


磺酰氟(SO2F)是一种重要的含氟结构单元, 由于其独特的稳定性和反应活性, 在药物化学、材料科学、有机合成等领域应用广泛[1-4]. 磺酰氟能提高化合物的生物利用度和药效, 增强化合物的代谢稳定性和药物安全性, 同时作为重要的中间体用于合成其他具有实际应用价值的分子结构[5-7]. 许多科学领域对磺酰氟的高度需求大大刺激了含磺酰氟官能团化合物合成策略的蓬勃发展[3,7-9].
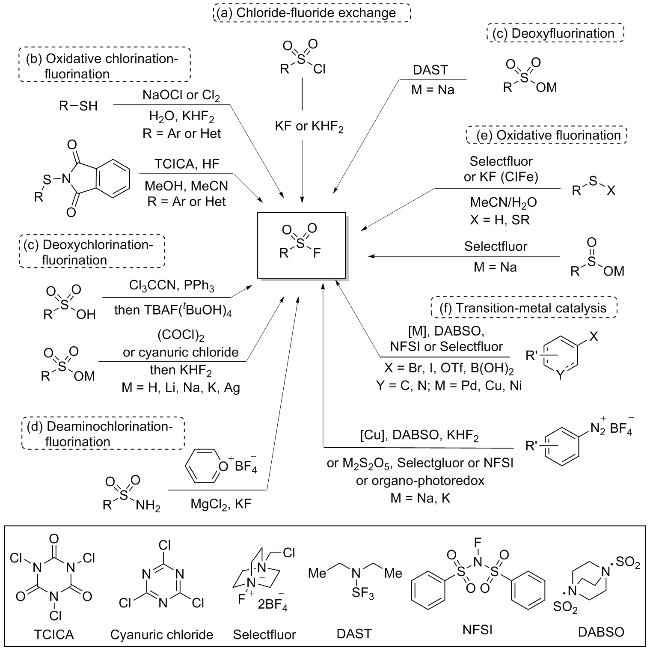
磺酰氟化合物常见的合成方法包括: 磺酰氯的氟-氯交换反应(Scheme 1a)[10-12]、硫醇氧化氯化氟化(Scheme 1b)[13-14]、磺酸及其共轭碱脱氧氯化氟化(Scheme 1c)[15-16]、磺酰胺脱氨基氯化氟化(Scheme 1d)[17-18]、硫醇或二硫化物氧化氟化(Scheme 1e)以及经过渡金属催化依次引入“SO2”和“F”(Scheme 1f)[19-27]. 尽管目前引入磺酰氟官能团的方法报道很多, 但大多受限于反应底物的适用范围和苛刻的反应条件[8].
Sharpless和董佳家[28]在2014年发表了关于点击化学的核心文章, 首次提出“六价硫氟交换[Sulfur (VI) Fluoride Exchange, SuFEx]”这一概念. 该反应以硫(Ⅵ)氟类化合物(硫酰氟)作为承载体可以通过简单高效的方式直接在分子结构中引入磺酰氟官能团. 这种高效高选择性反应的开发, 使得磺酰氟这种官能团的设计变得更加容易.
1 硫酰氟(SO2F2)
硫酰氟(Vikane, SO2F2)是SVI型氟氧化物家族中分子量最小的成员, 沸点为-55.2 ℃, 是一种无色、无味的气体, 比空气重3.5倍, 最早被广泛用作杀虫剂. 硫酰氟气体具有光热稳定性, 气体状态下保持相对惰性, 在干燥情况下400 ℃亦不分解; 在保持稳定性的同时, SO2F2又具有特定的反应性能.
1.1 硫酰氟参与酚类的点击反应
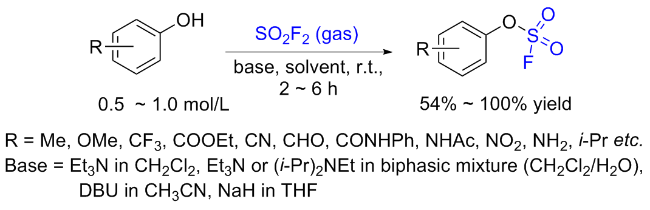
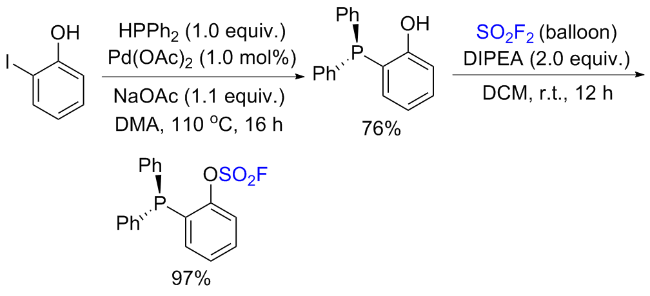
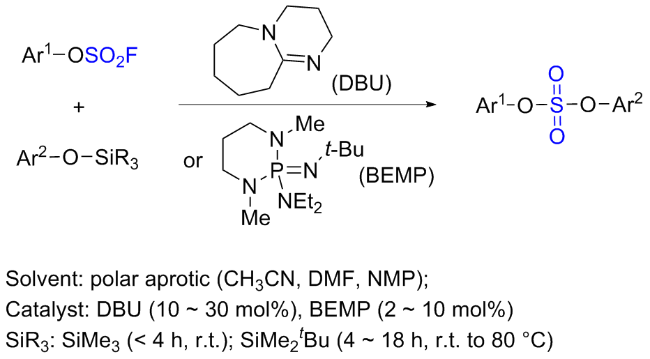
1.2 硫酰氟参与酚类的过渡金属催化反应
在硫酰氟气体应用于酚类合成氟磺酸酯之后, 关于这类拟卤代物过渡金属催化反应的相关研究不断增多, 这些反应使酚更容易完成各种化学转化变成结构复杂的分子.
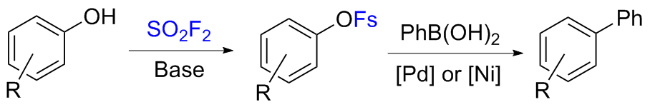
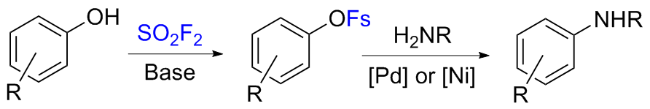
在2016年, Hanley课题组[37]报道了钯和镍催化芳基氟磺酸酯与芳香胺或烷基胺的胺化反应(Scheme 6). 通过钯金属和4,5-双二苯基膦-9,9-二甲基氧杂蒽(Xantphos)的组合催化, 将苯胺偶联到一系列不同的芳基氟磺酸盐上, 并与其他常见的芳基亲电试剂反应, 比较了芳基氟磺酸酯在Pd催化下胺化的相对反应活性. 此外, 作者通过与硫酰氟和碱反应原位生成芳基氟磺酸酯, 然后胺化形成新的C—N键, 从而实现苯酚的直接胺化. 最后, 作者还介绍了在乙腈存在下, Ni(COD)2和1,1'-双(二苯基膦)二茂铁(DPPF)联合催化芳基氟磺酸酯的胺化反应. 芳基氟磺酸酯亲电试剂与通用的钯和镍催化剂体系的高反应活性, 结合其由硫酰氟制备简单的特点, 使丰富的酚类原料实现商业化胺化反应成为可能.
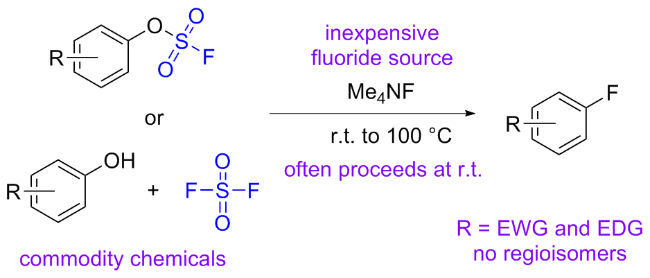

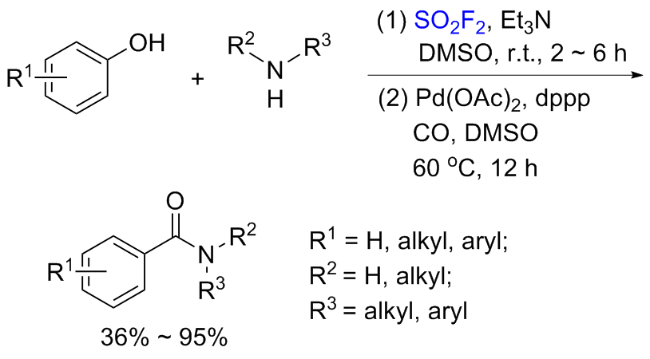
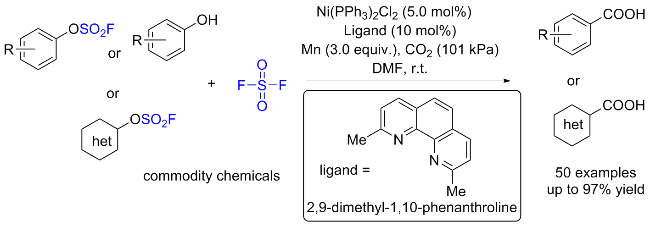
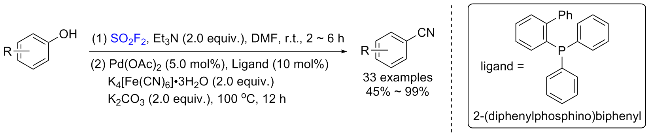
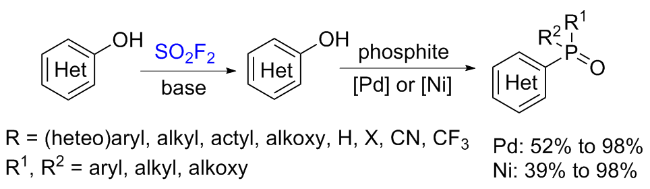
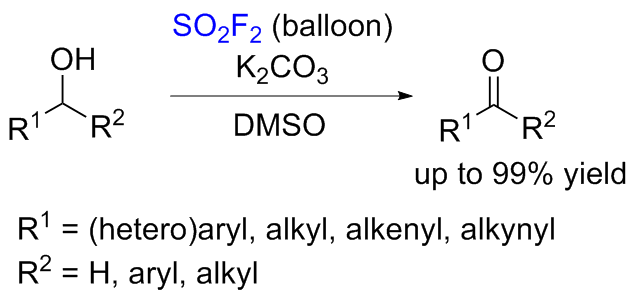
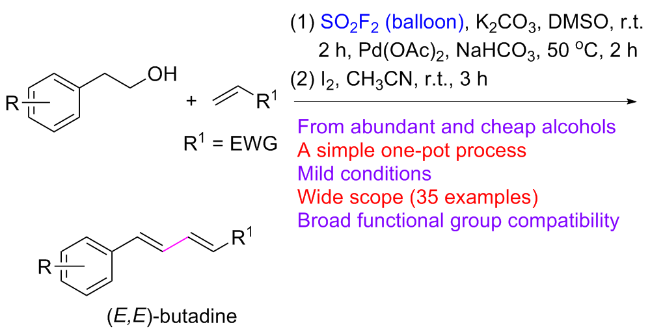
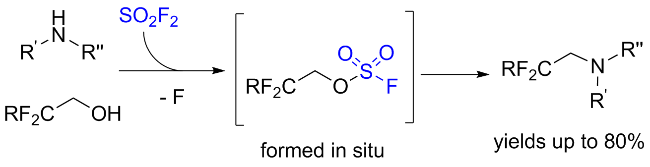
1.3 硫酰氟参与醇类化合物的化学转化
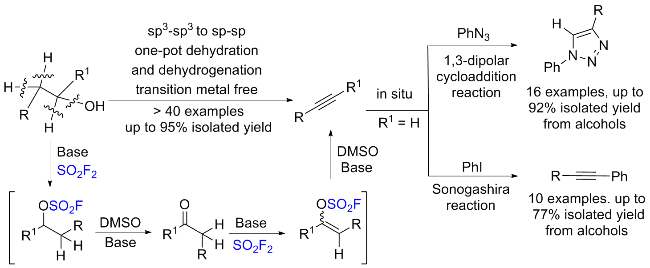
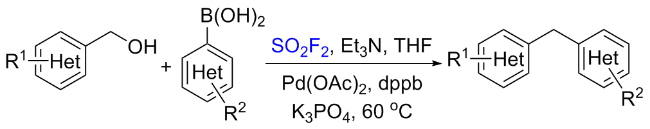

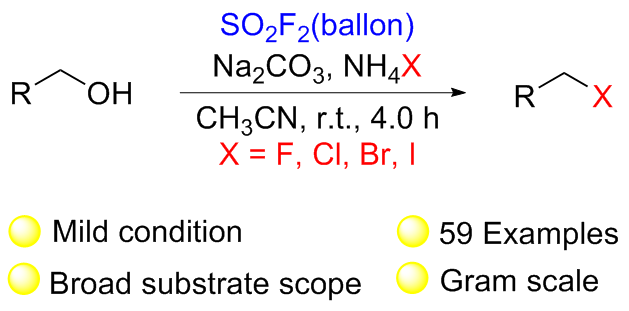
1.4 硫酰氟参与胺类化合物的化学转化
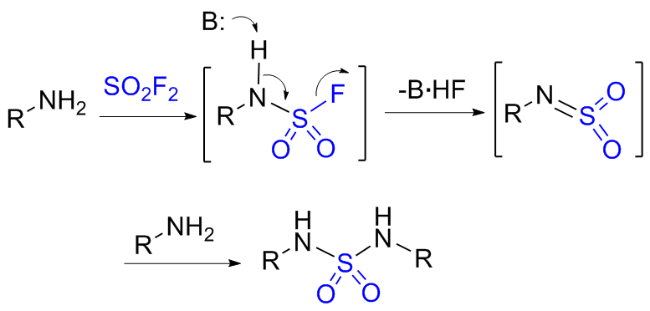
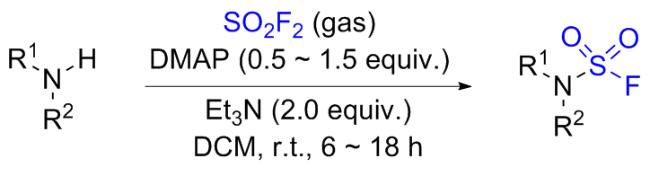
1.5 硫酰氟参与肟类化合物的化学转化
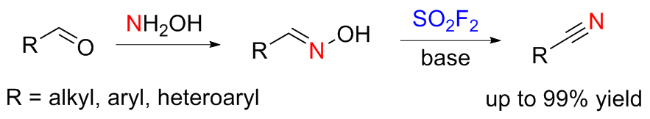
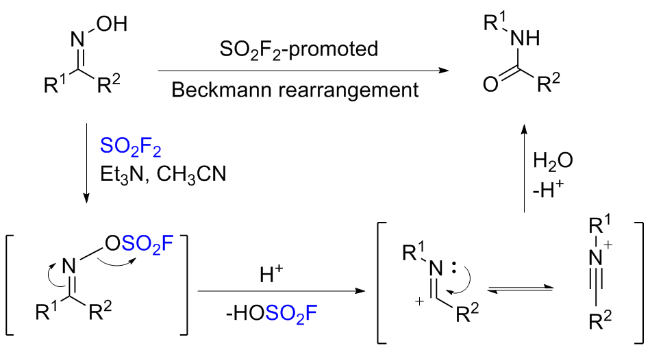
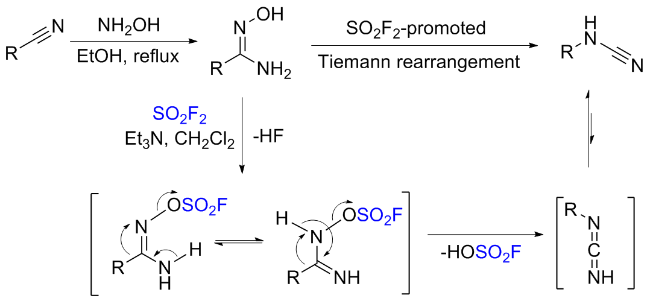
1.6 硫酰氟参与的其他反应
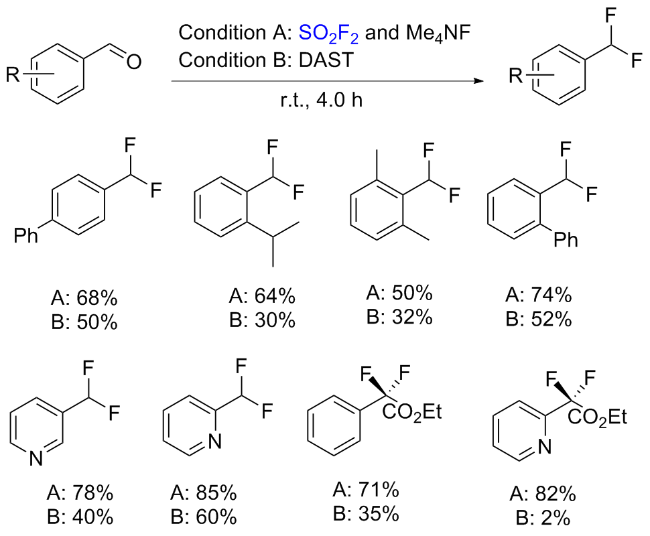

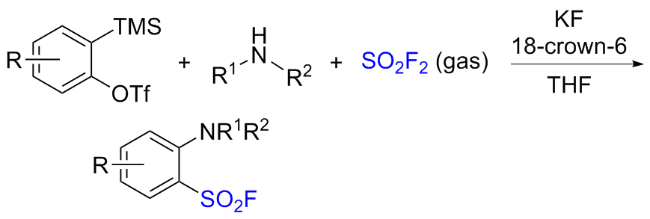

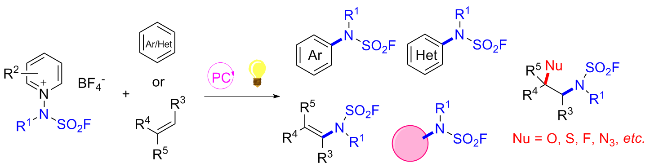
2 氟磺酰胺咪唑盐
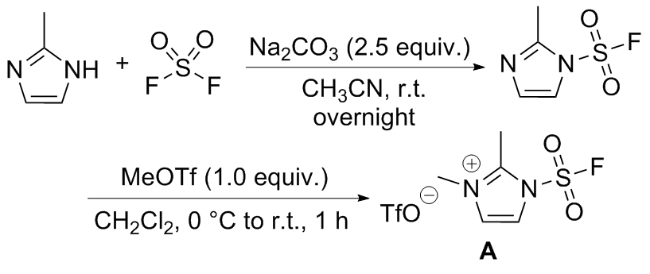
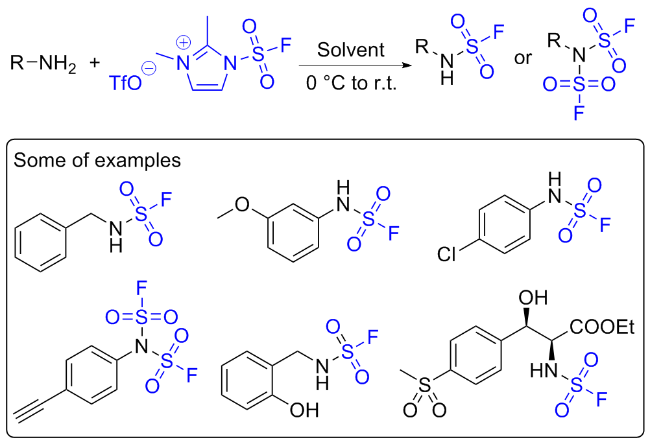
虽然SO2F2作为SuFEx试剂, 具有理想的反应性和选择性, 但在实验操作中, 作为温室气体需要注意气体泄漏的问题; 且与酚类反应相比, SO2F2与仲胺的反应较为缓慢, 生成氨磺酰氟的底物范围受限, 需要复杂的反应条件; 而与伯胺的反应得到氟磺酰胺后, 氮上氢质子由于在碱性条件下易发生消除, 进一步与伯胺反应形成对称取代的磺酰胺, 无法得到双氟磺酰胺[28,52]. 考虑到上述缺点, 且受磺酰基咪唑鎓盐试剂的启发[64-66], Sharpless和董佳家团队[33]开发了一种固态的氟磺酰试 剂氟磺酰胺咪唑盐. 以2-甲基咪唑为原料, 在碳酸钠作用下与硫酰氟发生SuFEx反应, 再在二氯甲烷溶剂中与三氟甲磺酸甲酯成盐, 获得1-(氟磺酰基)-2,3-二甲基- 1H-咪唑-3-鎓三氟甲磺酸盐(A) (Scheme 30). 该物质具有优异的热稳定性、反应选择性和高效性, 完美地克服了以上缺点. 下面对基于氟磺酰胺咪唑盐的化学转化做简单介绍.
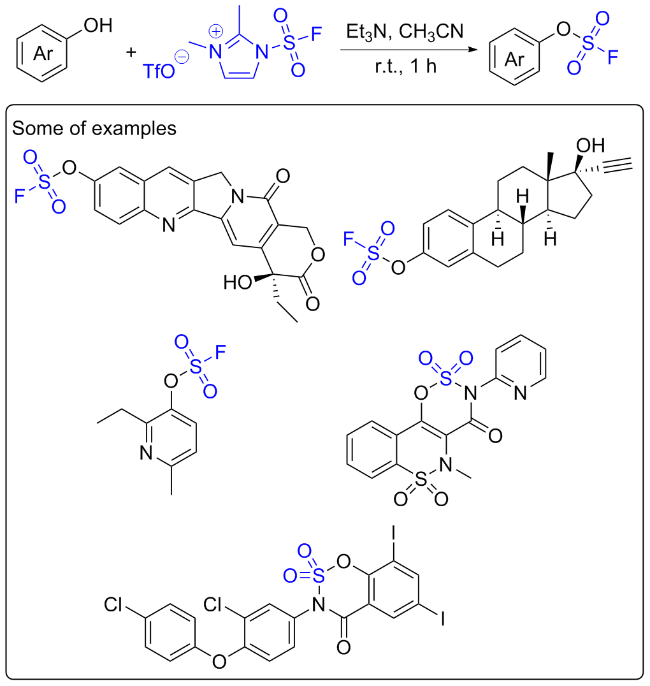
2.1 氟磺酰胺咪唑盐应用于酚的点击反应
2.2 氟磺酰胺咪唑盐作用于胺的反应
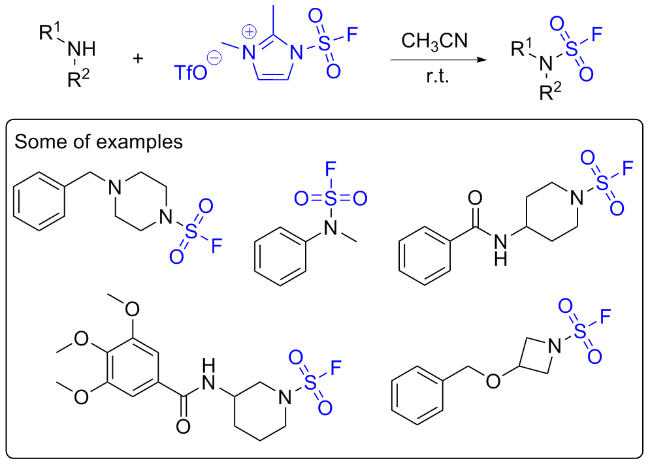
氟磺酰胺咪唑盐与伯胺独特的反应性能也为合成具有潜在生物活性的化合物N,N'-二取代的磺酰胺提供了新的途径(Scheme 34)[67]. 2021年, 徐汉虹课题组[67]利用此氟磺酰试剂, 通过硫(VI)氟交换点击反应高效合成了一系列新型的N,N'-二取代氨基磺酰胺衍生物作为农药候选物. 通过生物测定评估了目标化合物的杀虫和杀真菌活性. 初步结果表明, 靶分子具有良好的生物活性. 尤其是化合物D25对木霉菌的评估参数LC50为2.42 mg/L, 显著优于商用杀虫剂茚虫威(LC50=3.99 mg/L). 此外, 一些化合物对植物病原菌稻瘟病菌、灰葡萄孢菌和瓜亡革菌也表现出令人满意的杀菌活性. 该工作为杂环N,N'-二取代氨基磺酰胺作为新型农药的应用带来新的思路.
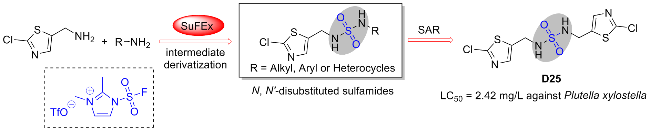
2.3 氟磺酰胺咪唑盐应用于叠氮化合物的合成
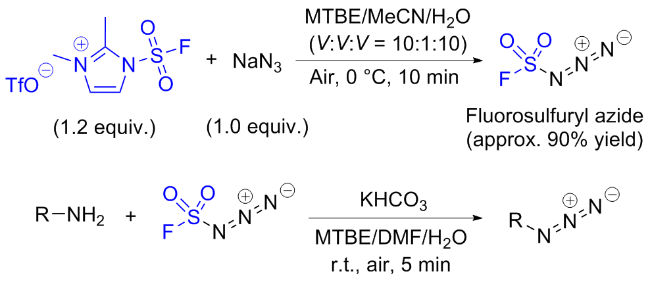
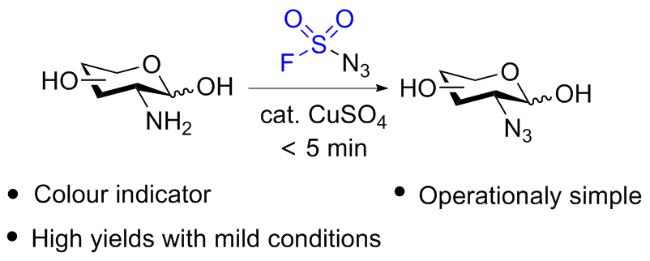
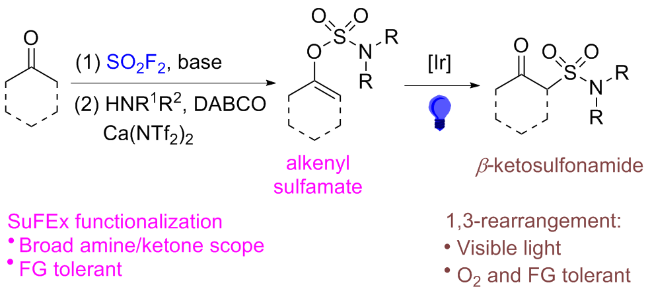
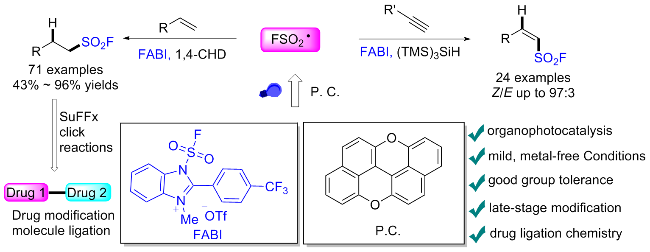
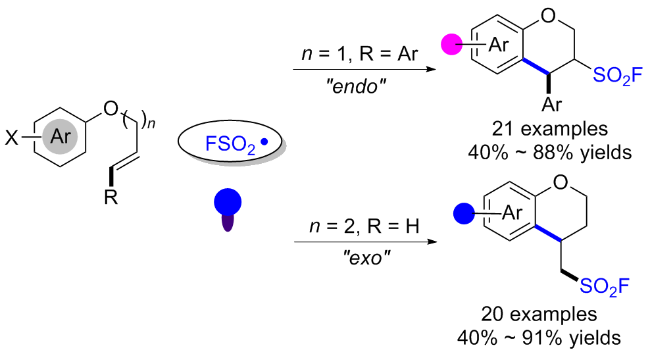
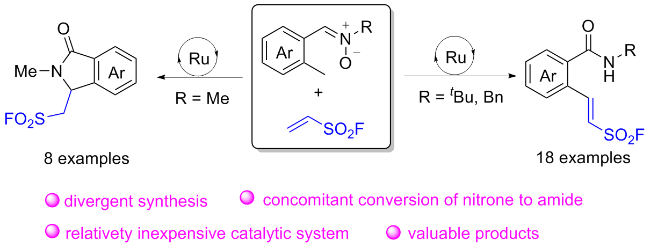
2.4 氟磺酰胺咪唑盐产生SO2F自由基的反应
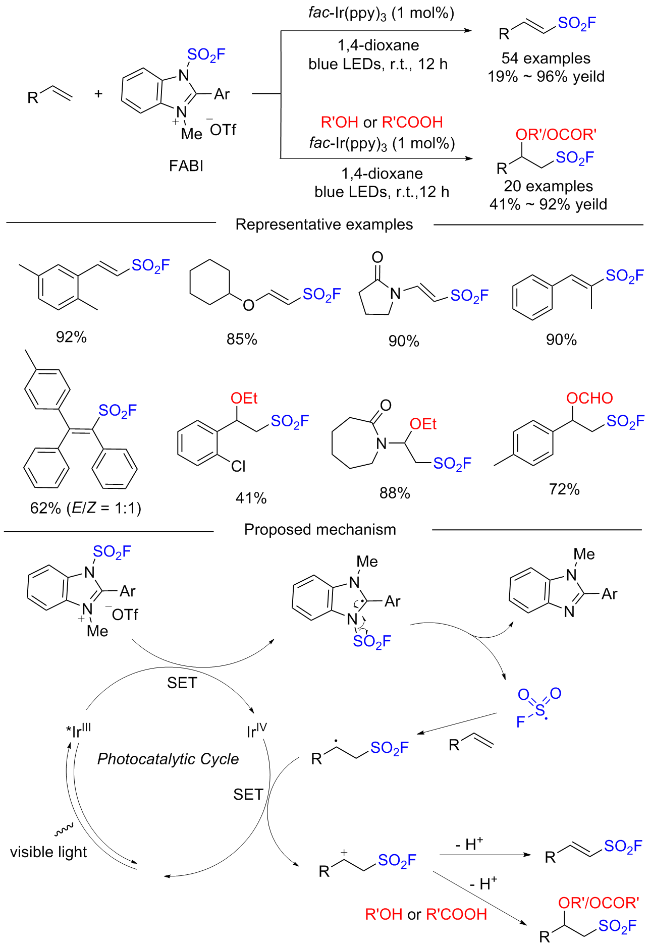
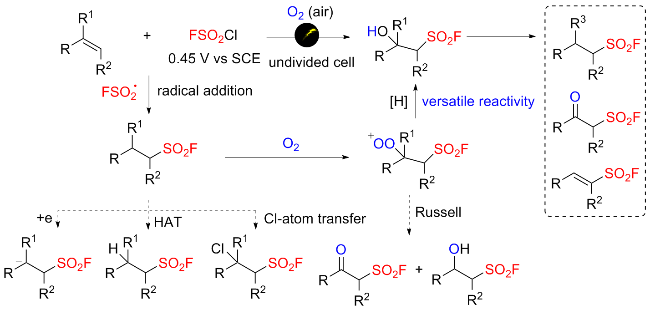
在此背景下, 廖赛虎及其同事[71]设计了固态氟磺酰试剂1-氟磺酰基苯并咪唑三氟甲磺酸盐(FABI)作为方便有效的氧化还原活性氟磺酰自由基前体, 以实现烯烃的自由基氟磺酰化(Scheme 37). 相较于已知的FSO2Cl, 该氟磺酰胺咪唑盐稳定且易于操作, 兼容多种烯烃底物, 充分证明了其优于FSO2Cl. 此外, 还开发了用FABI光氧化还原催化实现烯烃烷氧基-氟磺酰化反应. 通过使用合适的醇和酸作为亲核试剂, 可以得到相应的β-烷氧基磺酰氟, 收率中等到良好. 在实验结果的基础上, 提出了可能的反应机理, 即氧化还原活性自由基前体通过激发的Ir(Ⅲ)进行单电子转移, 得到中性自由基, 然后通过N—S键的均裂转化为氟磺酰自由基. 与加入的烯烃底物产生新的碳自由基, 该碳自由基经过Ir(Ⅳ)的单电子氧化形成碳阳离子中间体并再生形成基态Ir(Ⅲ)以完成光催化循环. 最终, 碳阳离子可以被去质子化或被亲氧试剂捕获得到相应的产物.
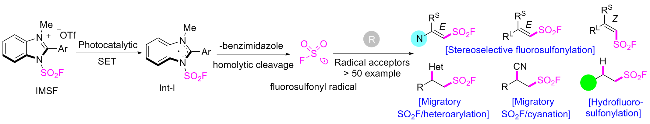
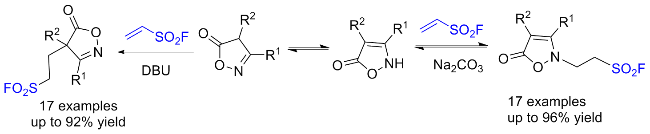
2.5 氟磺酰胺咪唑盐作用于肟类的反应
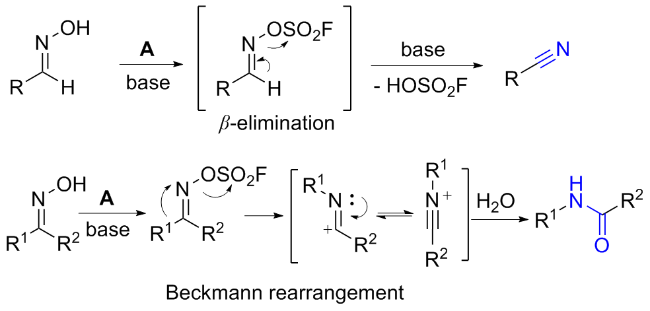
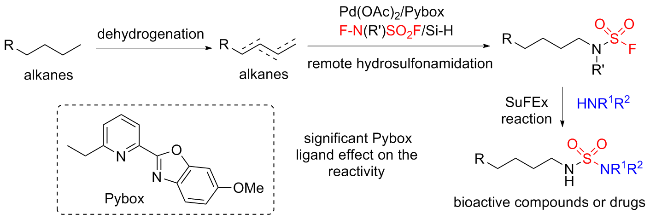
3 其他磺酰氟源
3.1 乙烯基磺酰氟(ESF)
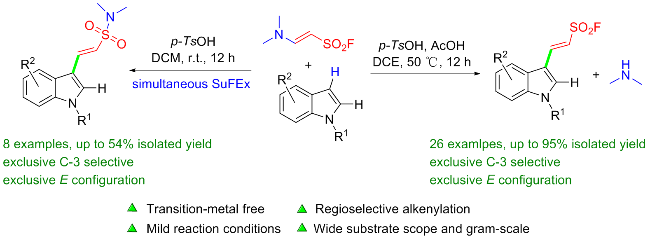
乙烯基磺酰氟(ESF)作为最重要的硫(VI)中心之一, 在硫点击化学和有机合成中表现出了非凡的反应活性. 2016年, Sharpless团队[76]通过氟氯交换先将2-氯乙烷磺酰氯转化成2-氯乙烷磺酰氟, 再经氧化镁实现β消除, 最终在水相中以高收率制得公斤级规模的ESF. 此方法大大降低了生产成本, 使作为SuFEx重要合成子之一的ESF在诸多领域的深入研究和大规模应用成为可能. ESF在点击化学、有机化学、材料科学、药物化学和许多其他领域有着重要的价值, 考虑已有相关综述详细介绍该试剂参与的反应[77], 因此, 我们仅对近几年来ESF的典型反应研究进行简要介绍, 其大多以Michael加成、Heck偶联和C—H键官能化等形式参与反应.
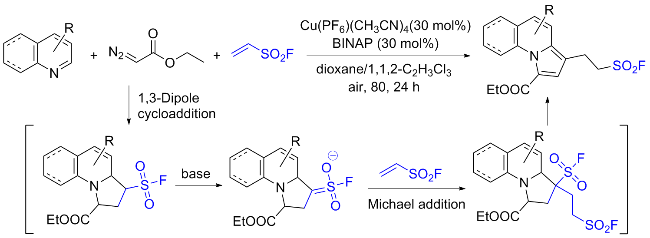
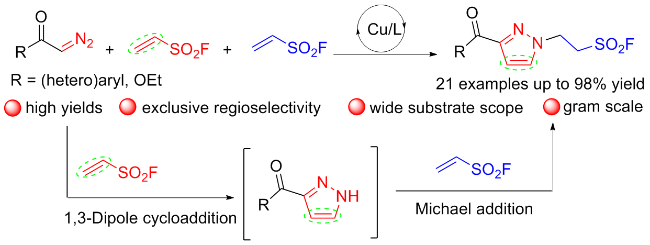
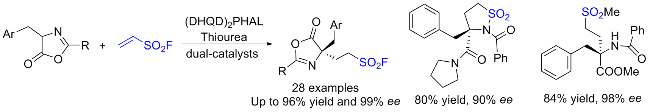
3.2 硫酰氟氯(ClSO2F)
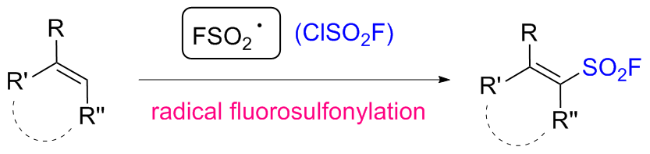
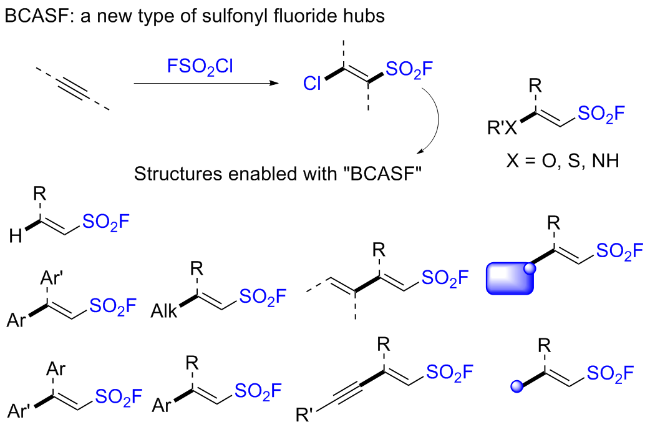
然而, 多取代烯烃的自由基氟磺酰化反应通常存在副反应、反应效率低、Z/E选择性低等问题. 为了解决这些问题, 该小组[71]随后开发了一种光氧化还原催化的炔烃自由基氯氟磺酰化反应, 用于构建β-氯烯基磺酰氟化合物(BCASF) (Scheme 50). 在最佳反应条件下, 端炔烃和内炔烃的转化反应顺利进行. 更重要的是, 该方法设计了一种新颖且功能强大的磺酰氟砌块, 可以转化成一系列官能化的烯基磺酰氟. BCASF分子可以在β-氯位点发生不同类型的转化, 同时保持磺酰氟基团的完整性, 例如铃木偶联, Sonogashira偶联, 选择性还原以及与氮、氧和硫亲核试剂的亲核取代. 此外, 这种多功能砌块还可以应用于多肽和药物的后期修饰.


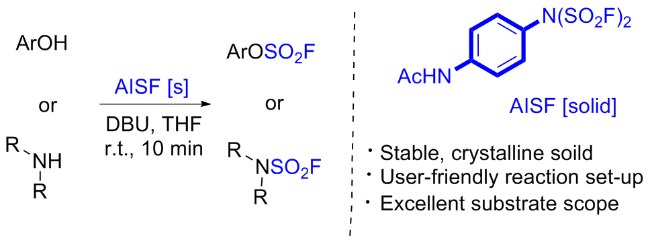
3.3 双氟磺酰亚胺类
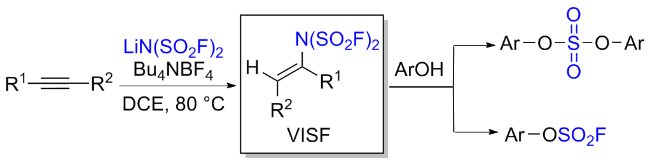
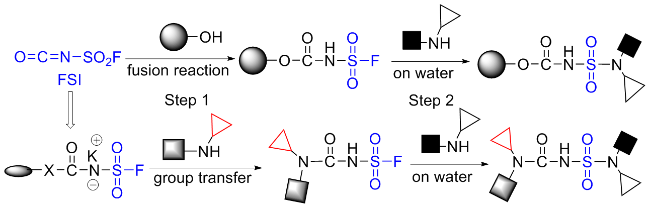
3.4 氟磺酰基异氰酸酯(FSI)
3.5 2-取代炔基磺酰氟(SASF)
除乙烯基磺酰氟外, 炔基磺酰氟也是一种良好的磺酰氟引入试剂. 2020年, Sharpless和Moses团队[88]描述了2-取代炔基-1-磺酰氟化物(SASFs)作为一种新型的连接枢纽, 并与1,3、1,5-耦极和二烯的环加成反应相结合, 经多样性导向的点击反应(diversity oriented clicking, DOC), 仅需最少的合成步骤, 即可构建一个由173种独特功能分子组成的多样化点击库(Scheme 57). 通过对侧链磺酰氟基团进行SuFEx点击衍生化, 利用96孔板实现了分子库的扩展, 形成278种离散化合物, 展示了DOC方法在合成多样化功能结构方面的灵活性. 2022年, 他们[89]还通过扩展SASF枢纽的点击路径, 成功将胺、1H-1,2,3-三氮唑、卤化物和羧酸的Michael加成反应结合, 进一步证明了DOC方法是一种有效探索功能分子的策略(Scheme 58). 利用DOC方法, 作者高效地合成了一系列前所未有的β-取代乙烯基硫酰氟, 反应过程仅产生单一的立体异构体. 与SuFEx反应相比, β-取代的乙烯基硫酰氟的反应活性可以通过选择不同的β-取代基来调节, 因此具有开发共价抑制剂的潜力. 此外, 作者还展示了SASF衍生物作为人类中性粒细胞弹性蛋白酶潜在共价抑制剂的生物活性.
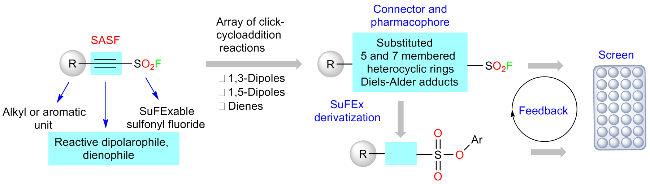
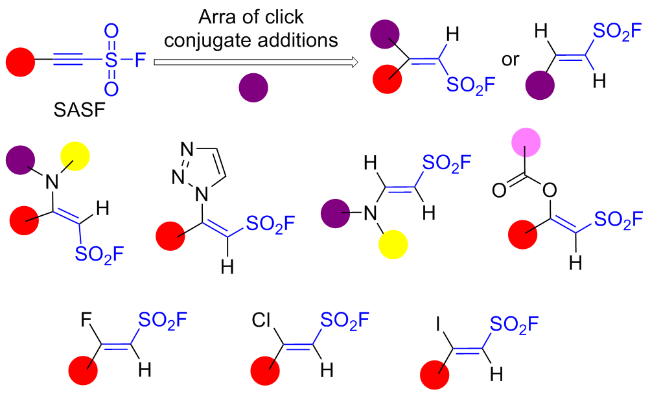
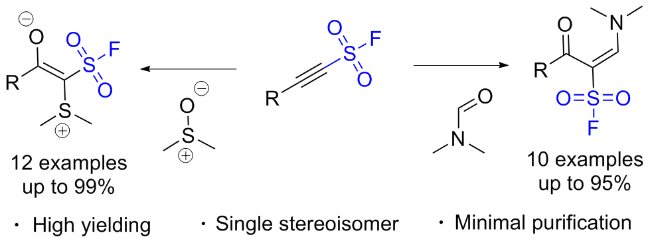
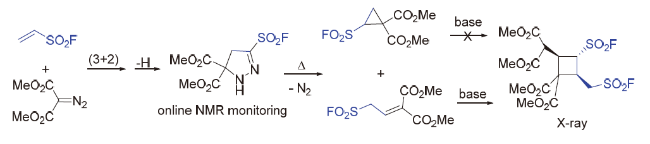
3.6 β-亚胺磺酰氟
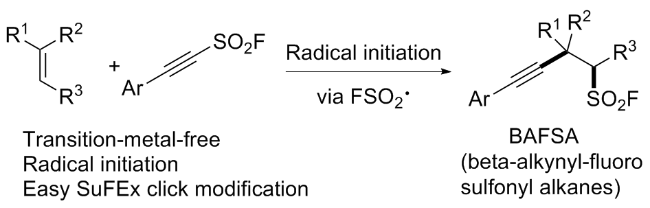
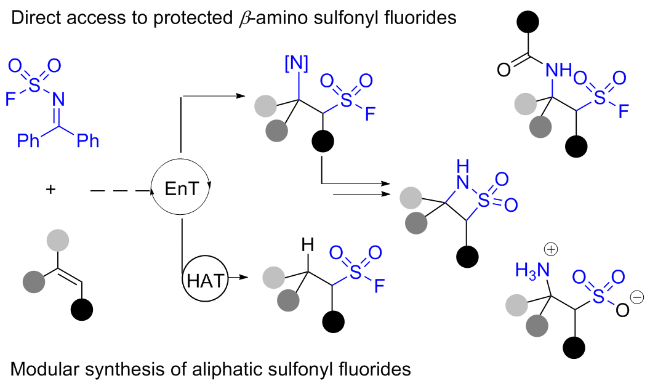
近来, Glorius团队[92]报道了一种基于自由基的过程合成含有脂肪族碳硫键结构砌块的方法(Scheme 61). 该过程中磺酰氟自由基来源于双官能化试剂β-亚胺磺酰氟, 它是一种能够实现长期储存、可在可见光诱导和光敏剂作用下发生σ键自由基解离的稳定氟磺酰试剂, 该试剂可以与烯烃反应形成一系列保护的β-氨基磺酰氟, 也可以通过单电子转移实现氢化. 值得注意的是, 该策略为药物化学相关的多肽磺酰氟化合物的合成提供了一种有效途径, 为从烯烃实现β-磺酸铵和β-磺内酰胺的合成提供了新的思路. 机理研究表明, 此转化涉及能量转移(EnT)介导的过程, 瞬态磺酰氟自由基加成至烯烃后, 分别经自由基-自由基偶联或氢原子转移(HAT)等过程获得相应的目标产物.
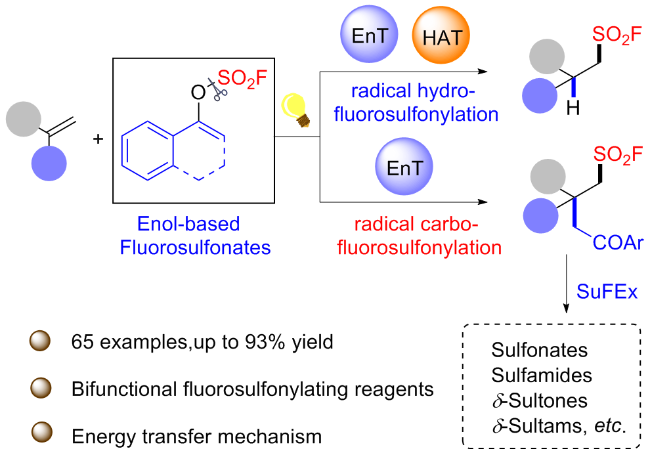
3.7 烯醇衍生的氟磺酸酯
3.8 N-氟磺酰基化合物
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
尽管近年来开发的多种磺酰氟源试剂应用于构建磺酰氟官能团, 但其往往受限于特有的底物及苛刻的反应条件, 且磺酰氟源试剂涉及温室气体, 或本身不稳定, 制备方式较复杂, 原子利用率较低. 因此, 有待发展更高反应活性和底物普适性的磺酰氟源, 同时兼顾绿色化学和原子经济性, 并进一步实现其更广泛的应用. 该综述仅对近年来有机合成领域进行整理总结, 磺酰氟化合物同样在材料科学、生物学、环境科学和药物化学等领域起着至关重要的作用, 对相关领域的归纳综述有利于多学科领域的发展. 磺酰氟化合物具有广阔的应用前景, 需要化学家们更多的努力以促进氟化学的长足发展.
(Cheng, F.)