Acta Chimica Sinica ›› 2026, Vol. 84 ›› Issue (3): 305-315.DOI: 10.6023/A25110385 Previous Articles Next Articles
Article
彭天资a,†, 沈嘉克a,†, 郭淑雅c, 夏潇潇d,*( ), 李炜a,b,*()
), 李炜a,b,*()
投稿日期:2025-11-27
发布日期:2026-02-02
基金资助:
Peng Tianzia, Shen Jiakea, Guo Shuyac, Xia Xiaoxiaod,*(), Li Weia,b,*()
Received:2025-11-27
Published:2026-02-02
Contact:
*E-mail: 2320232062@nue.edu.cn;
weili@jnu.edu.cn
About author:† These authors contributed equally to this work.
Supported by:Share
Peng Tianzi, Shen Jiake, Guo Shuya, Xia Xiaoxiao, Li Wei. Transfer Learning Predicted the Self-Diffusion Coefficients of Light-Gas in Metal/Covalent Organic Frameworks[J]. Acta Chimica Sinica, 2026, 84(3): 305-315.
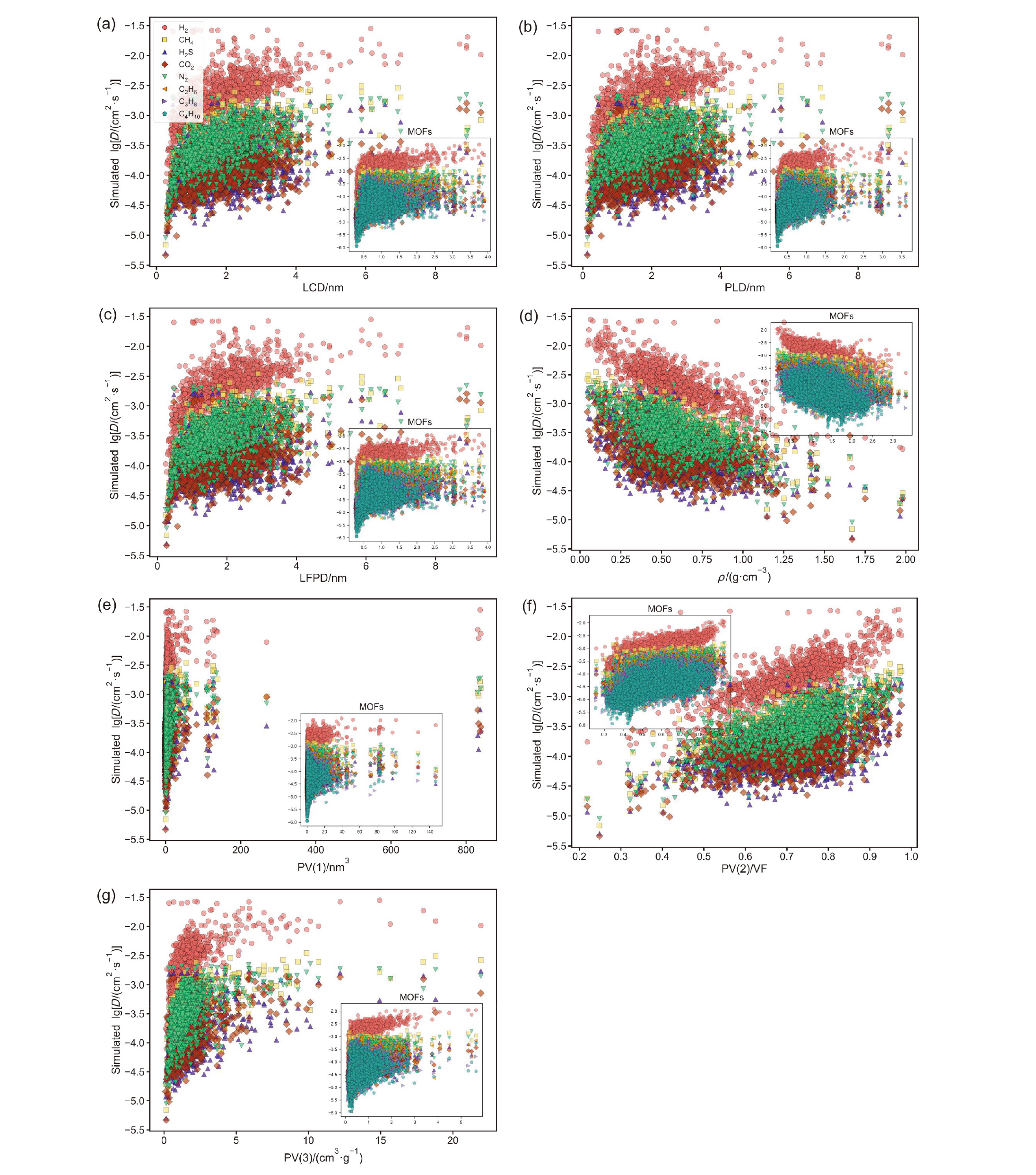
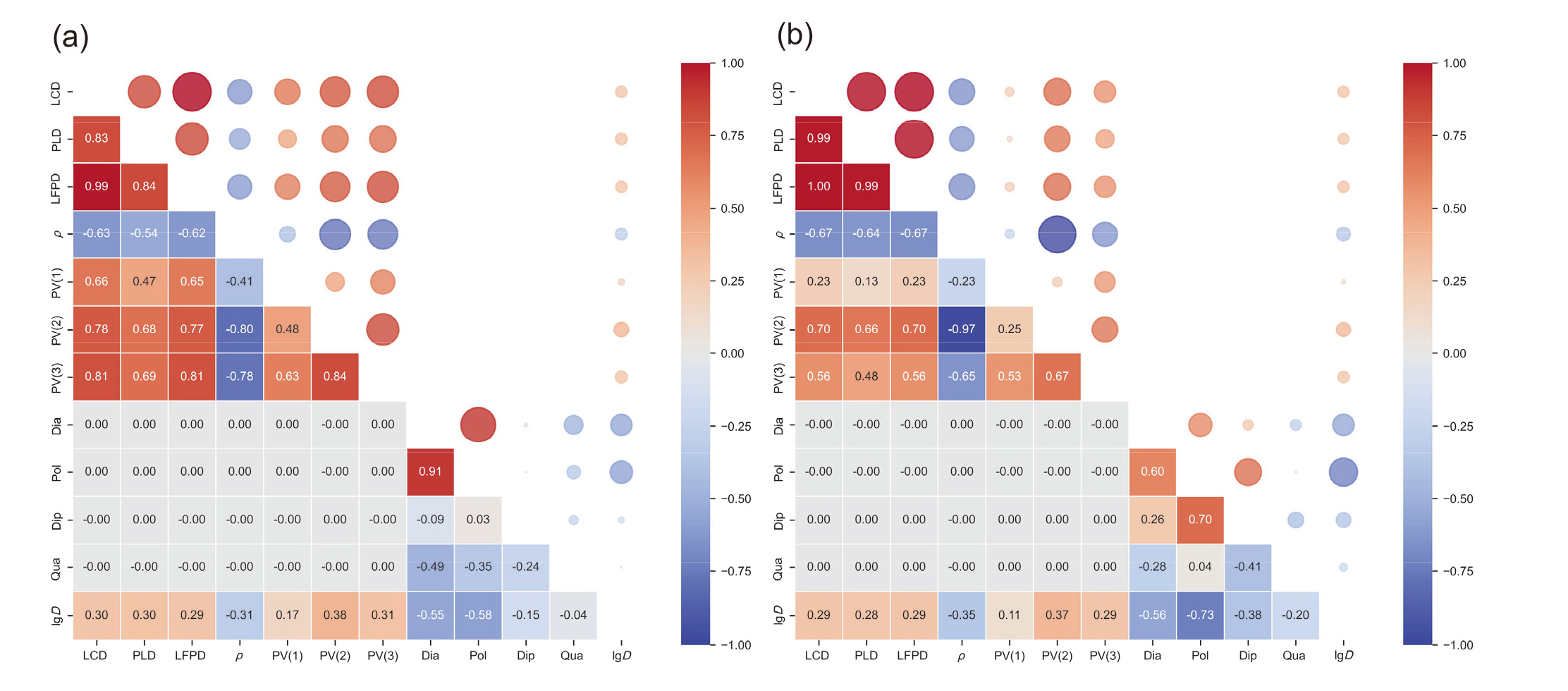
| Descriptors | Abbreviations | Units |
|---|---|---|
| Largest Cavity Diameter | LCD | nm |
| Pore Limiting Diameter | PLD | nm |
| Largest Free Path Diameter | LFPD | nm |
| Framework Density | ρ | g•cm−3 |
| Unit Cell Volume | PV(1) | nm3 |
| Porosity | PV(2) | — |
| Pore Volume per Unit Mass | PV(3) | cm3•g−1 |
| Kinetic Diameter | Dia | nm |
| Polarizability | Pol | nm3 |
| Quadrupole Moment | Qua | C•m2 |
| Dipole Moment | Dip | D |
| Descriptors | Abbreviations | Units |
|---|---|---|
| Largest Cavity Diameter | LCD | nm |
| Pore Limiting Diameter | PLD | nm |
| Largest Free Path Diameter | LFPD | nm |
| Framework Density | ρ | g•cm−3 |
| Unit Cell Volume | PV(1) | nm3 |
| Porosity | PV(2) | — |
| Pore Volume per Unit Mass | PV(3) | cm3•g−1 |
| Kinetic Diameter | Dia | nm |
| Polarizability | Pol | nm3 |
| Quadrupole Moment | Qua | C•m2 |
| Dipole Moment | Dip | D |
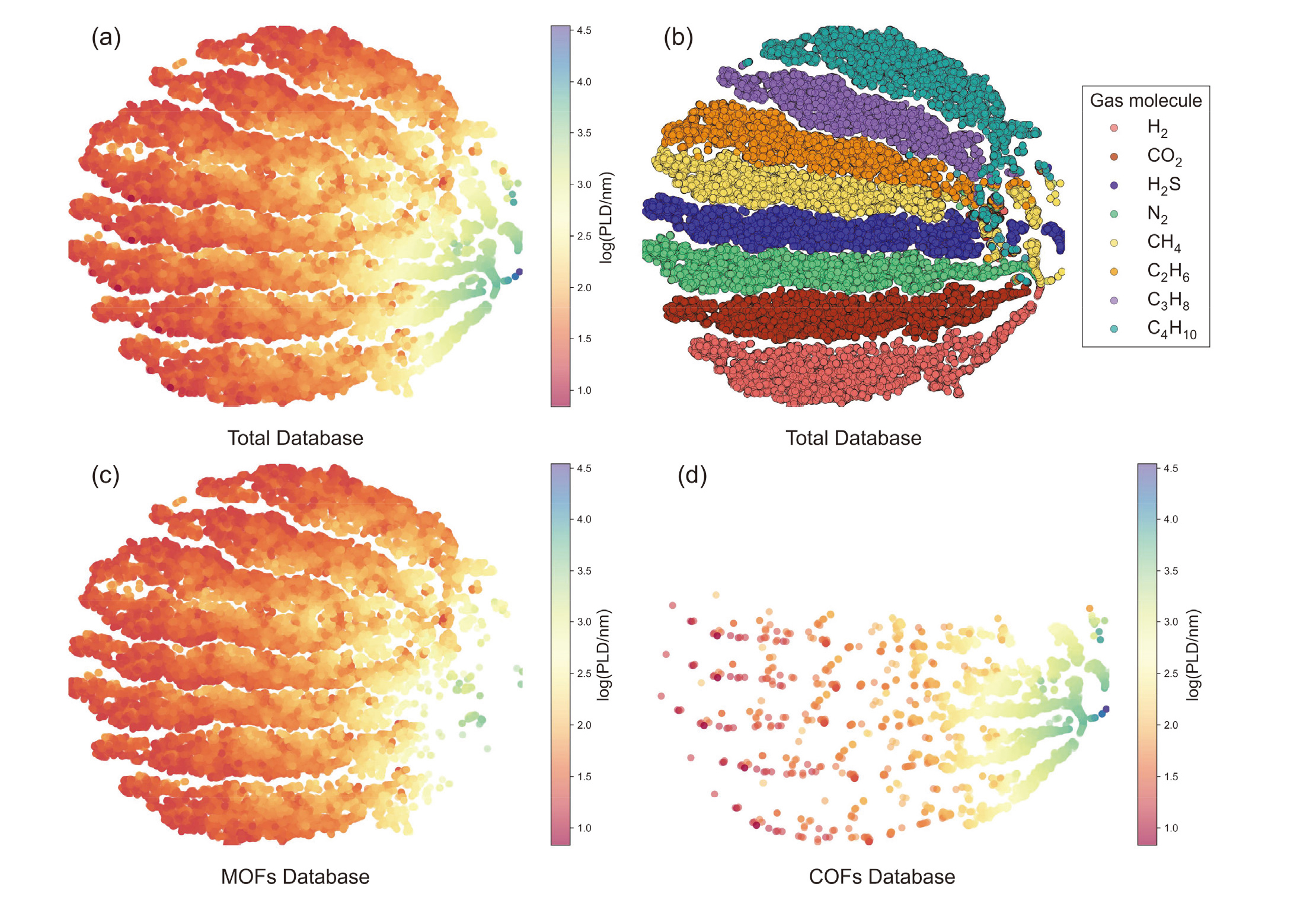

| Model | MOFs | COFs | |||||
|---|---|---|---|---|---|---|---|
| R2 | SRCC | MSE | R2 | SRCC | MSE | ||
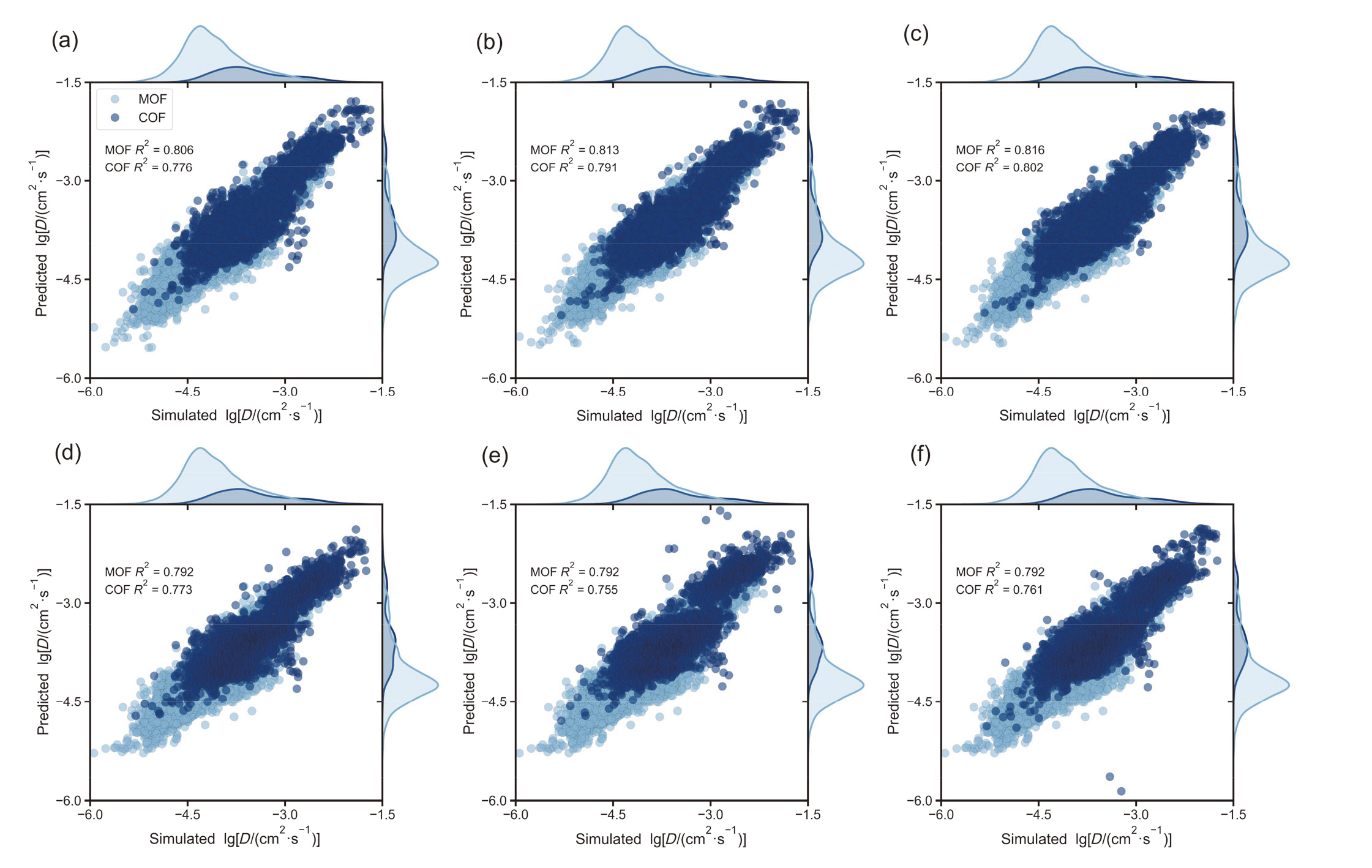
| RF | 0.806 | 0.855 | 0.051 | 0.716 | 0.805 | 0.091 | |
| XGBR | 0.813 | 0.862 | 0.049 | 0.666 | 0.785 | 0.108 | |
| LGBM | 0.817 | 0.863 | 0.048 | 0.683 | 0.792 | 0.102 | |
| DNN | 0.792 | 0.841 | 0.054 | 0.669 | 0.786 | 0.107 | |
| Model | MOFs | COFs | |||||
|---|---|---|---|---|---|---|---|
| R2 | SRCC | MSE | R2 | SRCC | MSE | ||
| RF | 0.806 | 0.855 | 0.051 | 0.716 | 0.805 | 0.091 | |
| XGBR | 0.813 | 0.862 | 0.049 | 0.666 | 0.785 | 0.108 | |
| LGBM | 0.817 | 0.863 | 0.048 | 0.683 | 0.792 | 0.102 | |
| DNN | 0.792 | 0.841 | 0.054 | 0.669 | 0.786 | 0.107 | |
| Learning Way | Model | COFs | ||
|---|---|---|---|---|
| R2 | SRCC | MSE | ||
| Direct Learning | RF | 0.809 | 0.852 | 0.063 |
| XGBR | 0.826 | 0.866 | 0.057 | |
| LGBM | 0.827 | 0.866 | 0.057 | |
| DNN | 0.781 | 0.841 | 0.072 | |
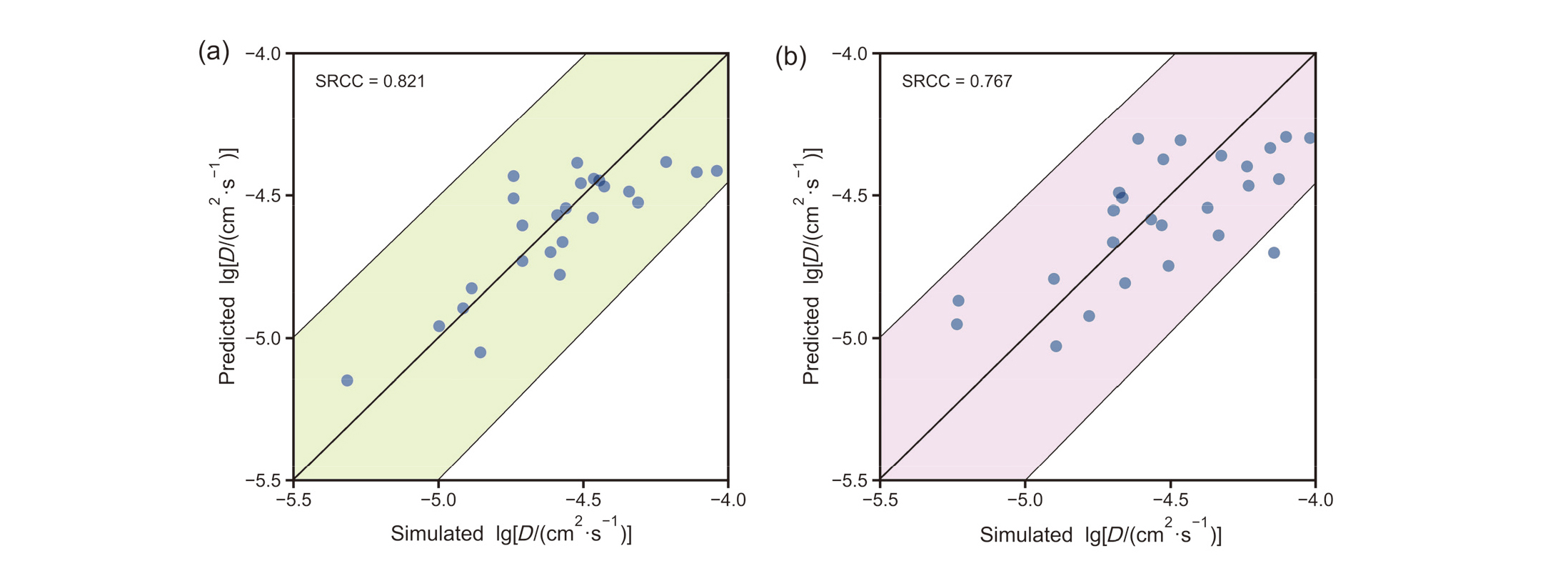
| Transfer Learning | RF | 0.775 | 0.822 | 0.071 |
| XGBR | 0.791 | 0.833 | 0.068 | |
| LGBM | 0.803 | 0.842 | 0.064 | |
| DNN_1 | 0.771 | 0.826 | 0.074 | |
| DNN_2 | 0.753 | 0.819 | 0.080 | |
| DNN_3 | 0.760 | 0.830 | 0.078 | |
| Learning Way | Model | COFs | ||
|---|---|---|---|---|
| R2 | SRCC | MSE | ||
| Direct Learning | RF | 0.809 | 0.852 | 0.063 |
| XGBR | 0.826 | 0.866 | 0.057 | |
| LGBM | 0.827 | 0.866 | 0.057 | |
| DNN | 0.781 | 0.841 | 0.072 | |
| Transfer Learning | RF | 0.775 | 0.822 | 0.071 |
| XGBR | 0.791 | 0.833 | 0.068 | |
| LGBM | 0.803 | 0.842 | 0.064 | |
| DNN_1 | 0.771 | 0.826 | 0.074 | |
| DNN_2 | 0.753 | 0.819 | 0.080 | |
| DNN_3 | 0.760 | 0.830 | 0.078 | |
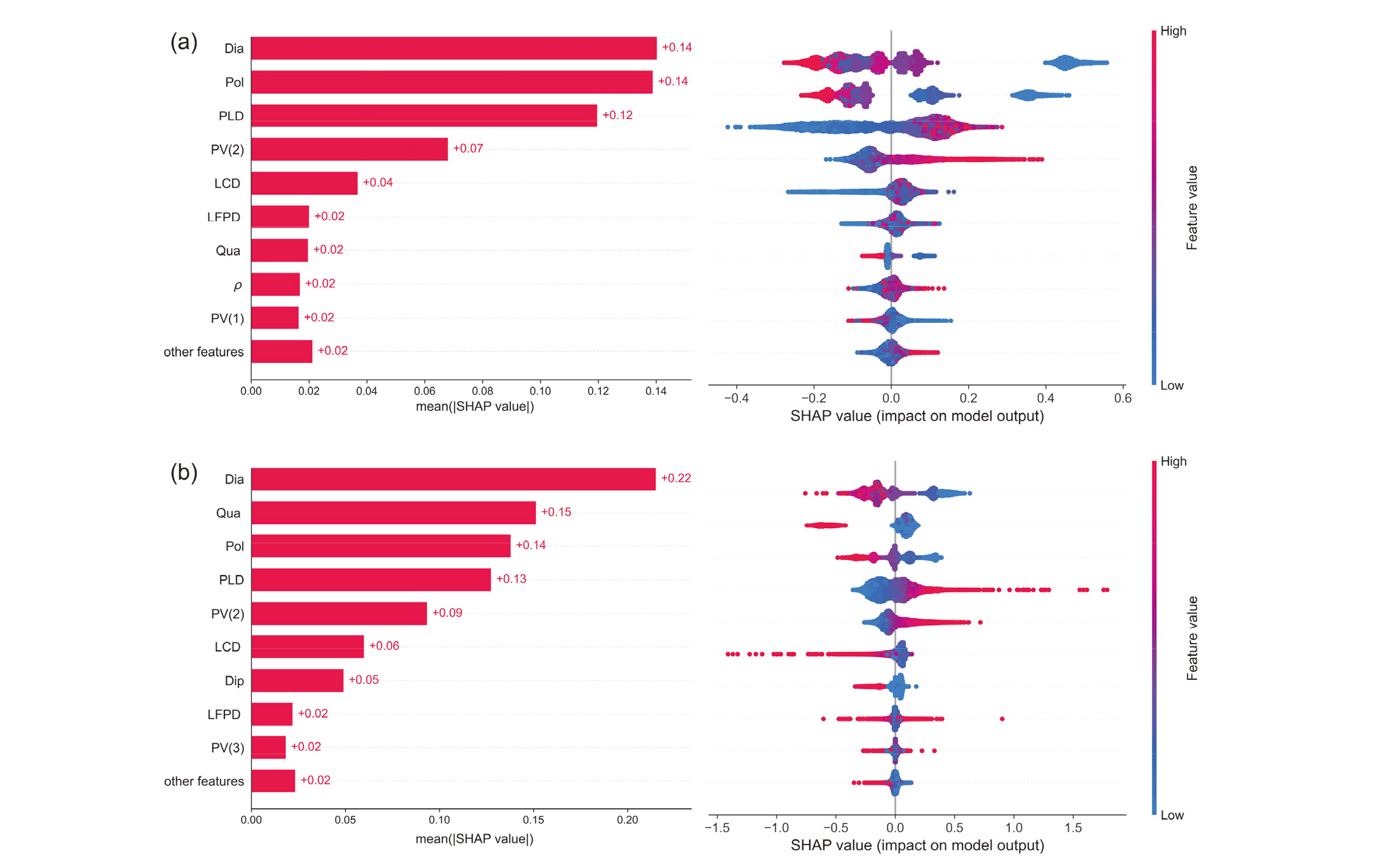

| [1] |
doi: 10.1021/ar800124u |
| [2] |
doi: 10.1039/C4CS90059F |
| [3] |
doi: 10.6023/A25040117 |
|
(高春, 张松涛, 庞欢, 化学学报, 2025, 83, 962.)
doi: 10.6023/A25040117 |
|
| [4] |
doi: 10.1039/c2cs35157a |
| [5] |
|
| [6] |
doi: 10.1039/C2CS35072F |
| [7] |
doi: 10.1021/ja9015765 |
| [8] |
doi: 10.1039/C2CS35251F |
| [9] |
doi: 10.1021/acsami.2c04746 |
| [10] |
doi: 10.6023/A25040140 |
|
(吴子林, 张璐, 陈杨, 李晋平, 李立博, 化学学报, 2025, 83, 917.)
doi: 10.6023/A25040140 |
|
| [11] |
doi: 10.1002/cjoc.v43.9 |
| [12] |
doi: 10.1039/c1cc11450f |
| [13] |
doi: 10.1039/b807083k |
| [14] |
doi: 10.6023/A25040135 |
|
(班渺寒, 双亚洲, 杨安平, 郑长勇, 张伟, 徐飞, 化学学报, 2025, 83, 1435.)
doi: 10.6023/A25040135 |
|
| [15] |
doi: 10.6023/A25060218 |
|
(李澄秋, 余嘉俊, 冯霄, 化学学报, 2025, 83, 1223.)
doi: 10.6023/A25060218 |
|
| [16] |
doi: 10.1002/cjoc.v43.1 |
| [17] |
doi: 10.6023/A25030098 |
|
(崔雨琪, 李军玉, 孙建科, 化学学报, 2025, 83, 624.)
doi: 10.6023/A25030098 |
|
| [18] |
doi: 10.1002/cjoc.v43.10 |
| [19] |
doi: 10.3866/PKU.WHXB20100616 |
|
(穆韡, 刘大欢, 阳庆元, 仲崇立, 物理化学学报, 2010, 26, 1657.)
|
|
| [20] |
doi: 10.1021/acs.jpclett.0c02745 |
| [21] |
doi: 10.1021/acs.langmuir.9b03802 |
| [22] |
doi: 10.1016/j.ces.2014.09.031 |
| [23] |
doi: 10.1021/acscatal.0c01432 |
| [24] |
|
|
(高正, 汪辉, 屈治国, 化工学报, 2025, 76, 4259.)
|
|
| [25] |
doi: 10.6023/A18120497 |
|
(刘治鲁, 李炜, 刘昊, 庄旭东, 李松, 化学学报, 2019, 77, 323.)
doi: 10.6023/A18120497 |
|
| [26] |
|
|
(李炜, 梁添贵, 林元创, 吴伟雄, 李松, 化学进展, 2022, 34, 2619.)
doi: 10.7536/PC220524 |
|
| [27] |
doi: 10.6023/A22010031 |
|
(王诗慧, 薛小雨, 程敏, 陈少臣, 刘冲, 周利, 毕可鑫, 吉旭, 化学学报, 2022, 80, 614.)
doi: 10.6023/A22010031 |
|
| [28] |
doi: 10.1021/acs.jpcc.4c00631 |
| [29] |
doi: 10.1002/advs.v10.21 |
| [30] |
doi: 10.1016/j.seppur.2025.134533 |
| [31] |
doi: 10.1021/jacsau.4c00618 |
| [32] |
doi: 10.1109/PROC.5 |
| [33] |
doi: 10.1021/acsami.3c10323 |
| [34] |
doi: 10.1016/j.matt.2025.102140 |
| [35] |
doi: 10.1016/j.ces.2017.05.004 |
| [36] |
doi: 10.1021/ja00051a040 |
| [37] |
doi: 10.1021/jp003882x |
| [38] |
doi: 10.1080/08927022.2015.1010082 |
| [39] |
doi: 10.1038/nchem.1192 |
| [40] |
doi: 10.1039/D1ME00060H |
| [41] |
doi: 10.1021/acs.jpclett.8b01749 |
| [42] |
doi: 10.1021/acs.iecr.4c03500 |
| [43] |
doi: 10.1016/j.micromeso.2011.08.020 |
| [44] |
doi: 10.1037/0033-2909.97.2.307 |
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
doi: 10.1016/0370-2693(95)00051-L |
| [49] |
|
| [50] |
|
|
(陈少臣, 程敏, 王诗慧, 吴金奎, 罗磊, 薛小雨, 吉旭, 张长春, 周利, 高等学校化学学报, 2023, 44, 1.)
|
|
| [51] |
doi: 10.1039/C6TA06262H |
| [52] |
doi: 10.3390/nano14131074 |
| [53] |
doi: 10.1016/j.seppur.2024.127752 |
| [1] | Ziyi Shui, Shurui Yin, Jintao Deng, Liuyun Xu, Li Guo. Polyvinylpyrrolidone-Assisted Synthesis of Metal-organic Framework-Derived Hierarchically Porous Carbon for Bifunctional Electrocatalysis [J]. Acta Chimica Sinica, 2026, 84(1): 53-63. |
| [2] | Huiqiong Weng, He Huang, Wenfei Wang, Heguo Li, Xiaopeng Li, Shouxin Zhang, Shuhua Li, Yue Zhao, Yufang Wu, Zhiwei Qiao. AI Big-Data Mining Empowered by MOFid for High-Performance Chemical Warfare Agent Adsorbents [J]. Acta Chimica Sinica, 2026, 84(1): 8-19. |
| [3] | Mengxi Zhang, Yuying Zhang, Jiaxuan Qin, Xiao Feng, Xueyan Li, Tong Chang, Haiying Yang. Electropolymerization of Novel Poly-m-phenylenediamine Membrame for H2/CO2 Separation [J]. Acta Chimica Sinica, 2025, 83(2): 132-138. |
| [4] | Ying Wei, Jiacheng Wang, Yue Li, Tao Wang, Shuwei Ma, Linghai Xie. Research Progress of Carbon-carbon Bond Linked Two-dimensional Covalent-Organic Frameworks [J]. Acta Chimica Sinica, 2024, 82(1): 75-102. |
| [5] | Bo Sun, Wenwen Ju, Tao Wang, Xiaojun Sun, Ting Zhao, Xiaomei Lu, Feng Lu, Quli Fan. Preparation of Highly-dispersed Conjugated Polymer-Metal Organic Framework Nanocubes for Antitumor Application [J]. Acta Chimica Sinica, 2023, 81(7): 757-762. |
| [6] | Junchang Chen, Mingxing Zhang, Shuao Wang. Research Progress of Synthesis Methods for Crystalline Porous Materials [J]. Acta Chimica Sinica, 2023, 81(2): 146-157. |
| [7] | Xiaojuan Li, Ziyu Ye, Shuhan Xie, Yongjing Wang, Yonghao Wang, Yuancai Lv, Chunxiang Lin. Study on Performance and Mechanism of Phenol Degradation through Peroxymonosulfate Activation by Nitrogen/Chlorine Co-doped Porous Carbon Materials [J]. Acta Chimica Sinica, 2022, 80(9): 1238-1249. |
| [8] | Xu Yan, Hemi Qu, Ye Chang, Xuexin Duan. Application of Metal-Organic Frameworks in Gas Pre-concentration, Pre-separation and Detection [J]. Acta Chimica Sinica, 2022, 80(8): 1183-1202. |
| [9] | Linan Cao, Min Wei. Recent Progress of Electric Conductive Metal-Organic Frameworks Thin Film [J]. Acta Chimica Sinica, 2022, 80(7): 1042-1056. |
| [10] | Fang Liu, Tingting Pan, Xiurong Ren, Weiren Bao, Jiancheng Wang, Jiangliang Hu. Research on Preparation and Benzene Adsorption Performance of HCDs@MIL-100(Fe) Adsorbents [J]. Acta Chimica Sinica, 2022, 80(7): 879-887. |
| [11] | Shihui Wang, Xiaoyu Xue, Min Cheng, Shaochen Chen, Chong Liu, Li Zhou, Kexin Bi, Xu Ji. High-Throughput Computational Screening of Metal-Organic Frameworks for CH4/H2 Separation by Synergizing Machine Learning and Molecular Simulation [J]. Acta Chimica Sinica, 2022, 80(5): 614-624. |
| [12] | Rong Zhang, Jiangping Liu, Ziyi Zhu, Shumei Chen, Fei Wang, Jian Zhang. Synthesis, Structure and Characterization of Two Ferrocene Functionalized Cadmium Metal Organic Frameworks※ [J]. Acta Chimica Sinica, 2022, 80(3): 249-254. |
| [13] | Xusheng Wang, Xu Yang, Chunhui Chen, Hongfang Li, Yuanbiao Huang, Rong Cao. Graphene Quantum Dots Supported on Fe-based Metal-Organic Frameworks for Efficient Photocatalytic CO2 Reduction※ [J]. Acta Chimica Sinica, 2022, 80(1): 22-28. |
| [14] | Yingzhe Du, Heng Zhang, Shiling Yuan. Molecular Dynamics Simulation of Thermal Conductivity of Al2O3/PDMS Composites [J]. Acta Chimica Sinica, 2021, 79(6): 787-793. |
| [15] | Yan-Wu Zhao, Xing Li, Fu-Qiang Zhang, Xiang Zhang. Precise Control of the Dimension of Homochiral Metal-Organic Frameworks (MOFs) and Their Luminescence Properties [J]. Acta Chimica Sinica, 2021, 79(11): 1409-1414. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
