


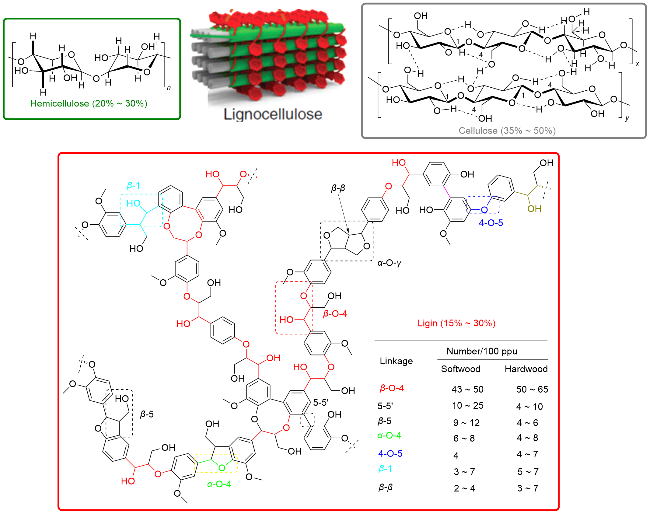
化石资源日益枯竭, 因此迫切需要开发可再生资源以替代化石资源促进社会经济可持续发展[1-3]. 非粮生物质是不与粮食产生冲突的可再生资源, 如农作物秸秆等, 年产量巨大, 约2000亿吨[4]. 然而, 目前仍以废弃为主[5]. 非粮生物质是由水和二氧化碳经过光合作用, 吸收太阳能形成的生物质能载体, 其主要成分为纤维素(约35%~50%)、半纤维素(约20%~30%)和木质素(约15%~30%), 统称为木质纤维素(图1), 具有转化为绿色能源和化学品的潜能[6-9]. 纤维素是由D-葡萄糖通过β-1,4-糖苷键连接而成的线性聚合物, 聚合度约800~10000[7], 具有归整的氢键网格结构. 半纤维素则是由多种不同类型的单糖, 如木糖、阿拉伯糖、半乳糖、甘露糖和葡萄糖等, 通过不同的糖苷键连接而成的聚合物[6-9]. 木质素是由芥子醇、松柏醇和对香豆醇三种单木酚, 通过C—C和C—O键连接而成的无定型三维网状聚合物. 连接单体的主要连接键如Scheme 1所示: 包括β-O-4、β-β、β-5、α-O-4、4-O-5、5-5和β-1[6]. 其中, β-O-4连接占主导地位, 其C—O键键能约290.56 kJ•mol-1, 在软木中占比约43%~50%, 在硬木中占比约50%~65%. 虽然4-O-5连接在木质素中含量较低, 但其C—O键能相对较高(约325.26 kJ•mol-1), 是影响木质素彻底降解的重要因素之一[10-11]. 由于纤维素和木质素的结构特征, 其在常规条件难以溶解, 导致与催化剂的有效接触严重受限, 因此, 非粮生物质的高效转化与利用受到大幅度限制. 已发展的从非粮生物质出发制备氢能[12-15]、碳基能源[16-20]和几类化学品, 包括糠醛[21-22]、5-羟甲基糠醛及衍生物[21-22]、苯乙酮[22-25]、苯甲醛[22-25]、苯甲酸[18]、苯酚[22-23,25]等的研究, 主要通过相对苛刻的热催化转化进行.
光催化, 尤其利用可见光进行催化转化, 因其在温和条件下能够产生活性物种, 促进高效、高选择性转化, 具有独特的优势[26-29]. 近年来, 光催化在生物质转化领域也得到了广泛应用, 通过在温和条件下产生碳中心或氧中心自由基, 诱导其β-位化学键键能降低, 发生断裂, 引发后续转化[7-8,30].
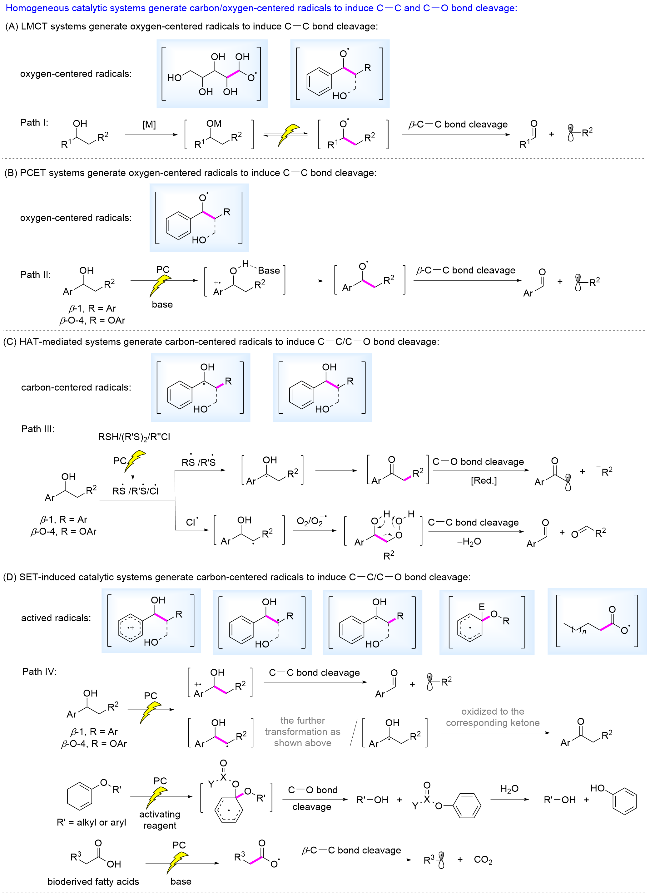
均相光催化条件下, 产生活性自由基诱导生物质相关转化的主要催化体系如Scheme 2所示: (1)通过配体到金属电子转移(ligand-to-metal charge transfer, 简称为LMCT)产生烷氧自由基. 转化路径概括为生物质结构单元/模型化合物中的醇羟基与过渡金属配位, 在可见光激发下发生LMCT, 产生烷氧自由基, 诱导β-C—C键键能显著降低, 发生断裂(Scheme 2A); (2)通过光催化剂与碱协同发生质子耦合电子转移(proton-coupled electron transfer, 简称为PCET)生成烷氧自由基. 转化路径概括为光催化剂氧化木质素结构单元/模型化合物中的芳环, 碱协同耦合苄位的羟基氢, 产生苄位烷氧自由基, 诱导β-C—C键键能显著降低, 发生断裂(Scheme 2B); (3)氢原子转移(hydrogen-atom-transfer, 简称为HAT)产生碳中心自由基. 转化路径概括为可见光照射下, 光催化剂氧化硫醇/氯负离子或光活化二硫醚等, 产生硫自由基/氯自由基, 与木质素β-1和β-O-4连接/模型分子发生HAT, 产生Cα自由基, 随后氧化为酮类中间产物, 诱导β-C—O键键能降低, 发生还原断裂; 或产生Cβ自由基, 被O2/$\mathrm{O}_{2}^{-·}$捕获, 中间体过氧键均裂诱导相应的C—C键断裂(Scheme 2C); (4)单电子转移(single-electron-transfer, 简称为SET)产生活性自由基. 转化路径概括为具有强氧化性的光催化剂通过SET过程氧化木质素结构单元中的芳环、β-O-4和β-1连接中的C—H键、辅助活化试剂及生物质衍生的脂肪酸负离子, 分别产生芳基自由基正离子、Cα自由基和Cβ自由基、去芳构化碳自由基、烷基羧基自由基, 进而活化β-化学键, 断裂相应的C—C和C—O键, 或Cα自由基进一步被氧化为相应的酮类中间体, Cβ自由基被O2/ 捕获, 进而发生上述类似的C—C键断裂(Scheme 2D).
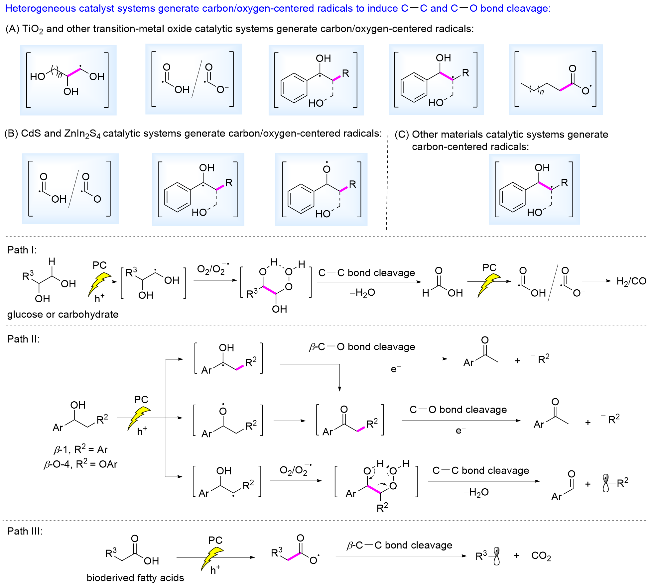
非均相光催化体系产生活性自由基诱导生物质相关转化的主要催化体系如Scheme 3所示: (1) TiO2及过渡金属氧化物光催化体系氧化产生多元醇的碳自由基、•CO2H、 、Cα自由基及Cβ自由基、烷基羧基自由基(Scheme 3A); (2) CdS及ZnIn2S4光催化体系氧化产生•CO2H、$\mathrm{CO}_{2}^{-·}$、Cα自由基和苄位氧自由基(Scheme 3B); (3)其他非均相光催化体系氧化产生Cβ自由基(Scheme 3C). 这些催化体系主要通过三条路径引发断裂相关化学键进行相应的催化转化. 在Path I中, 葡萄糖、多元醇等碳水化合物在价带氧化产生多元醇的碳自由基, O2/ 捕获该碳自由基, 产生的中间体的过氧键均裂引发C—C键断裂, 得到的主要产物为甲酸(HCO2H). 通过不同的光催化体系, HCO2H进一步在价带氧化产生•CO2H/ , 选择性生成H2或CO (Scheme 3, path I). 在Path II中, 木质素β-1和β-O-4连接/模型分子, 在价带氧化产生Cα自由基、苄位氧中心自由基、Cβ自由基. Cα自由基诱导β-C—O键键能显著降低, 发生Cβ—O键断裂, 得到芳基酮; Cα自由基及苄位氧中心自由基中间体也能够进一步被氧化为酮, 随后在价带还原断裂 Cβ—O键得到酮. Cβ自由基则被O2/ 捕获, 过氧键均裂引发相应的C—C键断裂, 得到醛类化合物(Scheme 3, path II). 在Path III中, 价带氧化烷基羧酸负离子, 脱羧产生烷基自由基, 转化为烷烃(Scheme 3, path III).
1 均相光催化生物质相关转化
均相催化体系在生物质转化过程中具有独特的优势. 例如, 均相催化剂能够进入到木质纤维素的稳固结构中, 通过密切接触充分展示其催化活性[8]. 此外, 均相催化剂的结构更容易解析, 反应中间体更容易表征, 有利于反应机理的探究. 已发展的均相光催化体系主要包括: 通过LMCT和PCET过程产生烷氧自由基, 断裂生物质中C—C键; 通过HAT选择性地产生碳中心自由基, 断裂生物质中C—C或C—O键; 通过光催化剂与模型分子发生直接或间接SET过程, 产生活性自由基, 断裂生物质中C—C或C—O键. 本节将系统介绍以上四种均相光催化体系, 阐述在光照下产生关键自由基引发化学键断裂进行生物质相关转化的过程.
1.1 LMCT光催化体系
LMCT过程涉及配体中的电子跃迁到金属中心空轨道, 实现电子从配体到金属中心的转移[31]. 由于金属的轨道为空轨道时, 电子从配体轨道跃迁到金属轨道需要克服的能垒较低, 因此, 这种激发态通常出现在带有亲电性、高价金属中心的配合物中, 例如, Fe(III)、V(V)、Ce(IV)等[31-32]. 另一方面, 由于配体在电子跃迁过程中充当内部电子源, 因此, 富含电子的σ或σ+π配体(如卤化物、羧酸盐或叠氮化物)有利于在相对较低能量下发生LMCT跃迁[31]. 这种独特的电子转移模式为温和条件下的化学转化开辟了新路径. 近年来, 该模式已经用于生物质转化研究中. 相关转化主要利用金属催化剂及生物质结构中的羟基配位, 在可见光照射下发生LMCT过程生成烷氧自由基, 诱导β-位化学键键能降低, 发生β-C—C键断裂, 进行后续相关转化. 利用该策略已经实现了将葡萄糖、纤维素及麦秆转化至HCO2H[33], 以及木质素β-O-4模型中Cα—Cβ键断裂[34-35], 这些催化体系充分展现了LMCT过程在促进生物质高效转化方面的潜力. 本小节将对Fe(III)、V(V)、Ce(IV)三类过渡金属催化剂的LMCT催化体系展开综述.
1.1.1 Fe(III)催化的LMCT反应体系
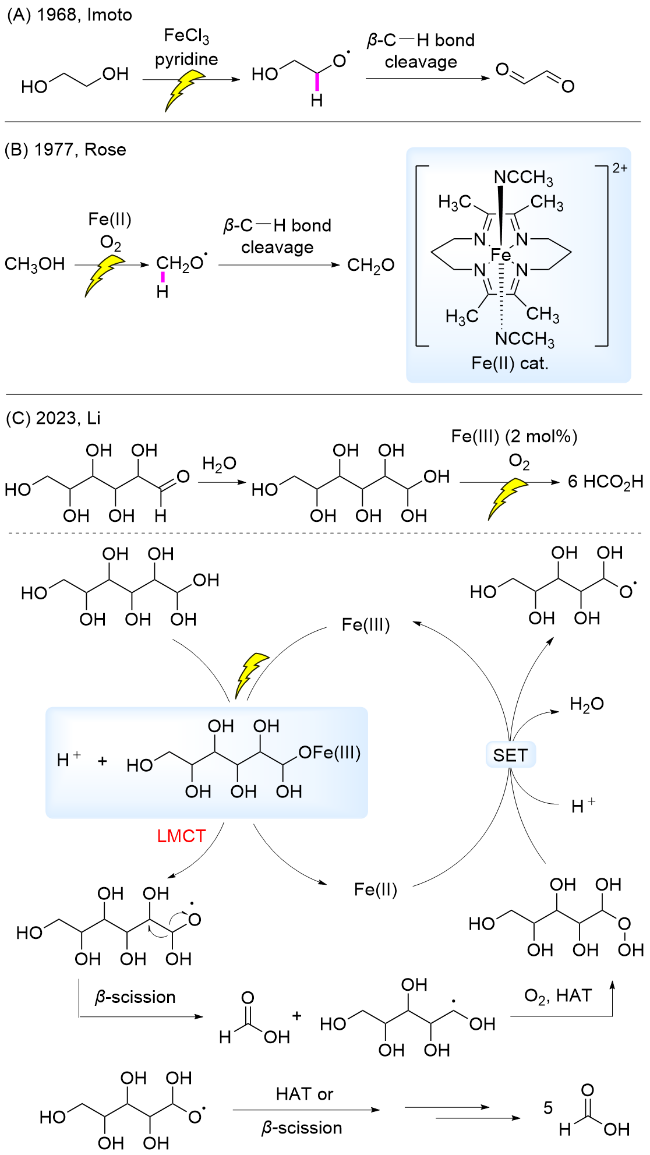
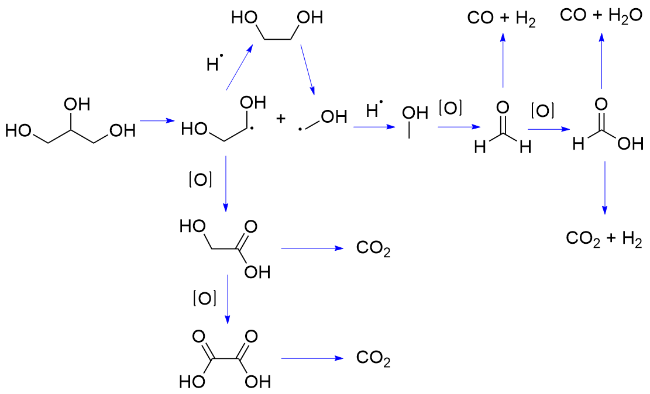
2023年, 我们课题组[33]报道了以Fe(NO3)3•9H2O为催化剂, O2为氧化剂, 通过LMCT过程实现了室温下葡萄糖C—C键逐级断裂, 产生6分子HCO2H. 反应机理为: Fe(III)与水合葡葡糖配位, 在465~470 nm波长光照射下发生LMCT过程, 产生烷氧自由基, 诱导β-C—C键断裂, 随后经历O2捕获碳自由基、HAT、Fe(III)发生的SET, 及HAT或β-C—C键断裂, 随后经历类似的系列催化过程, 生成了6分子HCO2H (Scheme 4C). 根据同位素效应实验推测HAT为该转化的决速步骤. 随后, 研究了该催化体系对原生生物质的降解, 由于其稳固的结构较难以溶解, 通过在反应体系中加入LiCl, 破坏纤维素的氢键网络结构; 通过增加氧气压力及催化剂浓度提高反应体系中生成的羟基自由基浓度, 同时加入质量分数为5%的H2SO4, 促进断裂纤维素β-1,4-糖苷键; 从而使纤维素和麦秆展示出一定的反应活性, 以其为原料得到HCO2H的产率分别为31%和12%. 该方法展示了LMCT催化体系在葡萄糖及纤维素转化中的应用潜力.
Fe(III)催化的LMCT过程也应用到木质素β-O-4模型分子转化中. 2021年, 曾荣课题组[42]报道了以FeCl3和CeCl3为共催化剂, O2为氧化剂, 在390 nm波长光照射下, 通过LMCT过程催化1-苯基环己醇及衍生物产生烷氧自由基, 诱导β-C—C键断裂, 通过O2进一步氧化中间体生成苯甲酸. 作者将该催化体系应用于木质素β-O-4模型Cα—Cβ键断裂中, 生成苯甲酸和甲酸苯酯. 最近, 刘守新、田冰和陈志俊团队[43]报道了以FeCl3/ NaCl或FeCl3/nBu4Cl为催化剂, 在390 nm波长光照射下, 通过LMCT过程实现了木质素β-O-4和β-1模型分子C—C键断裂, 生成苯甲醛和碳中心自由基, 该碳中心自由基被体系中偶氮类或烯烃类化合物捕获, 生成含氮化合物或烯烃双键加成产物. 通过光开关实验、自由基捕获实验及控制实验, 推测反应通过LMCT过程实现转化. 该反应体系能够用于溶剂解木质素的降解, 主要得到两种芳基醛类单体化合物, 总收率为8.7%.
1.1.2 V(V)催化的LMCT体系
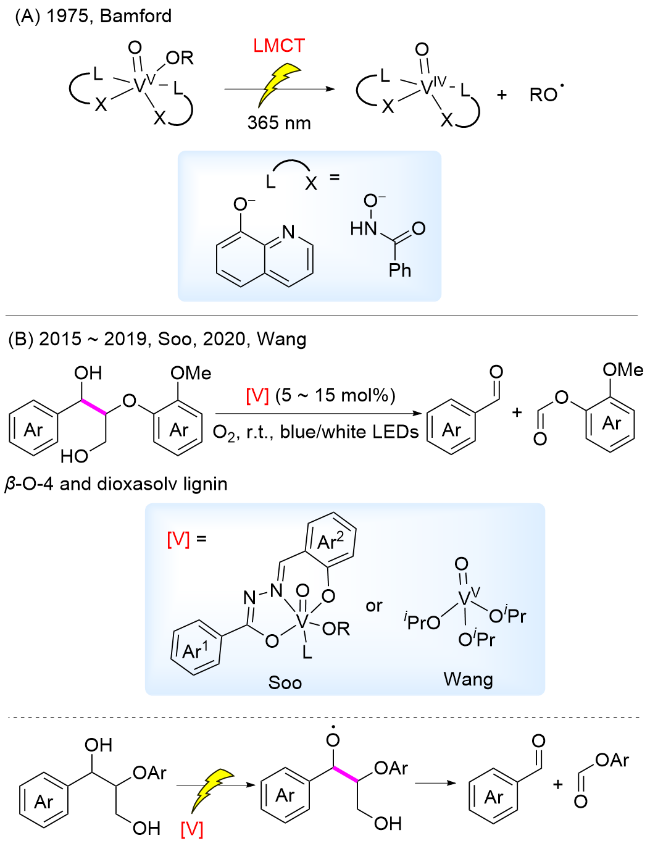
2015年, Soo课题组[45]合成了一种以腙酰胺为配体的钒氧络合物, 该络合物与反应物中醇羟基配位后具有显著的光学活性. 作者以该钒氧络合物为催化剂, 在可见光照射下实现了木质素β-O-4模型中Cα—Cβ键断裂. 通过机理实验及密度泛函理论(DFT)计算提出反应机理为: 催化剂和底物配位后, 在可见光照射下通过LMCT过程产生氧中心自由基, 进而诱导β-C—C键断裂, 产生苯甲醛和碳中心自由基, 碳自由基被O2捕获, 经过过氧键均裂、β-消除, 形成稳定的产物甲酸芳基酯类化合物和HCO2H, 完成催化循环(Scheme 5B). 为了进一步探究不同配体对LMCT过程的影响, 2017年, Soo课题组[46-47]合成了同系列含腙酰胺配体的钒催化剂, 发现在钒催化剂配体中Ar1芳环引入五个氟原子、Ar2芳环氧原子的对位引入硝基吸电子基团, 能够提升催化效率及反应产率. 作者通过动力学、光谱学和电化学实验推测, 催化剂配体中引入吸电子基团有利于促进LMCT过程, 并利用DFT计算对其进行验证. 2020年, 王峰课题组[34]报道了以商业可得的VO(i-Pr)3为光催化剂, 通过LMCT过程实现了室温下木质素β-O-4模型中Cα—Cβ键断裂. 该催化体系能够用于溶剂解木质素的降解, 白桦木溶剂解木质素降解, 得到芳基单体收率为1.39%.
1.1.3 Ce(IV)催化的LMCT体系
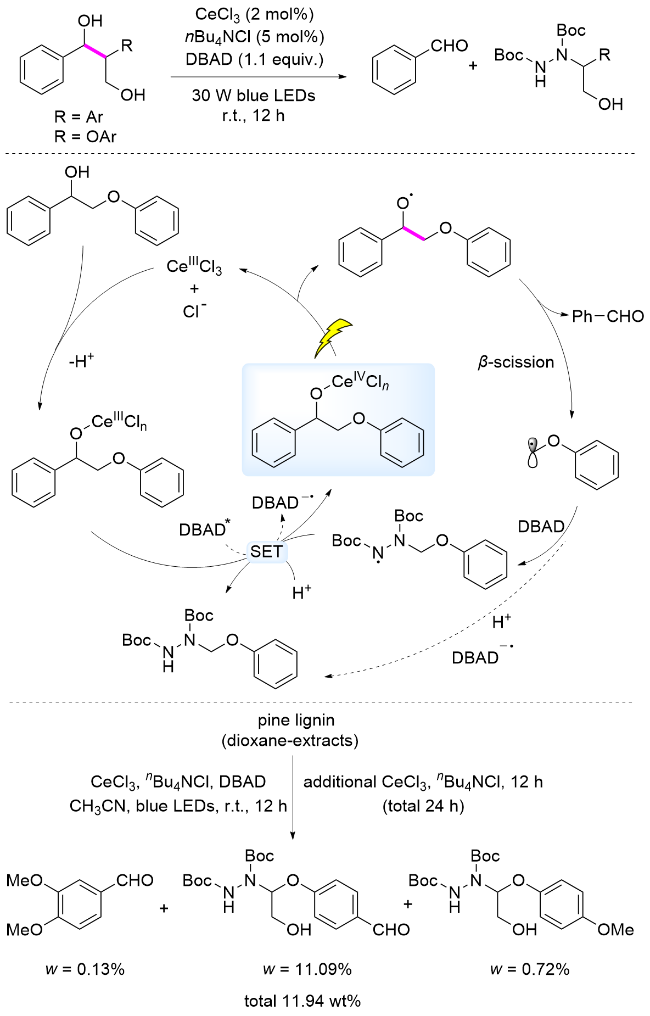
铈(Ce)是一种丰富的稀土元素, Ce(III)化合物在有机合成中得到了广泛应用. CeCl3作为一种廉价的路易斯酸, 在光氧化还原催化中发挥着重要作用[48]. 2016年, 左智伟课题组[49]报道了以CeCl3为催化剂, 在可见光照射下CeCl3与1-甲基环己醇中羟基基团配位, Ce(III)配合物被氧化为Ce(IV), 通过LMCT过程产生烷氧自由基, 诱导β-C—C键断裂, 产生的碳中心自由基被体系中二叔丁基偶氮二甲酸酯(DBAD)捕获, 转化为有机含氮化合物. 该研究首次详细描述了CeCl3与醇羟基配位形成配合物, 在可见光照射下Ce(III)配合物在体系中被氧化至Ce(IV), 发生LMCT过程.
木质素结构中含有丰富的醇羟基. 2020年, 张越涛课题组[35]报道了以CeCl3为催化剂, 在可见光照射下, 通过Ce(IV)配合物发生LMCT过程, 实现了木质素β-O-4和β-1模型Cα—Cβ键断裂, 生成芳基醛和碳中心自由基, DBAD捕获碳中心自由基转化为含氮化合物. 反应机理为: Ce(III)与底物形成配合物, 该配合物被激发态二叔丁基偶氮二甲酸酯(DBAD*)氧化为Ce(IV)配合物, 在可见光照射下发生LMCT过程生成烷氧自由基, 诱导β-C—C键断裂, 实现木质素模型中Cα—Cβ键断裂, 生成芳基醛和碳中心自由基. 该碳中心自由基被DBAD捕获生成有机含氮化合物, 完成催化循环(Scheme 6). 此外, 体系中生成的烷基自由基还可能在质子存在下与DBAD-•发生自由基偶联反应, 生成含氮产物. 该方法能够用于溶剂解松木木质素的解聚, 获得芳基单体化合物的产率为11.94%.
1.2 PCET光催化体系
光催化条件下, 开发能够规避初始电子转移或质子转移所需高能垒的反应路径至关重要. PCET过程能够在热力学上避免单个化学位点的电荷积累, 同时能在动力学上避免高能中间体的形成.[50] PCET过程中, 单电子氧化剂和Brønsted碱以协同的方式从底物分子中分别获得一个电子和一个质子. 因为该体系中的单电子氧化剂与碱可以独立调控, 所以能够在较广的能量范围内调节该PCET过程的热化学性质, 以促进高键能化学键的高效断裂. 针对含有芳基的醇类化合物发生的PCET过程, Bacchioici、Bietti和Steenken等[51-53]利用光谱实验证明, 与芳基自由基阳离子相邻的苄位羟基的质子以接近扩散的速率发生选择性地去质子化, 以产生游离的烷氧基自由基. Knowles课题组[54]通过实验表明了该过程为协同的PCET过程. 随后, 这种协同的PCET过程得到了广泛应用[55-56].
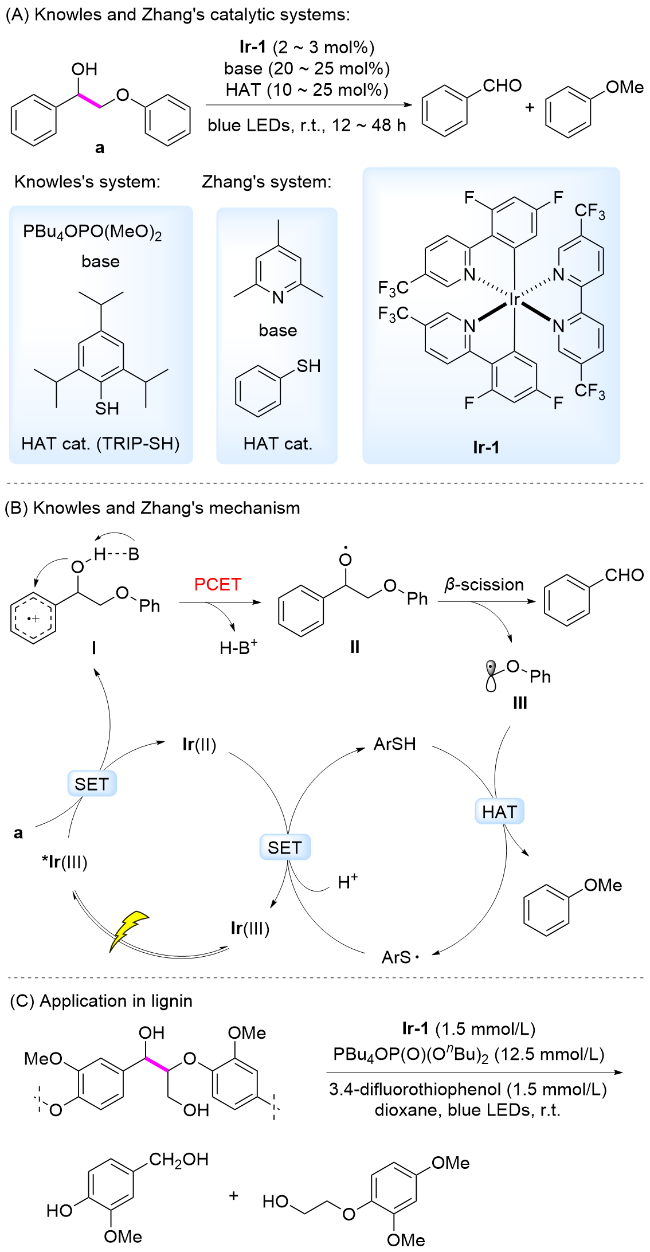
同年, 张越涛和何江华研究团队[58]以相同的Ir-1为光催化剂, 以2,4,6-三甲基吡啶(Collidine)为碱, 在可见光照射下, 通过PCET过程实现了木质素β-O-4和β-1模型中Cα—Cβ键断裂, 生成芳基醛和芳基甲醚类产物. 反应机理为: 在可见光照射下, 激发态光敏剂被木质素模型分子还原淬灭, 形成Ir(II)和阳离子自由基中间体I. 在Collidine存在下发生分子内协同的PCET, 生成苄位氧中心自由基, 诱导β-C—C键断裂, 生成苯甲醛和碳中心自由基中间体III. 该中间体III与HAT催化剂发生HAT过程, 形成另一产物苯甲醚, 完成催化循环(Scheme 7B). 由于酚羟基也是该PCET过程的较优基团, 木质素中含有大量的酚羟基, 其在降解过程中, 酚羟基与Cα—OH醇羟基存在竞争, 影响木质素降解反应顺利进行. 作者采用CH3I/K2CO3体系对提取的桦木木质素预处理, 将酚羟基保护后再降解, 得到芳基单体的收率为4.99%.
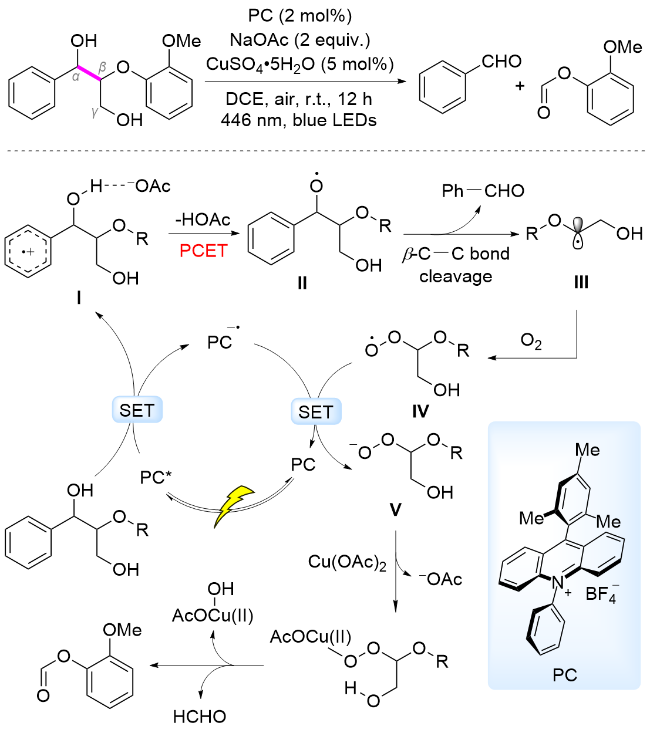
此外, Miura和Murakami课题组[60]发展了以吖啶盐为光催化剂, 以NaOAc为碱的PCET催化体系, 在可见光照射下实现了木质素β-O-4和β-1模型中Cα—Cβ键断裂, 生成芳基醛、甲酸芳基酯和甲醛(HCHO). 提出的反应机理为: 在446 nm波长光照射下, 激发态光催化剂被模型分子还原淬灭, 形成PC-•和芳基阳离子自由基中间体I, AcO-耦合羟基氢, 通过分子内PCET过程产生苄位氧中心自由基II, 诱导木质素β-C—C键断裂, 生成苯甲醛和碳中心自由基中间体III. 中间体III被体系中O2迅速捕获, 形成过氧自由基中间体IV, 随后与还原态光催化剂发生SET, 形成过氧阴离子V. 该过氧阴离子与Cu(OAc)2发生阴离子交换, 形成Cu(II)配合物, 促使过氧键均裂, 同时诱导β-C—C键均裂, 脱除一分子HCHO生成甲酸苯酯, 完成催化循环(Scheme 8).
2023年, Wallentin课题组[61]采用吖啶盐为光催化剂, 以Collidine为碱, 通过PCET过程实现了1-(4-甲氧基苯基)-2-苯氧基乙醇中C—O键断裂, 产生的碳中心自由基对烯烃双键亲电进攻, 生成加成产物(Scheme 9A). 随后, 刘守新和田冰研究团队[62]报道了类似的催化体系, 作者将底物拓展到更广的木质素β-O-4模型, 生成的碳中心自由基不仅能与烯烃类化合物发生亲电加成, 而且还能与偶氮类化合物反应生成含氮化合物(Scheme 9B). 反应机理为: 在可见光照射下, 通过光敏剂与碱协同作用产生苄位氧中心自由基, 诱导木质素β-C—C键断裂, 生成苯甲醛和碳中心自由基中间体. 该碳中心自由基与体系中烯烃类或偶氮苯类化合物反应, 生成相应的产物(Scheme 9C).
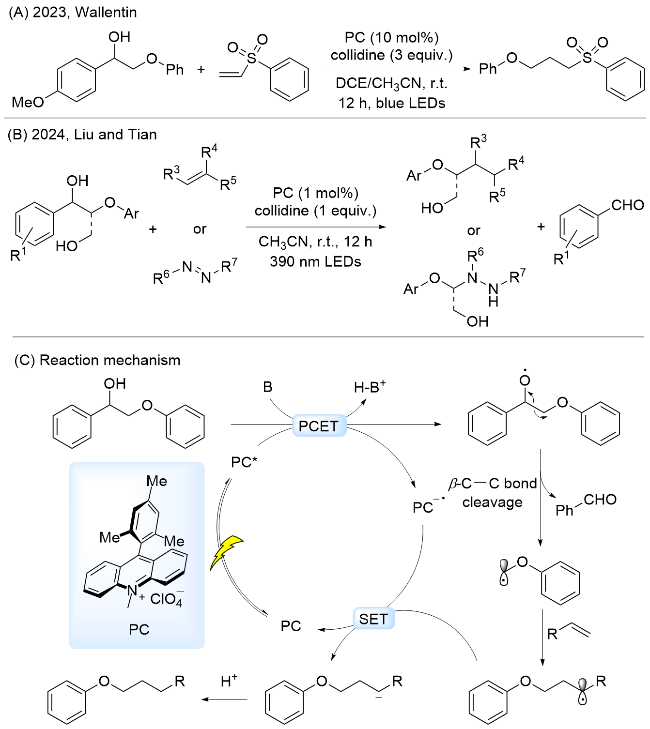
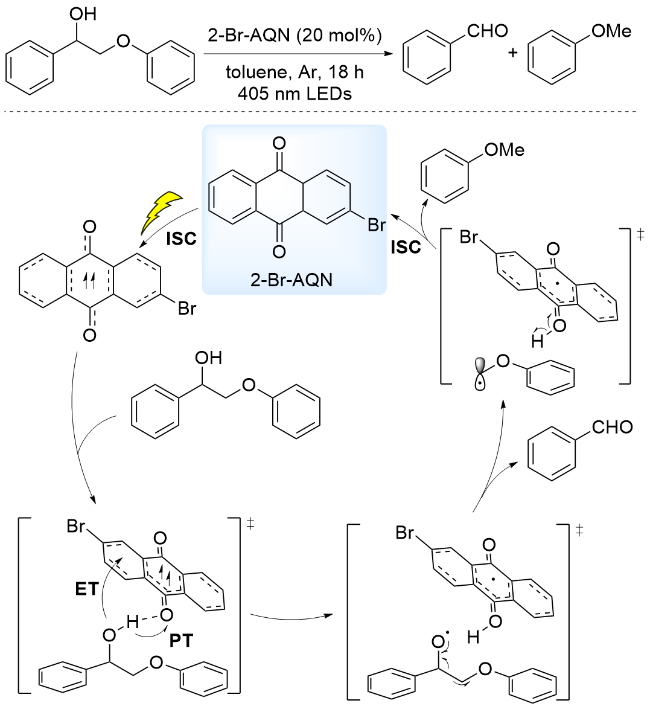
最近, 曾会应和李朝军研究团队[63]报道了以2-溴蒽醌(2-Br-AQN)为光催化剂, 在405 nm波长光照射下, 通过PCET过程实现了木质素β-O-4模型中Cα—Cβ键断裂, 生成芳基醛和芳基醚类产物. 通过机理实验和DFT计算提出的反应机理为: 在光照条件下, 激发态光催化剂与底物发生PCET过程, 促使底物形成氧中心自由基. 该PCET过程中光催化剂同时作为电子和质子受体, 从底物分子中得到一个质子和一个电子, 形成的氧中心自由基诱导其β-C—C键断裂, 生成苯甲醛和碳中心自由基, 进而转化为苯甲醚(Scheme 10). 该方法能够用于从松树、冷杉、白杨、栾树、桦树和小麦秸秆中提取的木质素, 芳基单体收率分别为: 3.67%、4.08%、4.40%、5.37%、3.86%、2.94%和2.35%.
1.3 HAT光催化体系
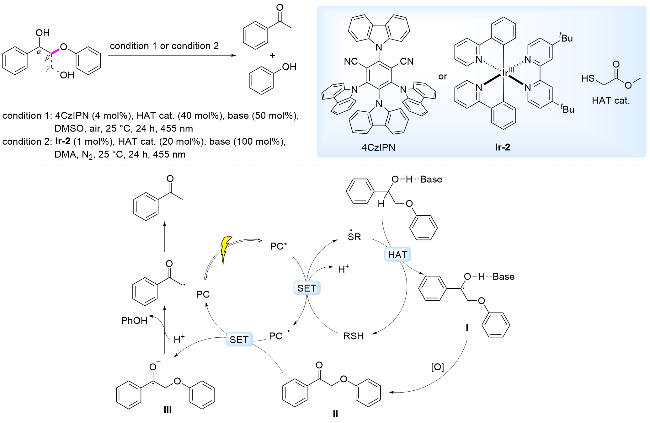
2019年, König课题组[65]报道了以4CzIPN或Ir-2为光催化剂, 以NaOP(O)(OBu)2为碱, 以HSCH2CO2Me为HAT催化剂, 在可见光照射下, 通过光催化剂与HAT催化剂协同催化, 实现了木质素β-O-4模型发生C—O键断裂, 生成苯乙酮和苯酚(Scheme 11). 提出的反应机理为: 在455 nm波长光照射下, HSCH2CO2Me还原淬灭激发态光催化剂, 去质子化产生硫中心自由基, 该硫自由基与木质素β-O-4模型中Cα—H发生HAT过程, 产生Cα自由基I. 在以4CzIPN为光敏剂的催化体系中, 自由基I需要在外加氧化剂条件下, 通过空气中O2将其氧化为酮类中间产物II; 在以Ir-2为光敏剂的催化体系中, 自由基I被激发态光敏剂氧化为酮类中间产物II. 形成的酮类化合物与还原态的光催化剂发生SET, 光敏剂回到基态, 同时形成阴离子自由基III, 诱导β-C—O键断裂, 生成苯酚和苯乙酮(Scheme 11).
2023年, 胡可课题组[67]报道了采用苝二酰亚胺(PDI)为光催化剂, nBu4Cl为HAT催化剂, 以空气中O2为氧化剂, 在可见光照射下, 通过光敏剂与HAT催化剂协同催化, 实现了木质素β-O-4和β-1模型中Cα—Cβ 键断裂, 生成芳基醛、芳基羧酸和甲酸芳基酯类化合物(Scheme 13A). 提出的反应机理为: 激发态光敏剂PDI*被nBu4Cl电离出的Cl-还原淬灭, 形成光敏剂阴离子自由基PDI-•和Cl•, PDI-•被O2氧化, 光敏剂回到基态, 同时生成超氧阴离子自由基( ). 形成的Cl•主要与底物分子中Cβ—H发生HAT, 形成Cβ自由基中间体I, 该中间体被 捕获形成过氧化物中间体II, 随后通过六元环过渡态发生 Cα—Cβ键断裂, 同时过氧键均裂脱除一分子H2O, 生成芳基醛和甲酸芳基酯类化合物(Scheme 13B). 由于该体系存在活性氧物种, 生成的芳基醛类化合物进一步被氧化为芳基羧酸.
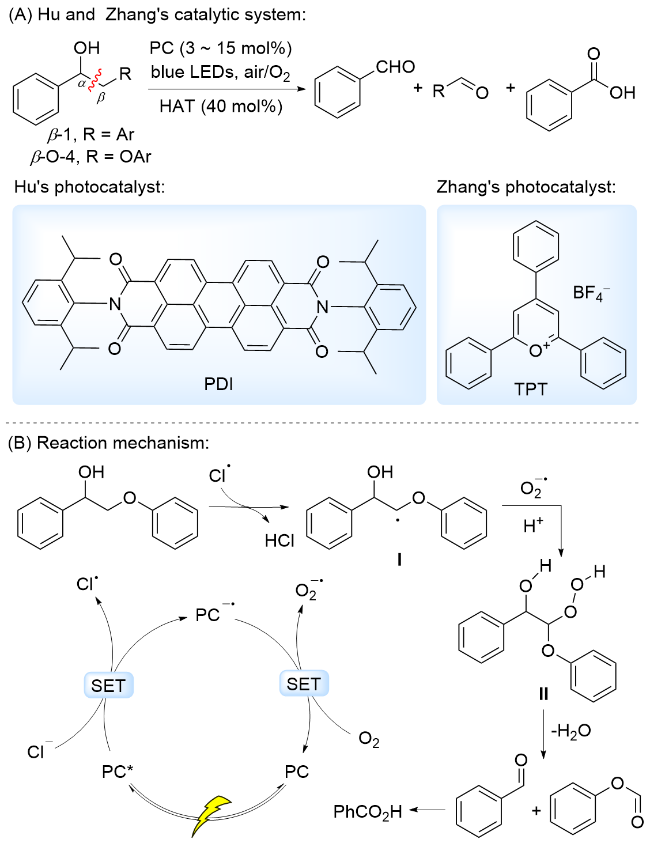
最近, 张越涛和何江华研究团队[68]采用商业可得的2,4,6-三苯基吡喃四氟化硼盐(TPT)为光催化剂, 以CaCl2为HAT催化剂, 以O2为氧化剂, 通过光催化剂与HAT催化剂协同催化, 实现了木质素β-O-4模型中 Cα—Cβ键断裂, 生成芳基醛、芳基羧酸和甲酸芳基酯类化合物(Scheme 13A). 作者提出了与PDI/nBu4Cl催化体系相似的反应机理(Scheme 13B). 此外, 该体系能够用于溶剂解木质素的解聚, 桦木和松木溶剂解木质素在该体系中降解, 产生芳基单体的收率分别为0.69%和0.42%.
1.4 SET光催化体系
光氧化还原中最普遍的SET过程可以通过激发态光催化剂得失电子而使底物分子产生自由基. 光催化剂与底物分子电位匹配及具有纳秒级荧光寿命是发生单电子转移的必要条件[26-27]. 光催化剂通过SET过程氧化芳环形成高活性的芳基阳离子自由基, 促使Cα—Cβ键断裂; 或选择性活化Cβ—H键, 产生活性Cβ自由基, 被O2/$\mathrm{O}_{2}^{-·}$捕获, 通过形成的六元环过氧化物中间体, 促使Cα—Cβ键断裂; 而对于惰性的4-O-5型模型分子中C—O键的活化, 通过外加或原位生成辅助试剂促使其形成高活性的去芳构化碳中心自由基中间体, 诱导C—O键的断裂; 或通过SET与氧原子转移(OAT)协同催化, 实现C—O键断裂, 其中SET是关键步骤. 来源于植物的一些脂肪酸类化合物通过SET过程较容易产生烷基羧基自由基, 脱羧后产生碳中心自由基, 能够进一步转化为高附加值产物.
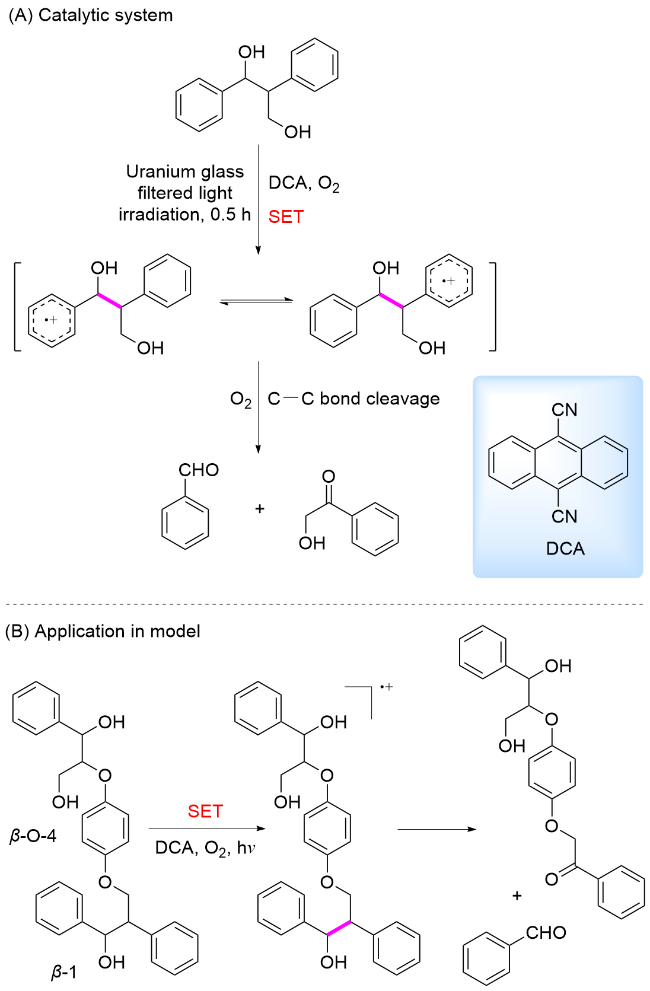
2011年, Yoon和Mariano研究团队[69]为了研究木质素过氧化酶对木质素的降解过程, 采用在酶降解过程中的电子受体9,10-二氰基蒽(DCA)为光催化剂, 以O2为氧化剂, 对木质素β-O-4和β-1模型中Cα—Cβ键断裂机理进行了模拟研究(Scheme 14A). DCA具有较强的氧化能力, 其激发态还原电势为+2.76 V vs Ag/AgCl. 反应机理为: 木质素β-O-4和β-1模型分子还原淬灭激发态光敏剂DCA*, 形成相应的芳基阳离子自由基, 诱导 Cα—Cβ键BDE显著降低, 导致芳基阳离子自由基中间体C—C键断裂. 在催化含有β-O-4与β-1键的四聚体木质素模型时, 由于它们的氧化电位接近, 激发态的DCA可以同时将四个芳环氧化为相应的阳离子自由基, 但四聚体底物总是优先在β-1单元发生C—C键断裂(Scheme 14B)[70], 详细的机制有待进一步探究. 尽管该方案能够有效降解木质素, 但由于强氧化性条件, 酚类也可能进一步氧化, 导致芳烃类化合物产率较低.
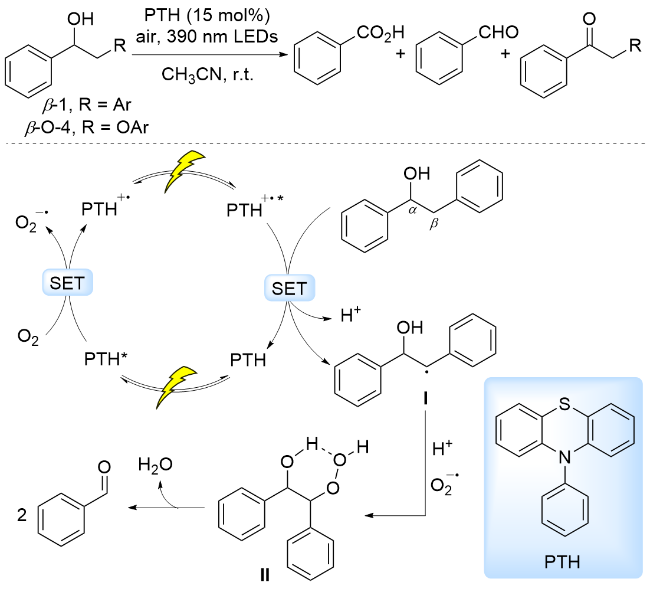
2023年, 胡可课题组[71]采用10-苯基吩噻嗪(PTH)为光催化剂, 空气为氧化剂, 在390 nm波长灯照射下实现了木质素β-O-4和β-1模型中Cα—Cβ键断裂, 生成芳基醛、芳基羧酸. 但该体系伴随生成的Cα—OH被氧化为羰基的转化, 因此有少量酮类副产物生成. 基于对照实验和瞬态光谱数据, 提出反应机理为: 光照条件下, PTH吸收第一个光子形成激发态PTH*, 被空气中O2氧化淬灭, 生成PTH+和$\mathrm{O}_{2}^{-·}$. PTH+的电势不足以氧化底物, 接着吸收第二个光子后, 形成的PTH+*是一种强氧化剂(+2.31 V vs NHE), 通过SET氧化C—H键, 去质子化形成Cβ自由基. Cβ自由基被$\mathrm{O}_{2}^{-·}$捕获形成六元环过氧化物中间体II, 过氧键均裂诱导Cα—Cβ键断裂, 并脱除一分子H2O, 生成苯甲醛(Scheme 15). 对照实验表明, 苯甲醛进一步被氧化为苯甲酸. 副产物酮的产生路径为: 部分底物首先被二次激发态光敏剂PTH+*单电子氧化, 质子离去后形成Cα自由基, 随后, Cα自由基被氧化生成酮.
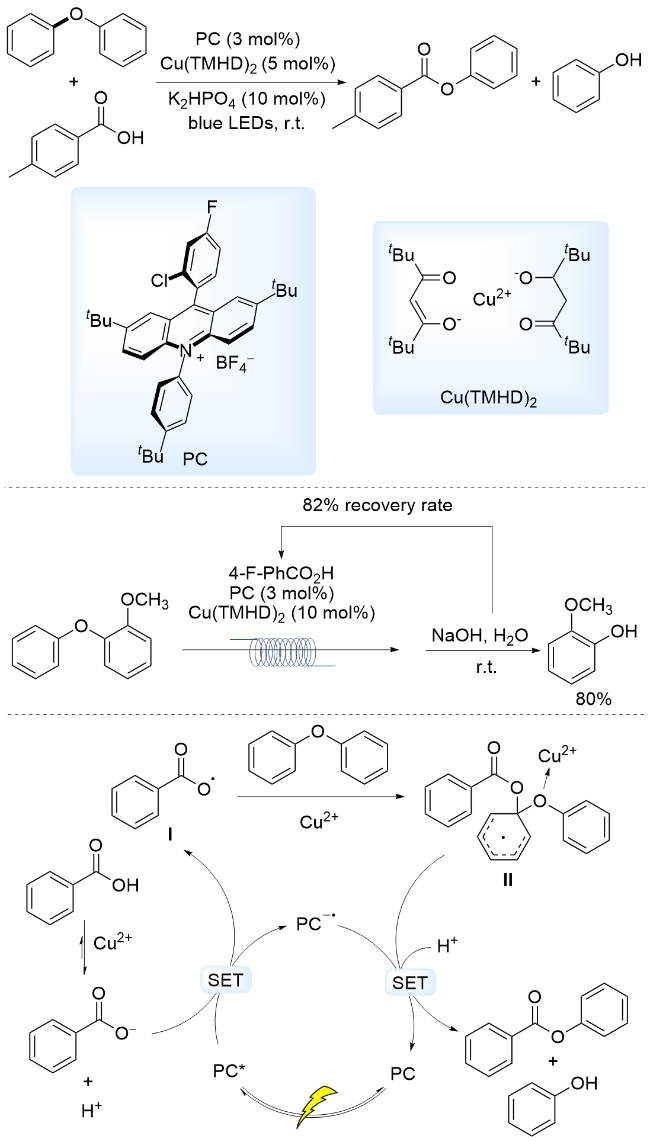
木质素4-O-5模型中C—O键的BDE约为326.26 kJ•mol-1, 难以在温和条件下直接发生活性自由基诱导的C—O键断裂, 长久以来是影响木质素彻底降解的因素之一[10-11]. 2017年, 我们课题组[72]采用商业可得的吖啶盐或PDI为光催化剂, 以邻苯氧基苯甲酸为底物, 在催化量碱作用下, 通过可见光诱导催化形成芳基羧基自由基, 发生分子内亲电取代反应, 生成邻羟基苯甲酸苯酯, 实现了室温断裂二芳基醚类C—O键, 并串连室温水解得到两种酚类化合物. 以上述工作为基础, 2020年, 我们[73]进一步通过修饰吖啶盐光敏剂, 在吖啶环上增加位阻, 在9位芳环上引入吸电子取代基, 发展了较稳定的光催化剂. 通过可见光诱导催化产生芳基羧基自由基, 与二芳基醚发生分子间亲电取代反应, 通过Cu(II)盐辅助, 断裂了4-O-5型C—O键, 随后室温水解得到两种酚类化合物(Scheme 16). 提出的反应机理为: 在可见光照射下, 激发态光敏剂PC*被苯甲酸负离子还原淬灭, 形成还原态光敏剂PC-•和芳基羧基自由基. 芳基羧基自由基对二芳基醚亲电进攻, 形成活性中间体自由基II, 在Cu(II)作为路易斯酸的协同作用下, 该活性中间体自由基与还原态光催化剂发生SET, 4-O-5型C—O键断裂, 生成苯甲酸苯酯和苯酚. 利用流动化学装置, 对木质素4-O-5简单模型分子进行转化, 并串联水解, 以80%的产率得到酚类产物, 芳基羧酸的回收率为82%.
无需化学计量的辅助试剂, 一步室温水解是断裂4-O-5型C—O键的理想方案. 2023年, 我们课题组[74]利用发展的吖啶盐光催化剂与V2O5催化剂, 在光照下实现了二芳基醚水解产生两分子酚的研究(Scheme 17). 机理实验与DFT计算表明反应机理为: 芳基醚还原淬灭激发态光敏剂PC*, 形成芳基阳离子自由基和PC-•. 同时, V2O5在碱性条件下原位产生H2VO4-, H2VO4-对二芳基醚阳离子自由基亲核进攻, 形成类Meisenheimer中间体. 该中间体自旋中心重新分布, 并在钒氧相互作用的促进下发生C—O键断裂, 生成钒酸苯酯和氧中心自由基. 其中氧自由基被淬灭生成酚或醇, 钒酸苯酯则原位水解生成苯酚和H2VO4-, 完成催化循环. 通过钒与光催化剂协同催化, 成功实现了4-O-5, α-O-4和β-O-4型C—O键断裂, 展现了该共催化体系潜在的应用价值.
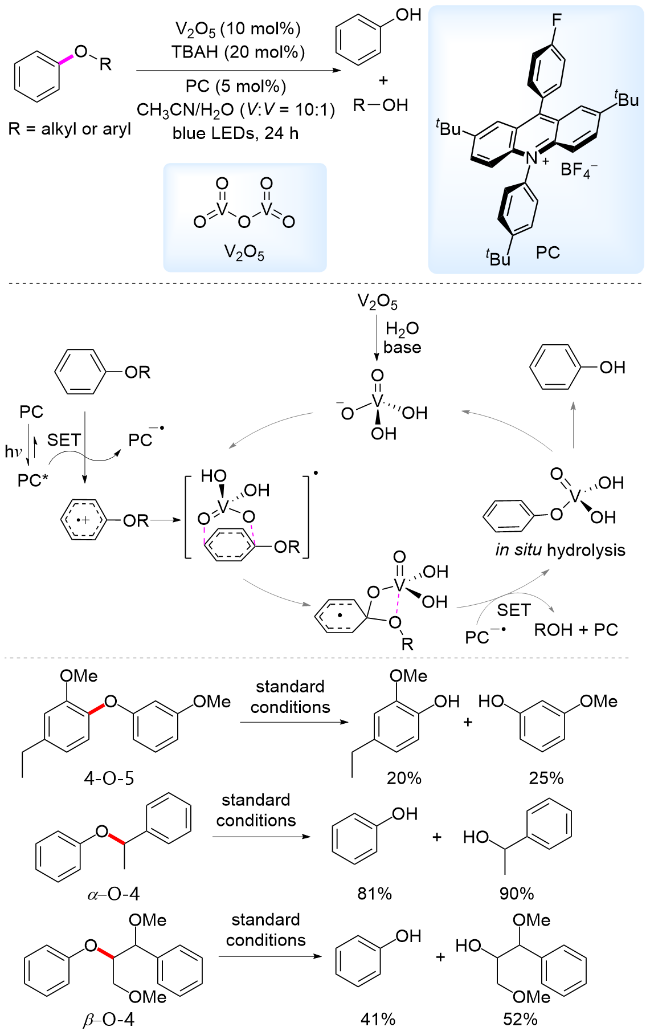
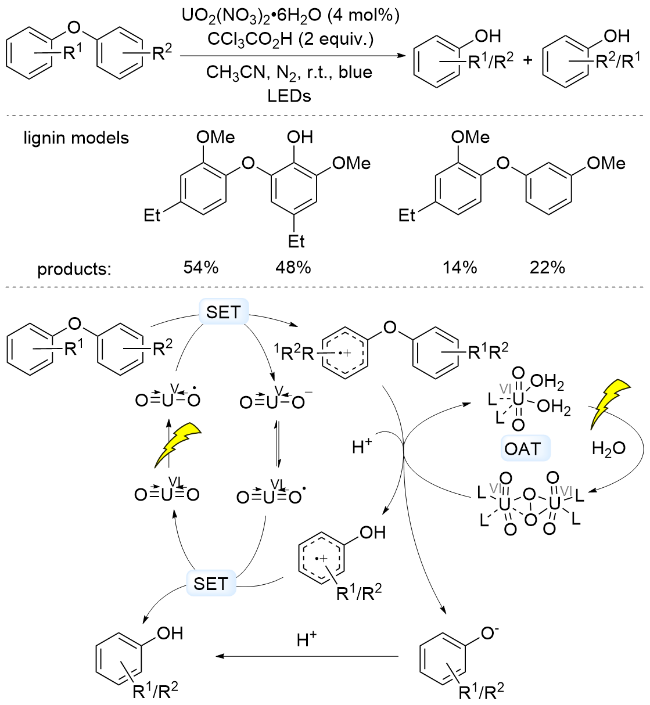
2021年, 姜雪峰课题组[75]采用UO2(NO3)2•6H2O为光催化剂, 在2 equiv. CCl3CO2H存在下酸性体系中, 实现了二芳基醚水解产生两分子酚类的转化. 作者通过自由基捕获、同位素标记等实验, 提出的反应机理为: 光照条件下, 二芳基醚还原淬灭铀酰阳离子(VUO2•), 形成芳基阳离子自由基, 随后二苯醚阳离子自由基与铀酰过氧化物发生OAT过程, 将水中的氧原子转移到产物中, 得到中间体芳基阳离子自由基和苯氧负离子. 芳基阳离子自由基单电子氧化VIUO2•转化为酚. 另一方面, 形成的苯氧负离子和质子结合也得到酚(Scheme 18). 该方法能够用于4-O-5连接的模型底物, 能够以低到中等收率得到酚类产物. 此外, 姜雪峰课题组[76]报道了采用UO2(OAc)2•2H2O为光催化剂, 在可见光照射下实现了苯基苄基醚C—O键断裂, 产生酚类产物. 以上研究体现了铀盐催化剂在C—O键活化方面的应用潜力.
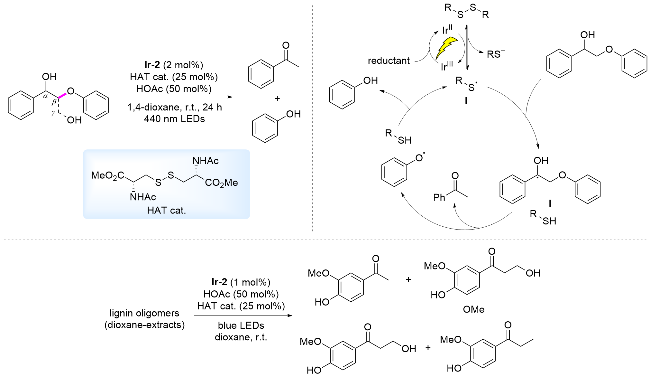
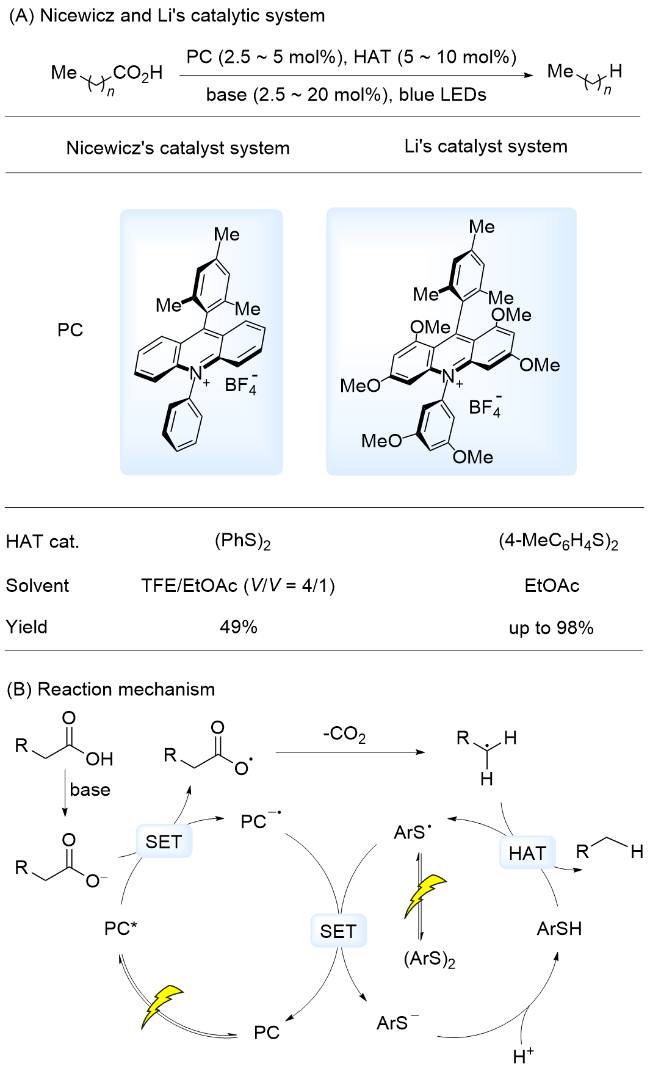
油料作物、动植物油通过衍生化能够得到大量的脂肪酸, 该脂肪酸的链状结构与柴油等燃料中烃类相似, 被视为生产可再生柴油和喷气燃料的理想原料[77]. 然而, 在热催化过程中, 通常需要苛刻的条件[t≥250 ℃, P(H2)≥2 MPa]才能将其转化为烃类燃料[78-79]. 光催化作为一种温和的转化方式, 通过SET过程, 脂肪酸可被氧化为相应的羧基自由基, 该自由基迅速脱除二氧化碳, 生成烷基自由基, 该自由基能够从体系中获得一个氢原子生成烷烃. 2015年, Nicewicz课题组[80]报道了以吖啶盐为光催化剂, 二苯基二硫醚(PhS)2为HAT催化剂, 对多种羧酸进行转化得到脱羧烷基化产物(Scheme 19A). 作者[80]以正十三烷基羧酸作为长链脂肪酸例, 生成相应烷烃的产率为49%. 2022年, 我们课题组[81]采用一种含有多个甲氧基的吖啶盐作为光催化剂和(4-Me- C6H4S)2作为HAT催化剂, 高效实现了脂肪族羧酸脱羧生成烷烃和氘代烷烃的转化, 产率可达98%. 提出的反应机理为: 碱促进羧酸产生羧酸阴离子, 在光照条件下, 该羧酸阴离子还原淬灭激发态光敏剂, 形成PC-•和羧基自由基, 羧基自由基诱导脱羧形成烷基自由基. 同时, (ArS)2在可见光照射下, 诱导硫硫键均裂生成ArS•, PC-•将硫自由基被还原为ArS-, 光催化剂再生. ArS-获得H+形成ArSH, 随后, 烷基自由基与ArSH发生HAT, 形成烷烃, 完成催化循环(Scheme 19B).
2 非均相光催化生物质相关转化
在非均相生物质催化转化体系中, 因光催化剂易于从反应体系中分离, 可实现循环利用, 因此, 能最大限度地降低能耗并减少废物产生等优点, 受到广泛关注[82-84]. 半导体材料因其具有适合生物质转化的带隙宽度, 被广泛应用于光催化领域[82-85]. 一般来说, 半导体吸收光能被激发, 电子从价带(VB)跃迁至导带(CB), 进而在价带处留下正电荷, 该正电荷也被称作空穴[82-85]. 随后, 电子和空穴转移至半导体表面, 并在表面发生氧化还原反应. 生物质中含有大量的C—H和O—H键, 在已报道的非均相催化转化过程中, 主要通过空穴或活性氧物种活化C—H和O—H键, 进而生成活性较高的碳中心或氧中心自由基, 随后引发一系列转化.
基于非均相催化体系相关研究更加关注光催化剂本身对生物质转化的影响, 本节将按照催化剂类型进行综述, 主要介绍TiO2等金属氧化物、CdS等金属硫化物及其他半导体材料在生物质转化中的研究进展.
2.1 TiO2等金属氧化物类光催化体系
早在1980年, Kawai与Sakata研究团队[89]报道了采用RuO2/TiO2/Pt为光催化剂, 在氙灯(500 W, λ<320 nm)照射下, 在6 mol/L NaOH溶液中, 首次实现了光催化纤维素水合制氢, H2产量为244 umol, 制氢效率为0.041 mmol•g-1•h-1, 该过程需要大量的RuO2/TiO2/Pt. 自此, 基于TiO2的金属氧化物类催化剂, 如Pt/TiO2[90-92]、Cellulose@TiO2(Pt)[93]、TiO2/NiOx@Cg[94]、TiO2/Pt[95-96]和TiO2-1[97]等广泛应用纤维素或木质纤维素制氢, 但均需高功率氙灯或紫外灯为光源, 且H2产量及制氢效率低. 为了实现温和条件下生物质高效制备能源物质及化学品, 大量研究者以多元醇或模型连接探究其反应路径[98-100].
2017年, 金放鸣课题组[101]报道了以纳米TiO2为光催化剂, 以125 W高压汞灯为光源, 在0.03 mol/L NaOH溶液中, 将葡萄糖转化为HCO2H, 产率为35%, 该方法也能够用于木糖到HCO2H的转化. 机理研究表明, 碱性条件下有利于活性氧物种($\mathrm{O}_{2}^{-·}$, •OH)的形成, 氢氧根还能调节TiO2表面的电荷分布, 并控制葡萄糖的吸附和HCO2H解吸.
2020年, 王峰和王敏研究团队[102]报道了采用TiO2纳米棒负载的铜Cu/TNR为光催化剂, 在365 nm LED灯照射下以H2O-CH3CN为混合溶剂, 将甘油转化为合成气(CO+H2)及甲醇. 多元醇和各种糖类均可转化为合成气及甲醇. 控制实验结果表明, 通过调控Cu的负载量, 能够有效调控产物分布. 以甘油为例, 提出的反应机理为: 在光照条件下, 甘油发生C—C键均裂, 产生乙二醇自由基和甲醇自由基, 进而转化成乙二醇和甲醇. 生成的乙二醇可以发生进一步催化发生C—C键均裂, 生成甲醇, 也可以被氧化为羟基乙酸和乙二酸. 羟基乙酸发生脱酸反应生成甲醇和CO2, 而乙二酸分解生成CO2和H2. 生成的甲醇在体系中进一步被氧化, 生成HCHO、HCO2H直至分解产生CO, CO2和H2 (Scheme 20).
2022年, 王敏和王峰研究团队[103]报道了TiO2的(101)和(001)晶面上光生电子向吸附物转移的机制, 揭示了TiO2材料催化过程中的电子转移具有表面依赖性. 即光生电子在TiO2的(101)晶面上被浅度捕获于Ti5c原子上, 而在(001)晶面上则被深度捕获于次层Ti6c原子上. 这种截然不同的电子陷阱状态对电子向吸附物的转移产生了重要影响. 基于这一特性, 作者通过调控TiO2晶面, 达到了调节糖类及多元醇在光催化氧化过程中选择性转化为CO或HCO2H. 推测体系中产生的HCO2H被光催化剂表面的空穴氧化形成•CO2H, 在(101)晶面上被浅度捕获的电子容易和质子结合产生活性氢, 易于结合•CO2H分解产生的•OH, 生成H2O, 从而促进HCO2H转化为CO; 而(001)晶面上深度捕获的电子则难以促进•CO2H向CO转化, 从而使HCO2H稳定存在.
2023年, 王敏课题组[104]采用TiO2为光催化剂, 通过晶相控制实现了多种生物多元醇选择性转化为HCO2H或CO. 在金红石相TiO2上生物多元醇容易氧化为HCO2H, 而HCO2H在TiO2锐钛矿相上进一步脱H2O生成CO. 作者通过原位傅里叶变换红外光谱(FTIR)和DFT计算研究了HCO2H在TiO2表面的分解过程, 揭示晶相依赖的选择性本质上源于HCO2H在不同晶面上吸附构型的差异. 该研究为设计用于生物质转化的光催化体系开辟了新的视野.
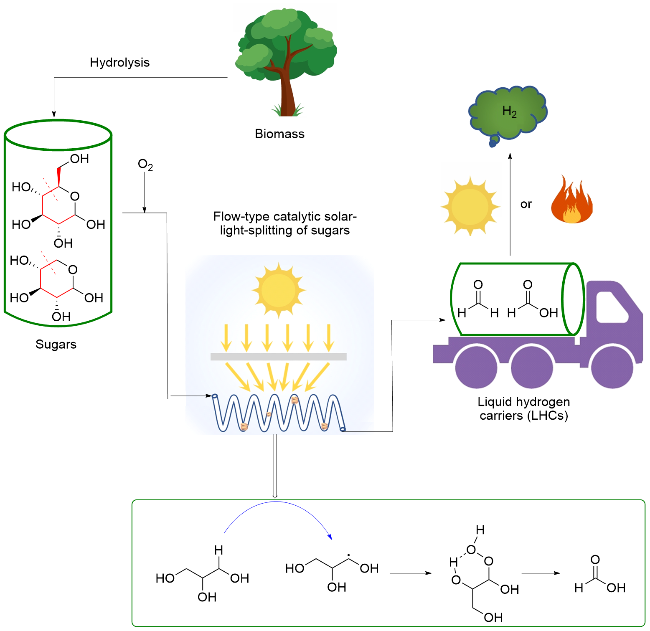
2023年, 王峰、罗能超和Fornasiero研究团队[105]报道了采用Ta掺杂的CeO2 (Ta-CeO2)为光催化剂, 优先断裂生物质衍生物C—C键, 将其转化为液态储氢载体(LHCs, 以HCO2H为主要成分, 含有少量HCHO), 之后按需通过光照或加热条件释放H2, 该转化实现了高效生物质制氢, 同时兼顾了储氢过程. 提出具体转化路径为: 在光热条件下, Ta-CeO2催化单糖、多元醇等碳水化合物形成相应的碳中心自由基, 进而被$\mathrm{O}_{2}^{-·}$捕获, 结合质子后形成高活性六元环过渡态, 其过氧键均裂引发 C—C键断裂, 同时脱除一分子水, 产生HCO2H. 在该光氧化过程中, 作者通过加热来抑制Ta-CeO2表面自由基偶联副反应, 随后通过Pt/P25光催化或Ru络合物热催化分解制氢(Scheme 21). 这种先将葡萄糖优先降解到LHCs, 再制氢的方法制氢产率高于直接光催化葡萄糖制氢产率.
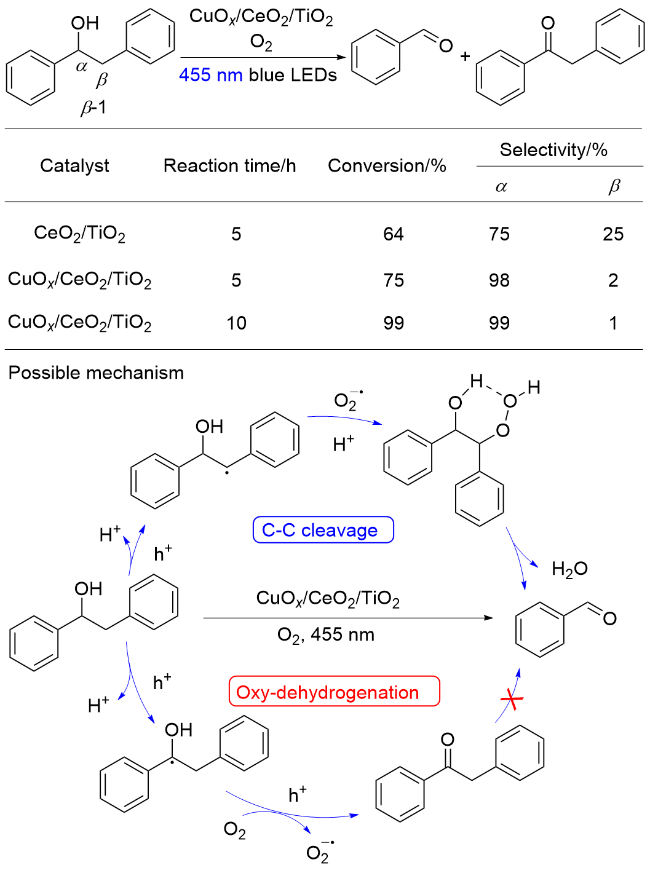
除了催化多元醇及纤维素制备燃料外, 基于TiO2的金属氧化物类催化剂在木质素模型分子转化方面也表现出一定的催化活性. 2017年, 王峰课题组[106]报道了以锐钛矿型CuOx/CeO2/TiO2为光催化剂, 以O2为氧化剂, 在455 nm波长的LED灯照射下, 实现了木质素β-1模型分子中C—C键断裂, 生成芳基醛类产物和苄位羟基被氧化的酮类副产物. 提出的反应机理为: 在光照条件下, 光催化剂选择性活化模型分子中Cβ—H键, 形成Cβ自由基, 该自由基被体系中形成的$\mathrm{O}_{2}^{-·}$捕获, 形成六元环中间体, 过氧键均裂引发Cα—Cβ键断裂, 消除一分子H2O, 生成苯甲醛(Scheme 22). 此外, 体系中的次要反应路径为优先活化Cα—H键, 随后被氧化得到酮类产物. 对催化剂进行研究表明, 催化剂价带最大值(VBM)主要由Cu的3d轨道构成, 光生空穴主要转移到CuOx纳米簇; CuOx纳米簇与CeO2紧密接触, 提高了CeO2上活性位点氧空位(VO)的浓度. 因此, CuOx/CeO2/TiO2催化剂在催化木质素β-1模型C—C键断裂时表现出显著的催化效率和选择性. 同时, 作者通过DFT计算对关键步骤进行了验证.
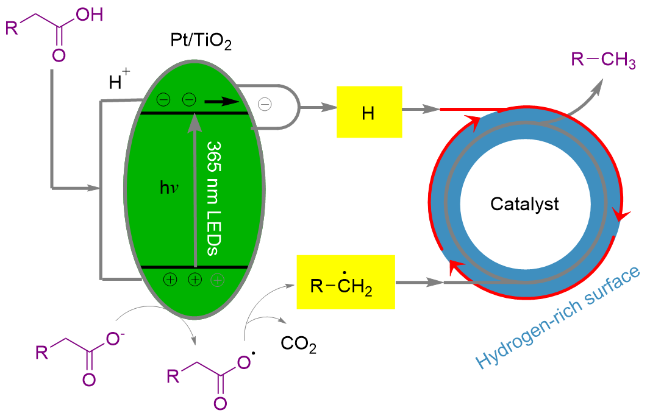
长链烷烃是石油和燃料的主要成分, 以生物质衍生的脂肪酸为原料制备长链烷烃有望缓解能源危机, 促进可持续发展[77]. 2020年, 王峰课题组[107]报道了以Pt/ TiO2为光催化剂, 以365 nm波长灯为光源, 在0.1~0.2 MPa H2氛围中实现了多种长链脂肪酸高效转化为相应的Cn-1烷烃. 提出的反应机理为: 在光照条件下, 体系中电离出的羧酸负离子被光催化剂氧化, 形成羧基自由基, 脱羧后产生烷基自由基. 另一方面, H2在该体系中被铂纳米颗粒活化解离, 在该体系中形成富氢表面, 生成的烷基自由基迅速加氢生成Cn-1烷烃, 抑制了副反应的发生(Scheme 23). 该方法能够应用于以大豆油精炼和制浆工业的副产品为原料制备长链烷烃.
2.2 金属硫化物类光催化体系
2017年, Reisner课题组[115]报道了采用CdS/CdOx为光催化剂, 在可见光照射下, 在10 mol/L KOH溶液中, 实现了光催化木质纤维素重整制氢, 产氢效率为2.570 mmol•g-1•h-1. 研究表明, 强碱性条件能够促进木质纤维素溶解, 同时还有助于在硫化镉表面形成氢氧化镉/氧化镉, 抑制了光催化剂发生光腐蚀, 显著提升了产氢速率. 该催化体系能够在太阳光照射下重整木质纤维素, 如木材和废纸, 发展了一种利用废弃生物质氧化驱动水合质子还原制氢气的途径.
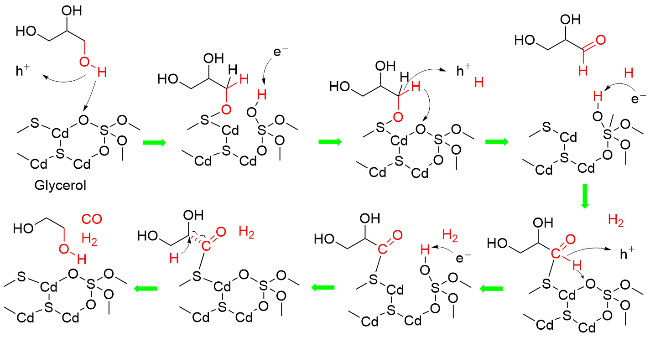
2021年, 王敏和王峰研究团队[116]报道了以[SO4]/ CdS为光催化剂, 以455 nm LED灯为光源, 催化甘油重整产生CO和H2, 气体生成速率分别为0.31和0.05 mmol•g-1•h-1. 该催化体系也能应用于糖类化合物, 如葡萄糖、果糖、麦芽糖、蔗糖、木糖、乳糖、淀粉等及胰岛素. 在催化剂制备中, 通过氧化CdS使其表面产生硫酸根离子[SO4], [SO4]不仅作为质子受体促进质子转移, 而且通过增加价带氧化电位促进了电子转移. 提出的反应机理为: 光照条件下, 以光催化剂产生的空穴为电子受体, 表面[SO4]为质子受体, 通过PCET过程活化甘油分子中O—H和C—H键, 使其转化为甘油醛. 同时, 质子被光生电子还原为氢原子, 转化为H2. 生成的甘油醛再次通过与光催化剂发生PCET过程, 活化其醛基C—H键, 形成酰基自由基. 酰基自由基脱羰基形成乙二醇和CO. 乙二醇被光催化剂吸附进行催化循环, 形成CO和H₂ (Scheme 24).
2022年, 王敏和王峰研究团队[117]以CdS@g-C3N4为光催化剂, 以O2为氧化剂, 在455 nm LED灯照射下催化甘油重整产生CO和H2, 气体生成速率分别为1.27和0.4 mmol•g-1•h-1, CO的产率可达到48%. 研究表明, 该催化剂在富氧条件下生成活性氧物种•OH, 有利于催化循环中醇羟基转化到醛基, 但浓度过高会使醛基氧化为羧基, 不利于CO的生成. 通过调节氧气与底物的比例, 既能加速反应进行, 又能避免底物过度氧化为CO2. 提出的反应机理为: 光照条件下, 甘油同时在空穴和•OH作用下形成甘油醛. 甘油醛在空穴氧化为酰基自由基, 脱除羰基产生乙二醇和CO. 乙二醇在催化剂空穴和•OH参与下被氧化为乙二醛, 在低氧浓度条件下, 乙二醛进一步被氧化转化为甲醛, 甲醛经空穴氧化产生CO和H2. 中间产物甘油醛、乙二醇、乙二醛和甲醛也能够被体系中•OH氧化, 最终分解为CO2和H2O (Scheme 25).
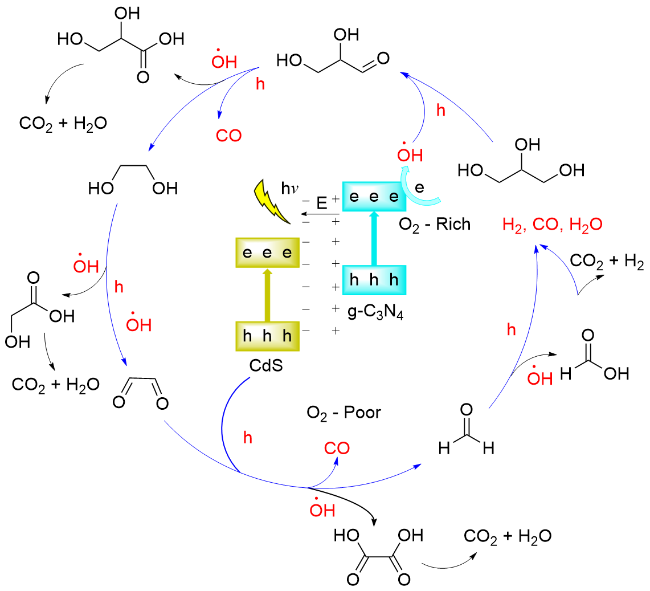
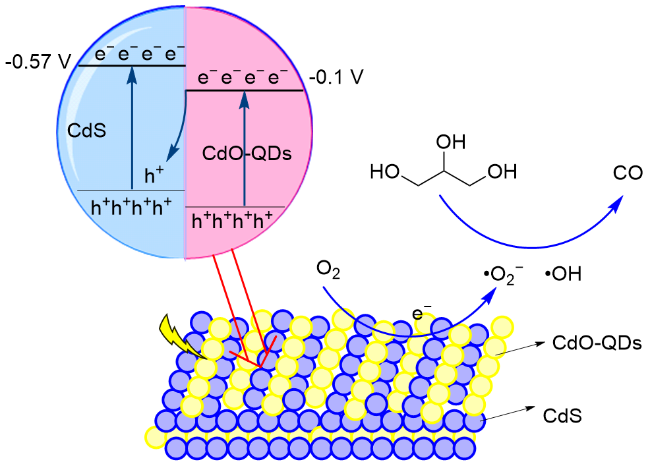
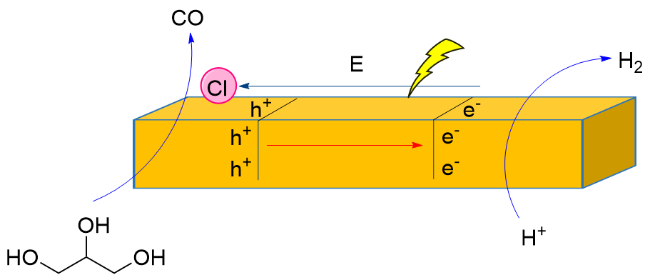
2023年, 王敏课题组[119]报道了以CdS-Cl为光催化剂, 以455 nm LED灯为光源, 催化甘油重整产生CO和H2, 气体生成速率分别为0.48和0.36 mmol•g-1•h-1. 研究表明, 通过向CdS表面吸附氯离子, 能够有效增加内部电场, 增强电荷分离并迁移至表面, 促进甘油重组制备合成气. 通过提高CdS内部电场强度, 增加生物质重整反应中光催化合成气产量. 提出的反应机理: 光照条件下会产生电子-空穴对. 空穴参与甘油的氧化以生成CO. 甘油的O—H或C—H键被活化并产生质子, 电子与质子相互作用生成H2 (Scheme 27). 氯化物吸附会产生内部电场, 从而增强光生载流子的分离, 促进了CdS- Cl上合成气的生成.
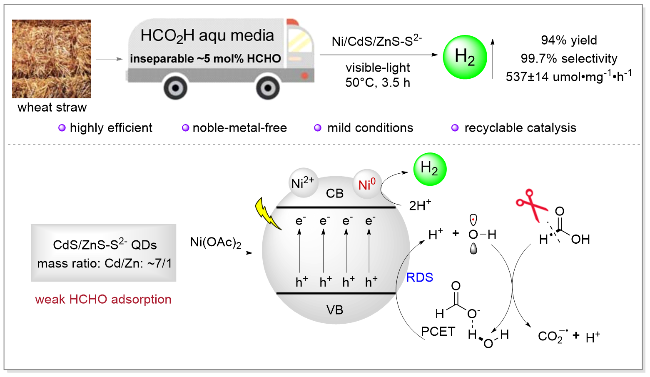
最近, 我们课题组[120]通过调控ZnS掺杂到CdS量子点表面的比例, 发展了能够兼容生物质HCO2H(从麦秆制备HCO2H)中微量甲醛的CdS/ZnS-S2-量子点光催化剂, 为生物质HCO2H制氢提供了必要条件(Scheme 28). 机理研究表明: 在可见光照射下, 价带通过PCET方式氧化H2O产生高活性•OH, 为反应的决速步骤, 产生的羟基自由基断裂生物质HCO2H的C—H键, 产生$\mathrm{CO}_{2}^{-·}$; Ni0作为主要活性位点还原H+产生氢气, 以537 mmol•g-1•h-1的制氢速率, 3.5 h达到94%氢气产率和99.7%的氢气选择性. 该催化体系无需贵金属、反应条件温和、催化体系通过光流体反应器可放大、可回收, 为开发利用原生生物质, 发展高效低成本的制氢体系提供了参考.
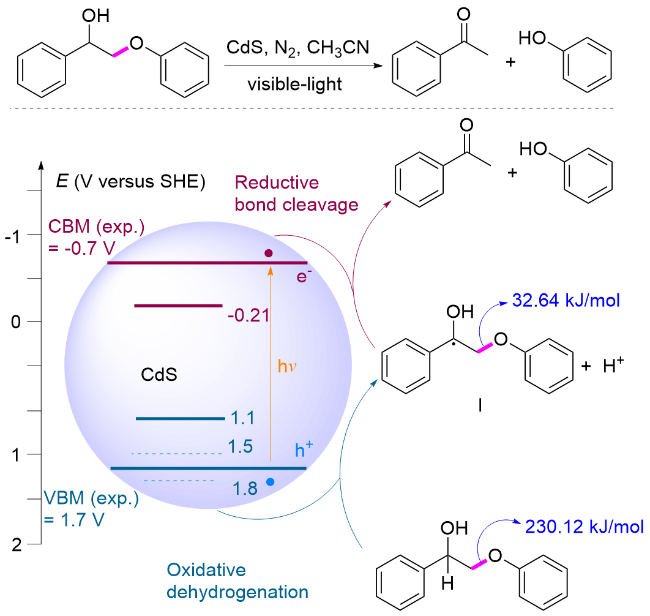
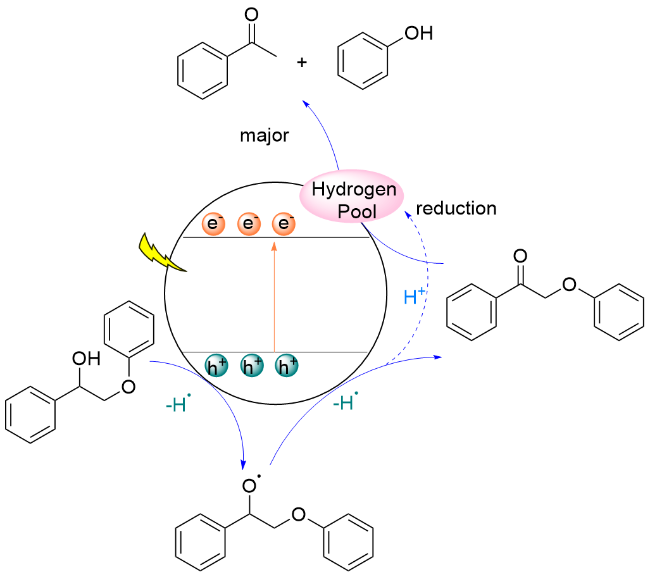
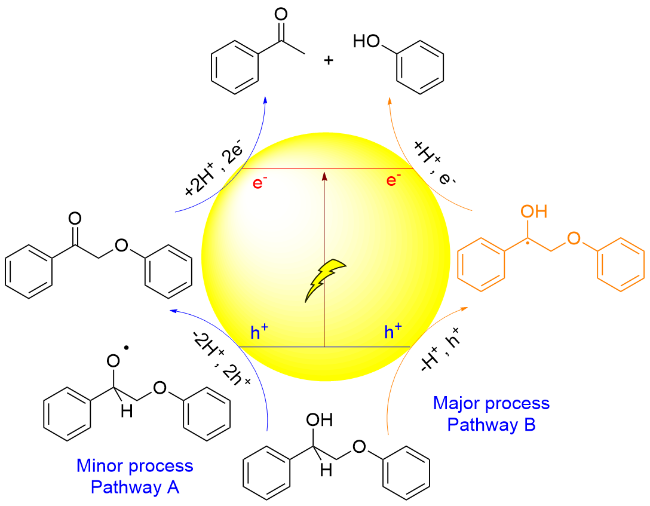
2018年, 王野、张庆红和程俊研究团队[127]报道了采用CdS QDs为光催化剂, 在可见光照射下实现了木质素β-O-4模型Cβ—O键断裂, 产生苯乙酮和苯酚类化合物. 作者提出了电子-空穴耦合(EHCO)的反应机理, 并通过机理实验及DFT计算进行验证, 具体过程为: 在光照条件下, 模型分子在催化剂形成的空穴被氧化, 形成Cα自由基中间体I, 该自由基诱导Cβ—O键键能大幅度降低; 同时, CdS的导带电位(-0.7 V vs SHE)和该反应还原电位(-0.21 V vs SHE)存在电位差, 促使Cα自由基中间体I在导带迅速还原断裂形成苯乙酮和苯氧负离子, 苯氧负离子得到质子形成苯酚(Scheme 29). 此外, 由于量子点的胶体特性使它能够和固体生物质紧密接触, 催化转化木质素, 同时几乎完好地保持了纤维素与半纤维素. 该催化体系能够用于木质素的转化, 以桦木木屑中提取的木质素为原料, 降解主要产生芳基酮类化合物, 收率达到27% (w). 该研究提供了一种选择性断裂β-O-4键的催化体系, 为在温和条件下通过“先木质素”策略实现木质纤维素的全面利用开辟了新的途径.
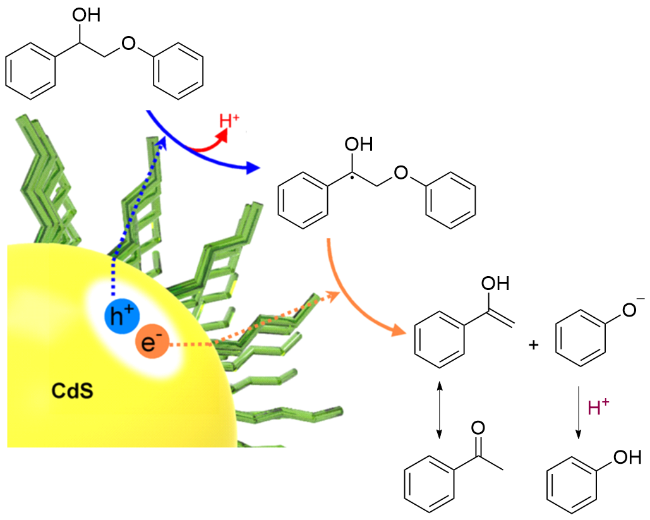
2019年, 王野、张庆红和王昭晖研究团队[128]进一步揭示了半导体CdS QDs的配体在生物质光催化转化中的重要作用. 以链状有机分子作为配体, 其亲水/疏水端促使CdS QDs形成胶体溶液, 使催化剂与原生木质素紧密接触, 从而促进其转化(Scheme 30). 配体的锚定基团和烷基链长度对催化体系中的电子传输有重要影响. 电子衰减过程的动力学研究表明: 配体中的电子转移通过电子隧穿路径进行, 随配体碳链长度的增加而呈指数下降趋势. 因此, 与其他不同锚定基团和不同长度烷基链的配体相比较, 以3-巯基丙酸为CdS QDs配体, 能够高效催化各种技术处理的木质素, 使其转化为芳香族化合物; 并且随β-O-4键含量的增加, 芳香化合物的产率呈现增长趋势.
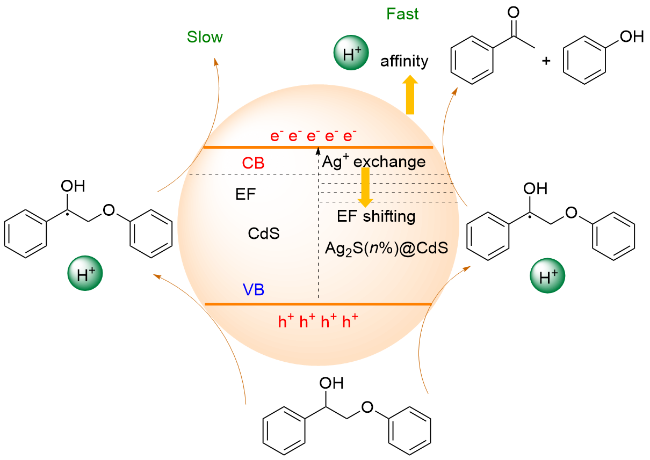
ZnIn2S4作为一种重要的三元硫化物半导体材料, 因其具有与可见光吸收相对应的带隙以及显著的化学稳定性, 已作为环保型光催化剂得到了广泛研究. 2016年, 王峰课题组[122]分别采用Pd/ZnIn2S4和TiO2为光催化剂, 通过切换两种光波长来实现β-O-4醇的串联氧化和加氢裂解反应. 自此, ZnIn2S4光催化剂在活化木质素β-O-4模型连接方面进一步得到应用.
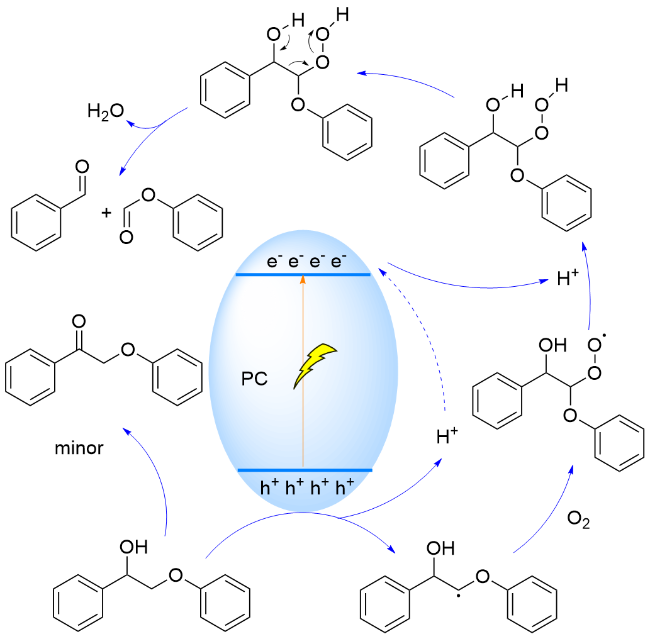
2019年, 王野和谢顺吉研究团队[132]合成了一系列ZnmIn2Sm+3催化剂, 通过调整Zn/In比例可以调节其能带结构. 研究表明, m值从1增加到6, 形成催化剂Znm-In2Sm+3的价带最大值(VBM)向更正的值移动, 从1.65 V vs. NHE提升至1.99 V vs. NHE, 而导带最小值(CBM)则向相反方向移动, 从-0.47 V vs. NHE变为-0.61 V vs. NHE. 因此, 在合成的催化剂中, Zn6In2S9因其具有最正的VBM和最负的CBM, 从而在395 nm光照射下对木质素β-O-4模型转化表现出高活性. 在EHCO机制中, β-O-4键的断裂需要光生空穴和电子的共同作用, 即光催化剂与木质素底物直接相互作用, 以产生关键的Cα自由基, 并驱动其后续转化(Scheme 34). 该研究为设计针对特定光催化反应的高效催化剂提供了重要的理论依据.
2.3 其他材料类光催化体系
随着光催化的发展, 开发高效、高选择性半导体材料作为光催化剂得到了广泛的关注. 石墨相氮化碳(g-C3N4)为一种能够吸收可见光且无金属的半导体材料, 因其成本低、面积大、吸光能力强及富含氮原子等特征, 已经应用于非均相光催化转化中. 由于木质素及模型分子结构较复杂且空间位阻较大, 将具有庞大苯环的分子吸附在刚性催化剂表面可能需要克服较高的能垒, 而有机二维g-C3N4的柔性则有可能缓解该问题. 2018年, 王峰课题组[133]报道了利用介孔石墨相氮化碳(mpg-C₃N₄)为光催化剂, 在455 nm波长的LED灯照射下, 有效断裂木质素β-O-4和β-1连接中的Cα—Cβ键(Scheme 35). 机理研究表明, g-C3N4表面与木质素模型分子之间存在紧密的π-π堆叠相互作用, 有助于光生空穴优先活化 Cβ—H键. 反应机理涉及光生空穴优先活化Cβ—H键生成Cβ自由基, 该自由基被O2捕获, 生成不稳定的过氧化物, 过氧键均裂引发Cα—Cβ键断裂, 同时消除一分子H2O, 生成苯甲醛和甲酸苯酯.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
聚酰亚胺作为一种非金属且低成本的聚合物光催化剂, 在能源和环保领域有着广泛的应用, 如光催化制氢和有机污染物的光降解等[134]. 与g-C3N4相比, 聚酰亚胺具有更深的价带位置, 表现出更强的氧化能力, 能够促进木质素转化中的脱氢过程, 实现木质素的高效降解[135]. 同时, 聚酰亚胺具有典型的供体-受体二元结构, 分别为富电子的胺和缺电子的酸酐, 这一结构对于防止电子-空穴快速复合起着至关重要的作用, 进而提高了其光催化性能[136-137]. 基于上述诸多优势, 聚酰亚胺在光催化活性木质素裂解方面具有较大的潜力. 2022年, 储升课题组[138]报道了利用聚酰亚胺(PI-BT)作为光催化剂, 在室温下通过可见光照射实现了β-O-4型木质素模型化合物C—C键的高效、选择性断裂. 机理研究表明, 这种具有高氧化能力的光生空穴主要引发优先生成Cβ自由基, 是整个转化过程中的决速步骤. 随后, Cβ自由基被O2捕获, 催化路径和上述过程类似(Scheme 35). 此外, 在转化过程中, 空穴可能与Cα—H或Cα—OH反应形成酮类化合物, 为反应的次要转化路径. 该催化体系对β-O-4模型底物C—C键的选择性和转化率大于99%.
3 总结和展望
随着化石资源日渐枯竭, 生物质作为一种大量丰富的可再生的资源, 其重要性日益凸显. 光催化具有绿色、温和的特征, 利用光催化进行生物质转化已受到广泛关注. 本文综述了近年来光催化转化生物质制备氢能、碳基能源及绿色化学品领域重要研究成果. 尽管已发展了众多催化剂与新反应路径, 取得了前所未有的发展, 但其催化效率及选择性仍难以满足实际应用需求, 因此未来的研究仍应积极致力于设计高效、高选择性的光催化体系, 以此提高反应转化效率, 增强应用潜能.
纤维素是非粮生物质中含量最高的成分. 然而, 由于其高聚合度及规整的氢键网络结构, 在温和条件下不溶于水、稀碱、稀酸及有机溶剂. 目前对纤维素的研究主要以单体葡萄糖为模板底物, 还需开发温和条件下纤维素解聚的高效方法, 为非粮生物质中纤维素的高效转化提供保障.
木质素也是非粮生物质中的一个主要成分, 富含芳香单元. 目前, 光催化断裂C—O或C—C键的研究主要集中于二聚体木质素模型化合物, 能够用于原生木质素高效转化的光催化体系十分有限. 此外, 现有的光催化体系通常只考虑C—O或C—C键的断裂, 限制了单体芳香族化合物的总产率. 为实现原生木质素的高效解聚利用, 未来应开发综合、高效的断键方法. 而且, 从原生木质素解聚得到的产品通常是多种成分的混合物, 分离纯产物消耗大量资源且成本高昂. 降低木质素产品复杂性的策略之一是将不同类型的混合物转化为具有相似功能或同类化学品, 为此需开发新的光催化体系以增强木质素转化的实用性.
随着光催化生物质高值化技术的蓬勃发展, 标准参数至关重要. 目前, 光催化生物质高值化的性能参数主要通过产品的收率和选择性来衡量. 量子效率也是评估光催化剂性能的重要参数, 报告量子效率有助于比较各种催化材料, 进一步指导光催化剂的设计和改进.
未来光催化生物质转化还需继续深入研究光催化机理, 通过理论计算与实验相结合的方式, 优化光催化剂的结构和性能. 例如, 设计具有特定能带结构和表面性质的光催化剂, 提高对可见光的吸收和利用效率. 另一方面, 加强光催化体系的集成与创新能够将光催化与其他技术(如生物技术、电化学技术等)相结合, 构建多功能的复合体系, 提高生物质转化的效率和选择性. 同时, 应注重开发绿色、可持续的反应介质和工艺条件, 减少对环境的影响. 此外, 还应加大对光催化设备的研发投入, 提高其稳定性和可靠性, 降低成本, 为光催化技术的大规模应用奠定基础.
(Cheng, F.)