuda, K. J. Chem. Theory Comput. 2008, 4,1230.
doi: 10.1021/ct8001046 pmid: 32379450 |
|
|
(c) Ufimtsev, I. S.; Martínez, T. J. J. Chem. Theory Comput. 2008, 4,222.
doi: 10.1021/ct700268q pmid: 32379450 |
|
|
(d) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theory Comput. 2009, 5,1004.
doi: 10.1021/ct800526s pmid: 32379450 |
|
|
(e) Luehr, N.; Ufimtsev, I. S.; Martínez, T. J. J. Chem. Theory Comput. 2011, 7,949.
doi: 10.1021/ct100701w pmid: 32379450 |
|
|
(f) Miao, Y.; Merz, K. M. J. Chem. Theory Comput. 2013, 9,965.
doi: 10.1021/ct300754n pmid: 32379450 |
|
|
(g) Manathunga, M.; Miao, Y.; Mu, D.; Götz, A. W.; Merz, K. M. J. Chem. Theory Comput. 2020, 16,4315.
doi: 10.1021/acs.jctc.0c00290 pmid: 32379450 |
|
|
(h) Gordon, M. S.; Barca, G.; Leang, S. S.; Poole, D.; Rendell, A. P.; Galvez Vallejo, J. L.; Westheimer, B. J. Phys. Chem. A 2020, 124,4557.
doi: 10.1021/acs.jpca.0c02249 pmid: 32379450 |
|
|
(i) Walker, R. C.; Gtz, A. W. Electronic Structure Calculations on Graphics Processing Units: From Quantum Chemistry to Condensed Matter Physics, Wiley, New York,2016.
pmid: 32379450 |
|
| [2] |
NVIDIA. CUDA. https://docs.nvidia.com/cuda/index.html (accessed Jan.20, 2021)
|
| [3] |
OpenCL. https://www.khronos.org/opencl/ (accessed Jan. 20, 2021)
|
| [4] |
(a) Kussmann, J.; Ochsenfeld, C. J. Chem. Theory Comput. 2017, 13,2712.
doi: 10.1021/acs.jctc.7b00515 pmid: 28605592 |
|
(b) Kussmann, J.; Ochsenfeld, C. J. Chem. Theory Comput. 2017, 13,3153.
doi: 10.1021/acs.jctc.6b01166 pmid: 28605592 |
|
|
(c) Kalinowski, J.; Wennmohs, F.; Neese, F. J. Chem. Theory Comput. 2017, 13,3160.
doi: 10.1021/acs.jctc.7b00030 pmid: 28605592 |
|
| [5] |
(a) Liu, W.-J.; Hong, G.-Y.; Dai, D. D.; Li, L.-M.; Dolg, M. Theor. Chem. Acc. 1997, 96,75.
doi: 10.1007/s002140050207 |
|
(b) Liu, W.-J.; Wang, F.; Li, L.-M. J. Theor. Comput. Chem. 2003, 02,257.
doi: 10.1142/S0219633603000471 |
|
|
(c) Liu, W.-J.; Wang, F.; Li, L.-M. Recent Advances in Relativistic Molecular Theory, Vol. 5, World Scientific, 2004, p.257.
|
|
|
(d) Zhang, Y.; Suo, B.-B.; Wang, Z.-K.; Zhang, N.; Li, Z.-D.; Lei, Y.-B.; Zou, W.-L.; Gao, J.; Peng, D.-L.; Pu, Z.-C.; Xiao, Y.-L.; Sun, Q.-M.; Wang, F.; Ma, Y.-T.; Wang, X.-P.; Guo, Y.; Liu, W.-J. J. Chem. Phys. 2020, 152,064113.
|
|
| [6] |
Becke, A. D. J. Chem. Phys. 1988, 88,2547.
|
| [7] |
Stratmann, R. E.; Scuseria, G. E.; Frisch, M. J. Chem. Phys. Lett. 1996, 257,213.
doi: 10.1016/0009-2614(96)00600-8 |
| [8] |
Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular Electronic-Structure Theory, Wiley, New York, 2000, p.336.
|
| [9] |
Almlöf, J.; Faegri Jr, K.; Korsell, K. J. Comput. Chem. 1982, 3,385.
doi: 10.1002/jcc.v3:3 |
| [10] |
McMurchie, L. E.; Davidson, E. R. J. Comput. Phys. 1978, 26,218.
doi: 10.1016/0021-9991(78)90092-X |
| [11] |
Obara, S.; Saika, A. J. Chem. Phys. 1988, 89,1540.
doi: 10.1063/1.455717 |
| [12] |
Head-Gordon, M.; Pople, J. A. J. Chem. Phys. 1988, 89,5777.
doi: 10.1063/1.455553 |
| [13] |
Dupuis, M.; Rys, J.; King, H. F. J. Chem. Phys. 1976, 65,111.
doi: 10.1063/1.432807 |
| [14] |
OpenMP. https://www.openmp.org/ (accessed Jan. 20, 2021)
|
| [15] |
AMD.RO. Cm. https://rocmdocs.amd.com/en/latest/ (accessed Jan. 20, 2021)
|
| [16] |
(a) Hehre, W. J.; Stewart, R. F.; Pople, J. A. J. Chem. Phys. 1969, 51,2657.
doi: 10.1063/1.1672392 |
|
(b) Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56,2257.
doi: 10.1063/1.1677527 |
|
|
(c) Dill, J. D.; Pople, J. A. J. Chem. Phys. 1975, 62,2921.
doi: 10.1063/1.430801 |
|
|
(d) Binkley, J. S.; Pople, J. A.; Hehre, W. J. J. Am. Chem. Soc. 1980, 102,939.
doi: 10.1021/ja00523a008 |
|
| [17] |
(a) NVIDIA, NVIDIA Tesla V 100 GPU Architecture.
|
|
(b) NVIDIA, NVIDIA A 100 Tensor Core GPU Architecture.
|
|
| [18] |
Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77,3865.
doi: 10.1103/PhysRevLett.77.3865 |
| [19] |
(a) Dunning, T. H. J. Chem. Phys. 1989, 90,1007.
doi: 10.1063/1.456153 |
|
(b) Woon, D. E.; Dunning, T. H. J. Chem. Phys. 1993, 98,1358.
doi: 10.1063/1.464303 |
|
| [20] |
(a) Becke, A. D. J. Chem. Phys. 1993, 98,5648.
doi: 10.1063/1.464913 |
|
(b) Kameo, H.; Ishii, S.; Nakazawa, H. Dalton Trans. 2012, 41,8290.
doi: 10.1039/c2dt30546a |
|
| [21] |
MPI. M. I. https://www.mpi-forum.org/ (accessed Jan. 20, 2021)
|
| [1] | Guanglong Huang, Xiao-Song Xue. Computational Study on the Mechanism of Chen’s Reagent as Trifluoromethyl Source [J]. Acta Chimica Sinica, 2024, 82(2): 132-137. |
| [2] | Xuefeng Liang, Jian Jing, Xin Feng, Yongze Zhao, Xinyuan Tang, Yan He, Lisheng Zhang, Huifang Li. Electronic Structure of Covalent Organic Frameworks COF66 and COF366: from Monomers to Two-Dimensional Framework [J]. Acta Chimica Sinica, 2023, 81(7): 717-724. |
| [3] | Lei Yang, Jiaoyang Ge, Fangli Wang, Wangyang Wu, Zongxiang Zheng, Hongtao Cao, Zhou Wang, Xueqin Ran, Linhai Xie. A Theoretical Study on the Effective Reduction of Internal Reorganization Energy Based on the Macrocyclic Structure of Fluorene [J]. Acta Chimica Sinica, 2023, 81(6): 613-619. |
| [4] | Jie Yang, Lin Ling, Yuxue Li, Long Lu. Density Functional Theory Study on Thermal Decomposition Mechanisms of Ammonium Perchlorate [J]. Acta Chimica Sinica, 2023, 81(4): 328-337. |
| [5] | Shaoqin Zhang, Meiqing Li, Zhongjun Zhou, Zexing Qu. Theoretical Study on the Multiple Resonance Thermally Activated Delayed Fluorescence Process [J]. Acta Chimica Sinica, 2023, 81(2): 124-130. |
| [6] | Jinjing Liu, Na Yang, Li Li, Zidong Wei. Theoretical Study on the Regulation of Oxygen Reduction Mechanism by Modulating the Spatial Structure of Active Sites on Platinum★ [J]. Acta Chimica Sinica, 2023, 81(11): 1478-1485. |
| [7] | Wenchao Bi, Linfeng Zhang, Jian Chen, Ruixue Tian, Hao Huang, Man Yao. Lithiation Mechanism and Performance of Monoclinic ZnP2 Anode Materials [J]. Acta Chimica Sinica, 2022, 80(6): 756-764. |
| [8] | Xuefei Luan, Congzhi Wang, Liangshu Xia, Weiqun Shi. Theoretical Studies on the Interaction of Uranyl with Carboxylic Acids and Oxime Ligands [J]. Acta Chimica Sinica, 2022, 80(6): 708-713. |
| [9] | Luocong Wang, Zhewei Li, Caiwei Yue, Peihuan Zhang, Ming Lei, Min Pu. Theoretical Study on the Isomerization Mechanism of Azobenzene Derivatives under Electric Field [J]. Acta Chimica Sinica, 2022, 80(6): 781-787. |
| [10] | Zhifan Wang, Bing He, Yanzhao Lu, Fan Wang. Single-precision CCSD and CCSD(T) Calculations with Density Fitting Approximations on Graphics Processing Units [J]. Acta Chimica Sinica, 2022, 80(10): 1401-1409. |
| [11] | Yinghui Wang, Simin Wei, Jinwei Duan, Kang Wang. Mechanism of Silyl Enol Ethers Hydrogenation Catalysed by Frustrated Lewis Pairs: A Theoretical Study [J]. Acta Chimica Sinica, 2021, 79(9): 1164-1172. |
| [12] | Qingmin Man, Zunyun Fu, Tiantian Liu, Mingyue Zheng, Hualiang Jiang. DFT Mechanism of Cu Catalyzed Coupling Reaction to Alkyl Aryl Ethers [J]. Acta Chimica Sinica, 2021, 79(7): 948-952. |
| [13] | Yu Mohan, Cheng Yuanyuan, Liu Yajun. Mechanistic Study of Oxygenation Reaction in Firefly Bioluminescence [J]. Acta Chimica Sinica, 2020, 78(9): 989-993. |
| [14] | Lu Xiaoqing, Cao Shoufu, Wei Xiaofei, Li Shaoren, Wei Shuxian. Investigation on Oxygen Reduction Reaction Mechanism on S Doped Fe-NC lsolated Single Atoms Catalyst [J]. Acta Chimica Sinica, 2020, 78(9): 1001-1006. |
| [15] | Yang Zhice, Tian Jianan, Cai Hongxue, Li Li, Pan Qingjiang. Theoretical Probe for Tris(aryloxide)arene Complexed Low-valent Actinide Ions and Their Structural/Redox Properties [J]. Acta Chimica Sinica, 2020, 78(10): 1096-1101. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
