Acta Chimica Sinica ›› 2024, Vol. 82 ›› Issue (2): 132-137.DOI: 10.6023/A23090434 Previous Articles Next Articles
Special Issue: 有机氟化学合集
Article
黄广龙, 薛小松*( )
)
投稿日期:2023-09-30
发布日期:2023-11-29
作者简介:基金资助:
Guanglong Huang, Xiao-Song Xue()
Received:2023-09-30
Published:2023-11-29
Contact:
E-mail: About author:Supported by:Share
Guanglong Huang, Xiao-Song Xue. Computational Study on the Mechanism of Chen’s Reagent as Trifluoromethyl Source[J]. Acta Chimica Sinica, 2024, 82(2): 132-137.


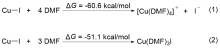
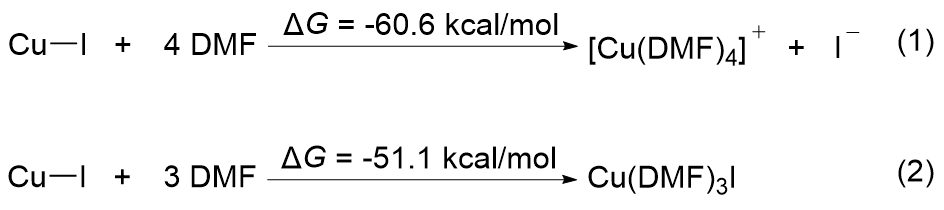
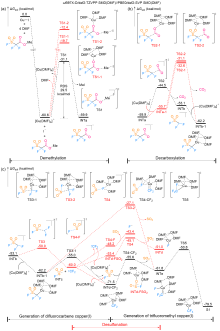

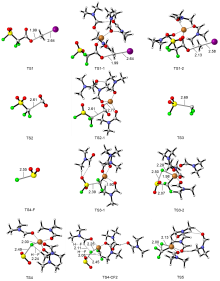

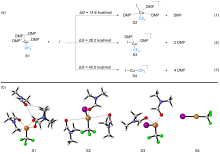
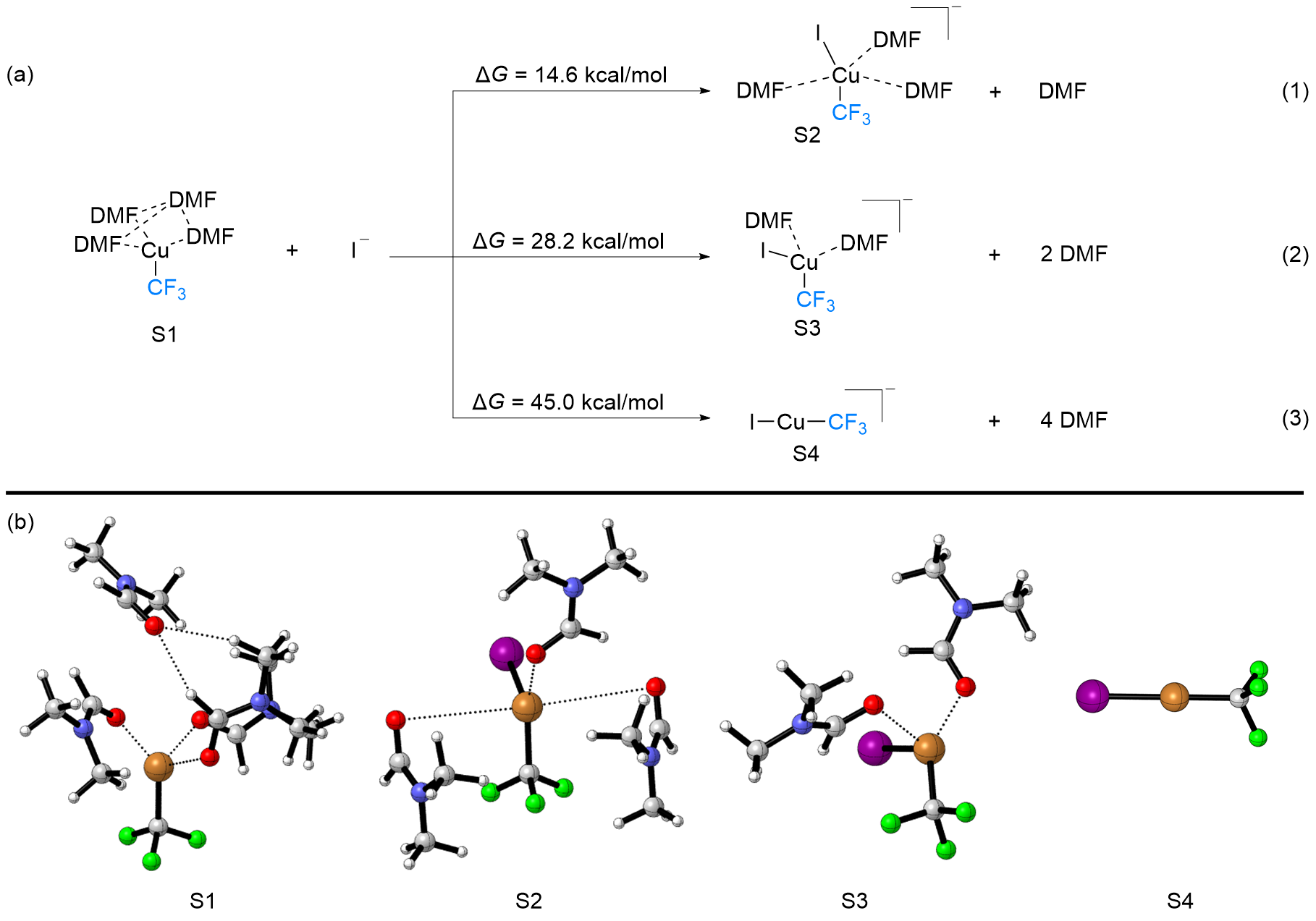
| [1] |
(a) Chen, Q.-Y.; Zhu, S. Z. Sci. China Chem. B 1986, 6, 561. (in Chinese)
|
|
(陈庆云, 朱仕正, 中国科学(B辑), 1986, 6, 561.)
|
|
|
(b) Chen, Q.-Y.; Wu, S.-W. J. Chem. Soc., Chem. Commun. 1989, 705.
|
|
| [2] |
(a) Jiang, X. K. Acta Chim. Sinica 1957, 5, 330. (in Chinese)
|
|
(蔣錫夔, 化学学报, 1957, 5, 330.)
|
|
|
(b) Olah, G. A.; Iyer, P. S.; Prakash, G. K. S. Synthesis 1986, 513.
|
|
|
(c) England, D. C. US 2852554, 1958.
|
|
|
(d) England, D. C.; Dietrich, M. A.; Lindsey, R. V. J. Am. Chem. Soc. 1960, 82, 6181.
doi: 10.1021/ja01508a051 |
|
|
(e) Dmitriev, M. A.; Sokol'skii, G. A.; Knunyants, I. L. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1960, 9, 792.
doi: 10.1007/BF01179175 |
|
|
(f) Dmitriev, M. A.; Sokol'skii, G. A.; Knunyants, I. L. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1960, 9, 966.
doi: 10.1007/BF00903970 |
|
|
(g) Zhao, G.; Wu, H.; Xiao, Z.; Chen, Q.-Y.; Liu, C. RSC Adv. 2016, 6, 50250.
doi: 10.1039/C6RA09011G |
|
|
(h) Zhang, C.-P.; Chen, Q.-Y; Guo, Y.; Xiao, J.-C.; Gu, Y.-C. Coord. Chem. Rev. 2014, 261, 28.
doi: 10.1016/j.ccr.2013.11.010 |
|
| [3] |
Chen, Q.-Y.; Yang, G.-Y.; Wu, S.-W. J. Fluorine Chem. 1991, 55, 291.
doi: 10.1016/S0022-1139(00)82357-X |
| [4] |
Zhao, S.; Guo, Y.; Han, E.-J.; Luo, J.; Liu, H.-M.; Liu, C.; Xie, W.; Zhang, W.; Wang, M. Org. Chem. Front. 2018, 5, 1143.
doi: 10.1039/C8QO00025E |
| [5] |
Chen, Q.-Y. J. Fluorine Chem. 1995, 72, 241.
doi: 10.1016/0022-1139(94)00414-B |
| [6] |
(a) Eusterwiemann, S.; Martinez, H.; Dolbier, W. R. J. Org. Chem. 2012, 77, 5461.
doi: 10.1021/jo300876z pmid: 22612642 |
|
(b) Thomoson, C. S.; Martinez, H.; Dolbier, W. R. J. Fluorine Chem. 2013, 150, 53.
doi: 10.1016/j.jfluchem.2013.02.026 pmid: 22612642 |
|
|
(c) Yu, W.; Xu, X.-H.; Qing, F.-L. Org. Lett. 2016, 18, 5130.
doi: 10.1021/acs.orglett.6b02580 pmid: 22612642 |
|
|
(d) Zhao, G.; Wu, H.; Xiao, Z.; Chen, Q.-Y.; Liu, C. RSC Adv. 2016, 6, 50250.
doi: 10.1039/C6RA09011G pmid: 22612642 |
|
|
(e) Liu, Y.; Wu, H.; Guo, Y.; Xiao, J.-C.; Chen, Q.-Y.; Liu, C. Angew. Chem., Int. Ed. 2017, 56, 15432.
doi: 10.1002/anie.v56.48 pmid: 22612642 |
|
| [7] |
(a) Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475.
doi: 10.1021/cr1004293 pmid: 21456523 |
|
(b) Clarke, S. L.; McGlacken, G. P. Chem. Eur. J. 2016, 22, 1.
doi: 10.1002/chem.v22.1 pmid: 21456523 |
|
|
(c) Xie, Q.; Hu, J. Chin. J. Chem. 2020, 38, 202.
doi: 10.1002/cjoc.v38.2 pmid: 21456523 |
|
|
(d) Chen, Q. Chin. J. Org. Chem. 2001, 21, 805. (in Chinese)
doi: 10.1002/cjoc.v21:7 pmid: 21456523 |
|
|
(陈庆云, 有机化学, 2001, 21, 805.)
pmid: 21456523 |
|
| [8] |
(a) Wiemers, D. M.; Burton, D. J. J. Am. Chem. Soc. 1986, 108, 832.
doi: 10.1021/ja00264a043 pmid: 25222650 |
|
(b) Dubinina, G. G.; Furutachi, H.; Vicic, D. A. J. Am. Chem. Soc. 2008, 130, 8600.
doi: 10.1021/ja802946s pmid: 25222650 |
|
|
(c) Morimoto, H.; Tsubogo, T.; Litvinas, N. D.; Hartwig, J. F. Angew. Chem., Int. Ed. 2011, 50, 3793.
doi: 10.1002/anie.v50.16 pmid: 25222650 |
|
|
(d) Litvinas, N. D.; Fier, P. S.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 536.
doi: 10.1002/anie.v51.2 pmid: 25222650 |
|
|
(e) Ni, C.; Hu, J. Synthesis 2014, 46, 842.
doi: 10.1055/s-00000084 pmid: 25222650 |
|
|
(f) Konovalov, A. I.; Lishchynskyi, A.; Grushin, V. V. J. Am. Chem. Soc. 2014, 136, 13410.
doi: 10.1021/ja507564p pmid: 25222650 |
|
|
(g) de Salinas, S. M.; Mudarra, Á. L.; Odena, C.; Belmonte, M. M.; Benet-Buchholz, J.; Maseras, F.; Pérez-Temprano, M. H. Chem. Eur. J. 2019, 25, 9390.
doi: 10.1002/chem.v25.40 pmid: 25222650 |
|
|
(h) Yu, W.; Ouyang, Y.; Xu, X.-H.; Qing, F.-L. Chin. J. Chem. 2018, 36, 1024.
doi: 10.1002/cjoc.v36.11 pmid: 25222650 |
|
|
(i) Zhang, W.; Lin, J.-H.; Wu, W.; Cao, Y.-C.; Xiao, J.-C. Chin. J. Chem. 2020, 38, 169.
doi: 10.1002/cjoc.v38.2 pmid: 25222650 |
|
| [9] |
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016.
|
| [10] |
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378.
doi: 10.1021/jp810292n |
| [11] |
(a) Zhao, Y.; Truhlar, D. G. J. Chem. Phys. 2006, 125, 194101.
doi: 10.1063/1.2370993 pmid: 16405344 |
|
(b) Quintal, M. M.; Karton, A.; Iron, M. A.; Boese, A. D.; Martin, J. M. L. J. Phys. Chem. A 2006, 110, 709.
pmid: 16405344 |
|
| [12] |
Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297.
doi: 10.1039/b508541a |
| [13] |
Chai, J.-D.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2008, 10, 6615.
doi: 10.1039/b810189b |
| [14] |
Weigend, F. Phys. Chem. Chem. Phys. 2006, 8, 1057.
doi: 10.1039/b515623h |
| [15] |
Funes-Ardoiz, I.; Paton, R. S., GoodVibes v2. 0.2, 2016, (http://doi.org/10.5281/zenodo.595246).
|
| [16] |
Legault, C. Y., CYLview, 1.0b, Université de Sherbrooke, 2009. (http://www.cylview.org).
|
| [1] | Xinpu Fu, Xiuling Wang, Weiwei Wang, Rui Si, Chunjiang Jia. Fabrication and Mechanism Study of Clustered Au/CeO2 Catalyst for the CO Oxidation Reaction★ [J]. Acta Chimica Sinica, 2023, 81(8): 874-883. |
| [2] | Guoqing Cui, Yiyang Hu, Yingjie Lou, Mingxia Zhou, Yuming Li, Yajun Wang, Guiyuan Jiang, Chunming Xu. Research Progress on the Design, Preparation and Properties of Catalysts for CO2 Hydrogenation to Alcohols [J]. Acta Chimica Sinica, 2023, 81(8): 1081-1100. |
| [3] | Luocong Wang, Zhewei Li, Caiwei Yue, Peihuan Zhang, Ming Lei, Min Pu. Theoretical Study on the Isomerization Mechanism of Azobenzene Derivatives under Electric Field [J]. Acta Chimica Sinica, 2022, 80(6): 781-787. |
| [4] | Siming Yang, Airong Liu, Jing Liu, Zhaoli Liu, Weixian Zhang. Advance of Sulfidated Nanoscale Zero-Valent Iron: Synthesis, Properties and Environmental Application [J]. Acta Chimica Sinica, 2022, 80(11): 1536-1554. |
| [5] | Yilong Hua, Donghan Li, Tianhang Gu, Wei Wang, Ruofan Li, Jianping Yang, Wei-xian Zhang. Enrichment of Uranium from Aqueous Solutions with Nanoscale Zero-valent Iron: Surface Chemistry and Application Prospect [J]. Acta Chimica Sinica, 2021, 79(8): 1008-1022. |
| [6] | Huang Rongyi, Shen Qiong, Zhang Chao, Zhang Shaoyong, Xu Heng. Studies on the Mechanism of the Transition Metal-Catalyzed Reaction of Organonitrile with Sodium Azide [J]. Acta Chimica Sinica, 2020, 78(6): 565-571. |
| [7] | Wu Weirong, Yuan Xiaomin, Hou Hua, Wang Baoshan. Theoretical Investigations on the Mechanisms for the Reactions of Sevoflurane Radicals[(CF3)2C(·)OCH2F, (CF3)2CHOC(·)HF] with O2and the OH· Radicals Regeneration [J]. Acta Chim. Sinica, 2018, 76(10): 793-801. |
| [8] | Mu Weihua, Ma Yao, Fang Decai, Wang Rong, Zhang Haina. Computational Insights into the Diels-Alder-alike Reactions of 1-Iodo-2-Lithio-o-Carborane with Fulvenes [J]. Acta Chim. Sinica, 2018, 76(1): 55-61. |
| [9] | Tang Jing, Tang Lin, Feng Haopeng, Dong Haoran, Zhang Yi, Liu Sishi, Zeng Guangming. Research Progress of Aqueous Pollutants Removal by Sulfidated Nanoscale Zero-valent Iron [J]. Acta Chim. Sinica, 2017, 75(6): 575-582. |
| [10] | Huang Xiao-yue, Wang Wei, Ling Lan, Zhang Wei-xian. Heavy Metal-nZVI Reactions: the Core-shell Structure and Applications for Heavy Metal Treatment [J]. Acta Chim. Sinica, 2017, 75(6): 529-537. |
| [11] | Ding Shuang, Ge Qingfeng, Zhu Xinli. Research Progress in Ketonization of Biomass-derived Carboxylic Acids over Metal Oxides [J]. Acta Chim. Sinica, 2017, 75(5): 439-447. |
| [12] | Yang Zhen, Xue Yijiang, He Yuanhang. Thermal Sensitivity of CL20/DNB Co-crystal Research via Molecular Dynamics Simulations [J]. Acta Chim. Sinica, 2016, 74(7): 612-619. |
| [13] | Ma Jun, Li Rong, Ren Kuiwei, Ma Xilong, Zhu Kaili, Geng Zhiyuan. Mechanism and Spin-orbit Coupling Study for Two State Reaction Ni2+ with Cyclohexane [J]. Acta Chim. Sinica, 2015, 73(5): 431-440. |
| [14] | Wang Ying, Zhang Limin, Hu Tianjun. Progress in Oxygen Reduction Reaction Electrocatalysts for Metal-Air Batteries [J]. Acta Chim. Sinica, 2015, 73(4): 316-325. |
| [15] | Tian Yana, Fu Yao, Zhang Qi, Yu Haizhu, Shi Jing. Theoretical Study on the Mechanism of the Imine-induced Tetrapeptides Cyclization [J]. Acta Chimica Sinica, 2014, 72(8): 935-941. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
