Acta Chimica Sinica ›› 2021, Vol. 79 ›› Issue (4): 472-480.DOI: 10.6023/A20100489 Previous Articles Next Articles
Special Issue: 纪念南开大学化学学科创建100周年
Review
付浩浩a, 陈淏川a, 张宏a, 邵学广a,b,*( ), 蔡文生a,*()
), 蔡文生a,*()
投稿日期:2020-10-24
发布日期:2020-12-02
通讯作者:
邵学广, 蔡文生
作者简介: |
付浩浩, 本科和博士均毕业于南开大学化学系. 现为南开大学博士后、助理研究员. 研究方向为增强采样算法开发、蛋白质-配体准确结合自由能计算策略研究和复杂体系中高度耦合的运动研究. |
 |
邵学广, 南开大学教授, 博士生导师, 1992年获中国科学技术大学中日联合培养博士学位. 2002 年获教育部第三届高校青年教师奖, 2003 年获国家自然科学基金委杰出青年基金. 主要从事化学计量学及近红外光谱分析方面的研究工作. 建立了小波变换和免疫算法用于复杂信号解析和在线处理的新方法以及一系列用于近红外光谱信号处理和建模的化学计量学方法. |
 |
蔡文生, 南开大学教授, 博士生导师. 1994年获中国科学技术大学博士学位. 主要从事分子模拟与理论化学计算领域的研究工作, 包括优化算法、自由能计算方法、分子模拟及理论化学计算在蛋白质-配体、药物载体、分子机器中的应用研究. |
基金资助:
Haohao Fua, Haochuan Chena, Hong Zhanga, Xueguang Shaoa,b,*(), Wensheng Caia,*()
Received:2020-10-24
Published:2020-12-02
Contact:
Xueguang Shao, Wensheng Cai
About author:Supported by:Share
Haohao Fu, Haochuan Chen, Hong Zhang, Xueguang Shao, Wensheng Cai. Accurate Estimation of Protein-ligand Binding Free Energies Based on Geometric Restraints[J]. Acta Chimica Sinica, 2021, 79(4): 472-480.

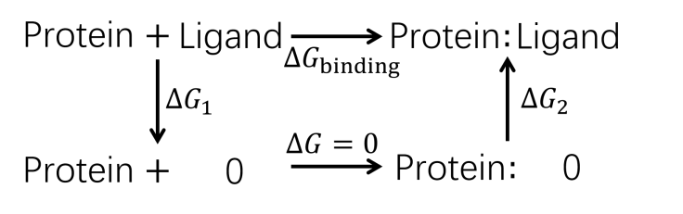


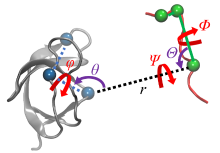
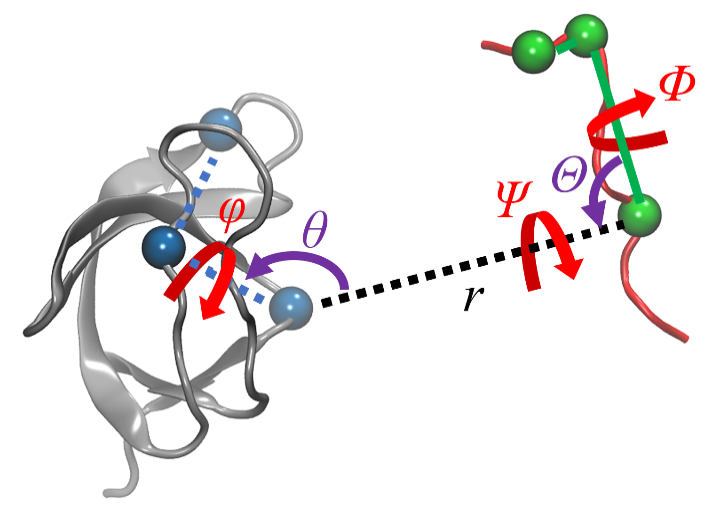
| 步骤 | 对应项 | 集合变量 | 约束 | 体系 |
|---|---|---|---|---|
| 1 | RMSD | - | 蛋白质-配体 | |
| 2 | RMSD | 蛋白质-配体 | ||
| 3 | RMSD, | 蛋白质-配体 | ||
| 4 | RMSD, , | 蛋白质-配体 | ||
| 5 | RMSD, , , | 蛋白质-配体 | ||
| 6 | RMSD, , , , | 蛋白质-配体 | ||
| 7 | RMSD, , , , , | 蛋白质-配体 | ||
| 8 | RMSD | - | 配体 | |
| 9 | - | - | 解析计算 |
| 步骤 | 对应项 | 集合变量 | 约束 | 体系 |
|---|---|---|---|---|
| 1 | RMSD | - | 蛋白质-配体 | |
| 2 | RMSD | 蛋白质-配体 | ||
| 3 | RMSD, | 蛋白质-配体 | ||
| 4 | RMSD, , | 蛋白质-配体 | ||
| 5 | RMSD, , , | 蛋白质-配体 | ||
| 6 | RMSD, , , , | 蛋白质-配体 | ||
| 7 | RMSD, , , , , | 蛋白质-配体 | ||
| 8 | RMSD | - | 配体 | |
| 9 | - | - | 解析计算 |
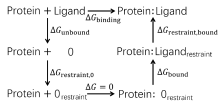
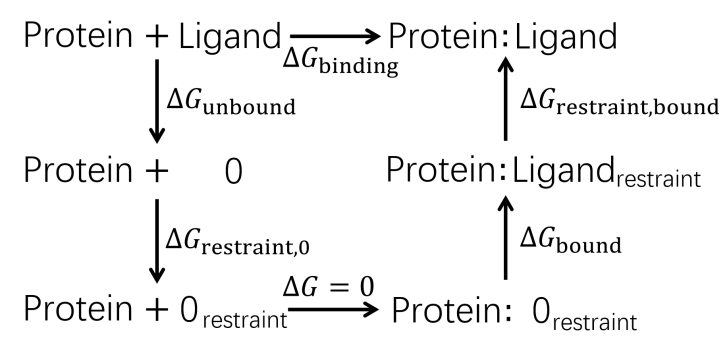
| 步骤 | 对应项 | 消失/生长部分 | 体系 |
|---|---|---|---|
| 1 | 配体 | 蛋白质-配体 | |
| 2 | 配体 | 配体 | |
| 3 | 七自由度约束 | 蛋白质-配体 | |
| 4 | (RMSD贡献) | RMSD约束 | 配体 |
| 5 | RMSD贡献以外部分) | 无 | 解析计算(式8) |
| 步骤 | 对应项 | 消失/生长部分 | 体系 |
|---|---|---|---|
| 1 | 配体 | 蛋白质-配体 | |
| 2 | 配体 | 配体 | |
| 3 | 七自由度约束 | 蛋白质-配体 | |
| 4 | (RMSD贡献) | RMSD约束 | 配体 |
| 5 | RMSD贡献以外部分) | 无 | 解析计算(式8) |
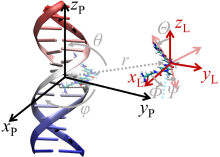
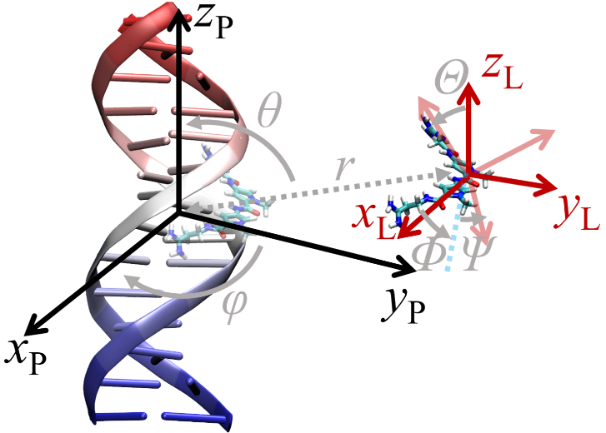
| 作者 | 使用策略 | 小分子配体个数 | 计算误差/ (kJ•mol–1) | 文献 |
|---|---|---|---|---|
| Aldeghi等 | alchemistry | 11 | MAE=2.5 | [ |
| Boyce等 | alchemistry | 13 | RMSE=7.5 | [ |
| Li等 | alchemistry | 100 | RMSE=2.59~6.44 | [ |
| Laury等 | alchemistry | 14 | MAE=5.02 | [ |
| Xie等 | alchemistry | 141 | RMSE=6.65 | [ |
| Deng等 | alchemistry/重要性采样 | 3 | AE=6.7~18 (alchemistry), 6.3~14 (重要性采样) | [ |
| Fu等 | 重要性采样 | 3(十肽) | AE<1.7 | [ |
| 作者 | 使用策略 | 小分子配体个数 | 计算误差/ (kJ•mol–1) | 文献 |
|---|---|---|---|---|
| Aldeghi等 | alchemistry | 11 | MAE=2.5 | [ |
| Boyce等 | alchemistry | 13 | RMSE=7.5 | [ |
| Li等 | alchemistry | 100 | RMSE=2.59~6.44 | [ |
| Laury等 | alchemistry | 14 | MAE=5.02 | [ |
| Xie等 | alchemistry | 141 | RMSE=6.65 | [ |
| Deng等 | alchemistry/重要性采样 | 3 | AE=6.7~18 (alchemistry), 6.3~14 (重要性采样) | [ |
| Fu等 | 重要性采样 | 3(十肽) | AE<1.7 | [ |
| 场景 | 建议 | 说明 |
|---|---|---|
| 基于数据库的药物筛选 | 分子对接 | 效率最高 |
| 结构相似配体结合能力的定性对比 | MM-G(P)BSA | 效率高, 使用简单 |
| 刚性小分子、结合位点在蛋白质表面 | 漏斗状约束或球状约束结合重要性采样方法 | 效率较高, 使用简单, 无需考虑小分子结构变化 |
| 刚性小分子、结合位点在蛋白质内部 | 基于六自由度约束的Alchemistry方法 | 无法使用重要性采样方法, 使用六自由度约束可以简单地解析计算约束贡献 |
| 柔性配体、结合位点在蛋白质表面 | 基于七自由度的重要性采样方法 | 同等条件下重要性采样算法收敛较快, 需要采用七自由度策略捕获柔性配体结构变化 |
| 柔性配体、结合位点在蛋白质内部 | 基于七自由度约束的Alchemistry方法 | 其他策略均不适合时最为“安全”的策略 |
| 场景 | 建议 | 说明 |
|---|---|---|
| 基于数据库的药物筛选 | 分子对接 | 效率最高 |
| 结构相似配体结合能力的定性对比 | MM-G(P)BSA | 效率高, 使用简单 |
| 刚性小分子、结合位点在蛋白质表面 | 漏斗状约束或球状约束结合重要性采样方法 | 效率较高, 使用简单, 无需考虑小分子结构变化 |
| 刚性小分子、结合位点在蛋白质内部 | 基于六自由度约束的Alchemistry方法 | 无法使用重要性采样方法, 使用六自由度约束可以简单地解析计算约束贡献 |
| 柔性配体、结合位点在蛋白质表面 | 基于七自由度的重要性采样方法 | 同等条件下重要性采样算法收敛较快, 需要采用七自由度策略捕获柔性配体结构变化 |
| 柔性配体、结合位点在蛋白质内部 | 基于七自由度约束的Alchemistry方法 | 其他策略均不适合时最为“安全”的策略 |
| [1] |
Chipot, C. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4,71.
|
| [2] |
Siebenmorgen, T.; Zacharias, M. WIREs Comput. Mol. Sci. 2019, 10,e1448.
|
| [3] |
de Ruiter A.; Oostenbrink C. Curr. Opin. Struct. Biol. 2020, 61,207.
|
| [4] |
Xu, J.; Wei, Y.; Wu, Z.; Yi, Z. Acta Chim. Sinica 2018, 76,408. (in Chinese)
|
|
( 徐婕, 魏雨晨, 伍智蔚, 易忠胜, 化学学报, 2018, 76,408.)
|
|
| [5] |
Cournia, Z.; Allen, B.K.; Beuming, T.; Pearlman, D.A.; Radak, B.K.; Sherman, W. J. Chem. Inf. Model. 2020, 60,4153.
|
| [6] |
Langan, R.A.; Boyken, S.E.; Ng, A.H.; Samson, J.A.; Dods, G.; Westbrook, A.M.; Nguyen, T.H.; Lajoie, M.J.; Chen, Z.; Berger, S.; Mulligan, V.K.; Dueber, J.E.; Novak, W.R. P.; El-Samad, H.; Baker, D. Nature 2019, 572,205.
|
| [7] |
Lemkul, J.A.; Huang, J.; Roux, B.; MacKerell, A.D. Chem. Rev. 2016, 116,4983.
|
| [8] |
Fan, Q.; Liang, H.; Xu, X.; Lv, S.; Liang, Z.; Yang, Y. Acta Chim. Sinica 2020, 78,547. (in Chinese)
|
|
( 范勤, 梁洪涛, 许贤祺, 吕松泰, 梁尊, 杨洋, 化学学报, 2020, 78,547.)
|
|
| [9] |
Nerenberg, P.S.; Head-Gordon, T. Curr. Opin. Struct. Biol. 2018, 49,129.
|
| [10] |
Chipot, C.; Pohorille, A. Free Energy Calculations, Springer, Berlin, Heidelberg, 2007.
|
| [11] |
Barducci, A.; Bonomi, M.; Parrinello, M. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1,826.
|
| [12] |
Laio, A.; Parrinello, M. Proc. Natl. Acad. Sci. 2002, 99,12562.
|
| [13] |
Fu, H.; Shao, X.; Cai, W.; Chipot, C. Acc. Chem. Res. 2019, 52,3254.
|
| [14] |
Fu, H.; Shao, X.; Chipot, C.; Cai, W. J. Chem. Theory Comput. 2016, 12,3506.
|
| [15] |
Chodera, J.D.; Mobley, D.L. Annu. Rev. Biophys. 2013, 42,121.
|
| [16] |
Zwanzig, R.W. J. Chem. Phys. 1954, 22,1420.
|
| [17] |
Kirkwood, J.G. J. Chem. Phys. 1935, 3,300.
|
| [18] |
Limongelli, V.; Bonomi, M.; Parrinello, M. Proc. Natl. Acad. Sci. 2013, 110,6358.
|
| [19] |
Raniolo, S.; Limongelli, V. Nat. Protoc. 2020, 15,2837.
|
| [20] |
Capelli, R.; Carloni, P.; Parrinello, M. J. Phys. Chem. Lett. 2019, 10,3495.
|
| [21] |
Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. J. Phys. Chem. B 2003, 107,9535.
|
| [22] |
Woo, H.-J.; Roux, B. Proc. Natl. Acad. Sci. 2005, 102,6825.
|
| [23] |
Fu, H.; Cai, W.; Hénin, J.; Roux, B.; Chipot, C. J. Chem. Theory Comput. 2017, 13,5173.
|
| [24] |
Torrie, G.M.; Valleau, J.P. J. Comput. Phys. 1977, 23,187.
|
| [25] |
Kästner, J. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1,932.
|
| [26] |
Zong, Z.; Li, Q.; Hong, Z.; Fu, H.; Cai, W.; Chipot, C.; Jiang, H.; Zhang, D.; Chen, S.; Shao, X. J. Am. Chem. Soc. 2019, 141,14451.
|
| [27] |
Deng, N.; Cui, D.; Zhang, B.W.; Xia, J.; Cruz, J.; Levy, R. Phys. Chem. Chem. Phys. 2018, 20,17081.
|
| [28] |
Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. J. Am. Chem. Soc. 2017, 139,946.
|
| [29] |
Boyce, S.E.; Mobley, D.L.; Rocklin, G.J.; Graves, A.P.; Dill, K.A.; Shoichet, B.K. J. Mol. Biol. 2009, 394,747.
|
| [30] |
Li, Z.; Huang, Y.; Wu, Y.; Chen, J.; Wu, D.; Zhan, C.G.; Luo, H.B. J. Med. Chem. 2019, 62,2099.
|
| [31] |
Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. Comput. Phys. Commun. 2014, 185,604.
|
| [32] |
Fiorin, G.; Klein, M.L.; Hénin, J. Mol. Phys. 2013, 111,3345.
|
| [33] |
Sidky, H.; Colón, Y.J.; Helfferich, J.; Sikora, B.J.; Bezik, C.; Chu, W.; Giberti, F.; Guo, A.Z.; Jiang, X.; Lequieu, J.; Li, J.; Moller, J.; Quevillon, M.J.; Rahimi, M.; Ramezani-Dakhel, H.; Rathee, V.S.; Reid, D.R.; Sevgen, E.; Thapar, V.; Webb, M.A.; Whitmer, J.K.; de Pablo, J.J. J. Chem. Phys. 2018, 148,44104.
|
| [34] |
Ibrahim, P.; Clark, T. Curr. Opin. Struct. Biol. 2019, 55,129.
|
| [35] |
Wang, J.; Deng, Y.; Roux, B. Biophys. J. 2006, 91,2798.
|
| [36] |
de Ruiter, A.; Oostenbrink, C. Curr. Opin. Chem. Biol. 2011, 15,547.
|
| [37] |
Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; Tsai, H.-C.; Xu, H.; Sherman, W.; York, D.M. J. Chem. Inf. Model. 2020, 60,5595.
|
| [38] |
Gumbart, J.C.; Roux, B.; Chipot, C. J. Chem. Theory Comput. 2013, 9,794.
|
| [39] |
Karplus, M.; Kuriyan, J. Proc. Natl. Acad. Sci. 2005, 102,6679.
|
| [40] |
Rizzi, A.; Grinaway, P.B.; Parton, D.L.; Shirts, M.R.; Wang, K.; Eastman, P.; Friedrichs, M.; Pande, V.S.; Branson, K.; Mobley, D.L.; Chodera, J.D. “YANK: A GPU-accelerated platform for alchemical free energy calculations,” can be found under getyank.org, 2020.
|
| [41] |
Mobley, D.L.; Chodera, J.D.; Dill, K.A. J. Chem. Theory Comput. 2007, 3,1231.
|
| [42] |
Henriksen, N.M.; Fenley, A.T.; Gilson, M.K. J. Chem. Theory Comput. 2015, 11,4377.
|
| [43] |
Heinzelmann, G.; Henriksen, N.M.; Gilson, M.K. J. Chem. Theory Comput. 2017, 13,3260.
|
| [44] |
Cruz, J.; Wickstrom, L.; Yang, D.; Gallicchio, E.; Deng, N. J. Chem. Theory Comput. 2020, 16,2803.
|
| [45] |
Coutsias, E.A.; Seok, C.; Dill, K.A. J. Comput. Chem. 2004, 25,1849.
|
| [46] |
Fu, H.; Gumbart, J.C.; Chen, H.; Shao, X.; Cai, W.; Chipot, C. J. Chem. Inf. Model. 2018, 58,556.
|
| [47] |
Xing, J.; Lu, W.; Liu, R.; Wang, Y.; Xie, Y.; Zhang, H.; Shi, Z.; Jiang, H.; Liu, Y.C.; Chen, K.; Jiang, H.; Luo, C.; Zheng, M. J. Chem. Inf. Model. 2017, 57,1677.
|
| [48] |
Wang, Y.; Li, L.; Zhang, B.; Xing, J.; Chen, S.; Wan, W.; Song, Y.; Jiang, H.; Jiang, H.; Luo, C.; Zheng, M. J. Med. Chem. 2017, 60,2026.
|
| [49] |
Zhang, X.; Li, X.; Wang, R. J. Chem. Inf. Model. 2009, 49,1033.
|
| [50] |
Pei, J.; Wang, Q.; Liu, Z.; Li, Q.; Yang, K.; Lai, L. Proteins Struct. Funct. Bioinforma. 2006, 62,934.
|
| [51] |
Liu, Y.; Xu, Z.; Yang, Z.; Chen, K.; Zhu, W. J. Mol. Model. 2013, 19,5015.
|
| [52] |
Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z. H.; Hou, T. Chem. Rev. 2019, 119,9478.
|
| [53] |
Ruiz-Blanco, Y.B.; Sanchez-Garcia, E. J. Chem. Theory Comput. 2020, 16,1396.
|
| [54] |
Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Chem. Sci. 2016, 7,207.
|
| [55] |
Li, Z.; Li, X.; Huang, Y.-Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; Xu, X.; Yu, K.; Zhang, Y.; Cui, J.; Zhan, C.G.; Wang, X.; Luo, H.B. Proc. Natl. Acad. Sci. 2020, 117,27381.
|
| [56] |
Qian, Y.; Cabeza de Vaca, I.; Vilseck, J.Z.; Cole, D.J.; Tirado-Rives, J.; Jorgensen, W.L. J. Phys. Chem. B 2019, 123,8675.
|
| [57] |
Gumbart, J.C.; Roux, B.; Chipot, C. J. Chem. Theory Comput. 2013, 9,3789.
|
| [58] |
Rizzi, A.; Jensen, T.; Slochower, D.R.; Aldeghi, M.; Gapsys, V.; Ntekoumes, D.; Bosisio, S.; Papadourakis, M.; Henriksen, N.M.; de Groot, B.L.; Cournia, Z.; Dickson, A.; Michel, J.; Gilson, M.K.; Shirts, M.R.; Mobley, D.L.; Chodera, J.D. J. Comput. Aided. Mol. Des. 2020, 34,601.
|
| [59] |
Zhang, H.; Gattuso, H.; Dumont, E.; Cai, W.; Monari, A.; Chipot, C.; Dehez, F. Molecules 2018, 23,228.
|
| [60] |
Du, S.; Wang, H.; Yang, Y.; Feng, X.; Shao, X.; Chipot, C.; Cai, W. J. Phys. Chem. C 2019, 123,922.
|
| [61] |
Liu, H.; Fu, H.; Shao, X.; Cai, W.; Chipot, C. J. Chem. Theory Comput. 2020, 16,6397.
|
| [62] |
Laury, M.L.; Wang, Z.; Gordon, A.S.; Ponder, J.W. J. Comput. Aided. Mol. Des. 2018, 32,1087.
|
| [63] |
Xie, B.; Nguyen, T.H.; Minh, D.D. L. J. Chem. Theory Comput. 2017, 13,2930.
|
| [64] |
Jiang, W.; Roux, B. J. Chem. Theory Comput. 2010, 6,2559.
|
| [65] |
Jo, S.; Jiang, W. Comput. Phys. Commun. 2015, 197,304.
|
| [66] |
Miao, Y.; Feher, V.A.; McCammon, J.A. J. Chem. Theory Comput. 2015, 11,3584.
|
| [67] |
Yang, Y.I.; Niu, H.; Parrinello, M. J. Phys. Chem. Lett. 2018, 9,6426.
|
| [68] |
Fu, H.; Zhang, H.; Chen, H.; Shao, X.; Chipot, C.; Cai, W. J. Phys. Chem. Lett. 2018, 9,4738.
|
| [69] |
Fu, H.; Chen, H.; Wang, X.; Chai, H.; Shao, X.; Cai, W.; Chipot, C. J. Chem. Inf. Model. 2020, 60,5366.
|
| [70] |
Perthold, J.W.; Oostenbrink, C. J. Chem. Theory Comput. 2017, 13,5697.
|
| [71] |
Pan, A.C.; Sezer, D.; Roux, B. J. Phys. Chem. B 2008, 112,3432.
|
| [72] |
Suh, D.; Jo, S.; Jiang, W.; Chipot, C.; Roux, B. J. Chem. Theory Comput. 2019, 15,5829.
|
| [73] |
Brotzakis, Z.F.; Limongelli, V.; Parrinello, M. J. Chem. Theory Comput. 2019, 15,743.
|
| [1] | Liu Zhenyu, Gan Li-Hua. Molecular Dynamics Simulation of Acetylene Pyrolysis into Fullerenes [J]. Acta Chimica Sinica, 2023, 81(5): 502-510. |
| [2] | Yizhi Han, Jianhui Lan, Xue Liu, Weiqun Shi. Advances in Molecular Dynamics Studies of Molten Salts Based on Machine Learning [J]. Acta Chimica Sinica, 2023, 81(11): 1663-1672. |
| [3] | Ke Zhao, Xiayu Cheng, Xuexue Ma, Minghui Geng. Mechanism of Two-photon Absorption Enhancement for a Piperazine-based Zinc Ion Probe [J]. Acta Chimica Sinica, 2023, 81(10): 1371-1378. |
| [4] | Yingzhe Du, Heng Zhang, Shiling Yuan. Molecular Dynamics Simulation of Thermal Conductivity of Al2O3/PDMS Composites [J]. Acta Chimica Sinica, 2021, 79(6): 787-793. |
| [5] | Chang-An Liu, Shi-Bo Hong, Bei Li. Molecular Dynamics Simulation of the Stability Behavior of Graphene in Glycerol/Urea Solvents in Liquid-Phase Exfoliation [J]. Acta Chimica Sinica, 2021, 79(4): 530-538. |
| [6] | Zun Liang, Xin Zhang, Songtai Lv, Hongtao Liang, Yang Yang. Crystal-Melt Interface Kinetics and the Capillary Wave Dynamics of the Monolayer Confined Ice-Water Coexistence Lines [J]. Acta Chimica Sinica, 2021, 79(1): 108-118. |
| [7] | Fan Qin, Liang Hongtao, Xu Xianqi, Lv Songtai, Liang Zun, Yang Yang. Study of the Dielectric Property of Monolayer Confined Water Using A Polarizable Model [J]. Acta Chimica Sinica, 2020, 78(6): 547-556. |
| [8] | Gao Simeng, Xia Kun, Kang Zhihong, Nai Yongning, Yuan Ruixia, Niu Ruixia. Molecular Dynamics Simulation of “Quasi-Gemini” Anionic Surfactant at the Decane/Water Interface [J]. Acta Chimica Sinica, 2020, 78(2): 155-160. |
| [9] | Yang, Pengli, Wang, Zhenxing, Liang, Zun, Liang, Hongtao, Yang, Yang. A Molecular Dynamics Simulation Study of the Effect of External Electric Field on the Water Surface Potential [J]. Acta Chimica Sinica, 2019, 77(10): 1045-1053. |
| [10] | Du Han, Liang Hongtao, Yang Yang. Molecular Dynamics Simulation of Monolayer Confined Ice-Water Phase Equilibrium [J]. Acta Chim. Sinica, 2018, 76(6): 483-490. |
| [11] | Yang Zhen, Xue Yijiang, He Yuanhang. Thermal Sensitivity of CL20/DNB Co-crystal Research via Molecular Dynamics Simulations [J]. Acta Chim. Sinica, 2016, 74(7): 612-619. |
| [12] | Zhang Chuan, Zhang Lujia, Zhang Yang, Huang He, Hu Yi. Study on the Stability and Enzymatic Property Improvement of Porcine Pancreas Lipase Modified by Ionic Liquids Using Molecular Simulation [J]. Acta Chim. Sinica, 2016, 74(1): 74-80. |
| [13] | Sun Ting, Liu Qiang, Xiao Jijun, Zhao Feng, Xiao Heming. Molecular Dynamics Simulation of Interface Interactions and Mechanical Properties of CL-20/HMX Cocrystal and Its Based PBXs [J]. Acta Chimica Sinica, 2014, 72(9): 1036-1042. |
| [14] | Wang Juan, Xia Shuwei, Yu Liangmin. Hydration Structure of Pb(II) from Density Functional Theory Studies and First-Principles Molecular Dynamics [J]. Acta Chimica Sinica, 2013, 71(9): 1307-1312. |
| [15] | Li Bo, Zhou Rui, He Gu, Guo Li, Huang Wei. Molecular Docking, QSAR and Molecular Dynamics Simulation on Spiro-oxindoles as MDM2 Inhibitors [J]. Acta Chimica Sinica, 2013, 71(10): 1396-1403. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
