


1 Introduction
The genus Rubus is one of the most species-rich (about 700 species) genera in the Rosaceae family, of which 194 species are found in the southwest of China and a few species have been used as medicinal herbs.[1] Rubus idaeus Linnaeus is a Chinese herbal medicine that has been widely used in China to nourish the liver, reinforce the kidney, reduce urination, and improve vision.[2] It’s not only a medicinal herb but also an ornamental. R. idaeus is a vine-like shrub that is 1~2 m high. The fruit of R. idaeus is nearly spherical, succulent and red with a diameter of 10~15 mm. Previous phytochemical investigations on R. idaeus have identified a diverse range of compounds, including flavonoids, phenolic acid, coumarins, tannins, lignans, terpenoids, alkaloids and steroids.[3-8] Among these compounds, many anti-inflammatory compounds were reported such as anthocyanins and ellagitannins.[9]
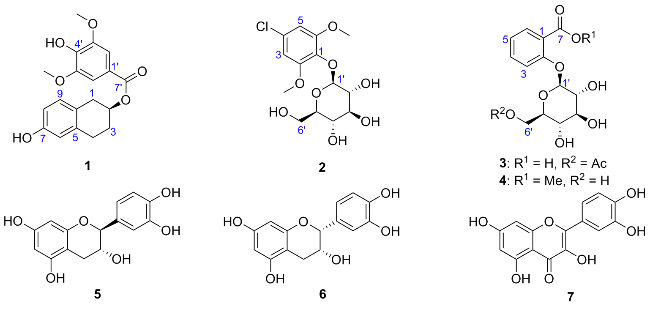
In order to search for significant bioactive natural products from the medicine-food plant R. idaeus, a new tetrahydrogenated naphthol syringic acid ester, named rubusno- licester (1), two new phenolic glycoside derivatives, 4-chloro-2,6-dimethoxylphenol-1-O-β-D-glucopyranoside (2) and salicylic acid-2-O-(6'-O-acetyl)-β-D-glucopyrano- side (3), together with one known salicylic acid glycoside derivative (4) and three known flavonoids derivatives (5~7), were isolated (Figure 1). Their structures were elucidated by HRESI-MS, NMR spectroscopy, and a comparison of optical rotation (OR). Herein, their isolation, structure elucidation and biological activities were reported.
2 Results and discussion
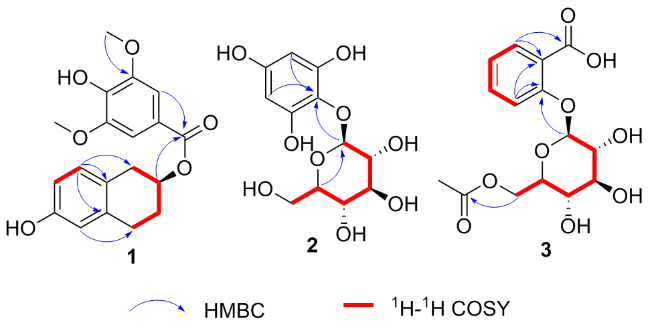
Compound 1 was obtained as white powder and had the molecular formula C19H20O6 based on the prominent signal at m/z 343.1185 [M-H]- in the HRESI-MS. The 1H NMR spectrum of 1 showed the presence of five aromatic protons in the downfield at δ 7.34 (s, 2H, H-2',6'), 6.86 (d, J=8.4 Hz, 1H, H-9), 6.54 (dd, J=8.4, 2.4 Hz, 1H, H-8), and 6.50 (d, J=2.4 Hz, 1H, H-6). In the upfield, one oxygenated methine proton at 3.97~4.05 (m, 1H, H-2), two methoxyl groups at δ 3.89 (s, 6H, 3'/5'-OMe), and three methylene groups at δ 2.94 (dd, J=15.6, 5.2 Hz, 1H, H-1a), 2.59 (dd, J=15.6, 8.4 Hz, 1H, H-1b), 2.82~2.88 (m, 1H, H-4a), 2.70~2.78 (m, 1H, H-4b), 1.98~2.04 (m, 1H, H-3a) and 1.67~1.75 (m, 1H, H-3b) were also observed. In addition, 19 carbon signals were detected in the 13C NMR spectrum (Table 1) including signals of two methoxy groups, three methylene carbons, and one oxygenated methine carbon, twelve aromatic carbons and one ester carbonyl carbon. These 1H NMR and 13C NMR spectroscopic features suggested that the structure of 1 comprised a syringic acid[10] subunit and a (R)-1,2,3,4-tetrahydro-6-hydroxynaphthalen- 2-ol[11] subunit. These assignments were confirmed by the following heteronuclear multiple-bond correlation (HMBC) data. The 1H-1H correlation spectroscopy (COSY) of H-1/ H-2, H-2/H-3, and H-3/H-4 combined with the HMBC from H-9 to C-1/5/7/10, H-6 to C-4/8/10 indicated the presence of the subunit (R)-1,2,3,4-tetrahydro-6-hydroxynaphthalen- 2-ol in 1 (Figure 2). The HMBC from H-2′/6′ to C-1'/3'/4'/7' (Figure 2) indicated the subunit syringic acid. Furthermore, in the HMBC spectrum, H-2 showed a correlation to C-7' (Figure 2). These data indicated that two subunits were joined via a C-2-O-C-7′ linkage. Compound 1 has the (2R)-form based upon the positive optical rotation of 1 and the reported similar compounds in literature.[11] Thus, the structure of compound 1 was identified as the Figure 1. compound 1 was named as rubusnolicester.
Table 1 1H NMR (400 MHz) and 13C NMR (100 MHz) spectral data of compounds 1~3 |
| Position | 1 (CD3OD) | 2 (CD3OD) | 3 (DMSO-d6) | |||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |||
| 1 | 2.94 (dd, 15.6, 5.2, H-1a) 2.59 (dd, 15.6, 8.4, H-1b) | 38.4 | — | 137.8 | — | 122.0 | ||
| 2 | 3.97~4.05 (m) | 76.0 | — | 154.8 | — | 155.7 | ||
| 3 | 1.98~2.04 (m, H-3a) 1.67~1.75 (m, H-3b) | 32.6 | 7.01 (s) | 103.3 | 7.17 (d, 8.4) | 116.8 | ||
| 4 | 2.82~2.88 (m, H-4a) 2.70~2.78 (m, H-4b) | 28.5 | — | 128.2 | 7.48 (dd, 8.4, 7.6) | 133.7 | ||
| 5 | — | 137.9 | 7.01 (s) | 103.3 | 7.05 (dd, 7.6, 7.6) | 122.3 | ||
| 6 | 6.50 (d, 2.4) | 115.6 | — | 154.8 | 7.59 (d, 7.6) | 130.8 | ||
| 7 | — | 156.3 | — | 167.6 | ||||
| 8 | 6.54 (dd, 8.4, 2.4) | 114.3 | ||||||
| 9 | 6.86 (d, 8.4) | 131.1 | ||||||
| 10 | — | 127.4 | ||||||
| 1' | — | 126.6 | 4.67 (d, 7.2) | 106.2 | 4.91 (d, 6.0) | 101.1 | ||
| 2' | 7.34 (s) | 108.3 | 3.46 (dd, 9.2, 7.2) | 75.8 | 3.24 (overlap) | 73.7 | ||
| 3' | — | 148.7 | 3.39 (dd, 9.2, 7.6) | 77.9 | 3.26 (overlap) | 76.7 | ||
| 4' | — | 140.9 | 3.41 (dd, 8.4, 7.6) | 71.4 | 3.15 (dd, 11.2, 7.6) | 70.2 | ||
| 5' | — | 148.7 | 3.18~3.21 (m) | 78.3 | 3.56 (dd, 11.2, 6.8) | 74.1 | ||
| 6' | 7.34 (s) | 108.3 | 3.80 (dd, 12.0, 6.4, H-6'a) 3.67 (dd, 12.0, 5.2, H-6'b) | 62.7 | 4.19 (d, 12.0, Ha) 4.04 (dd, 12.0, 6.8, Hb) | 63.8 | ||
| 7' | —— | 166.6 | — | 170.7 | ||||
| 8' | 1.96 (s) | 21.1 | ||||||
| 3'/5'-OMe | 3.89 (s) | 56.7 | ||||||
| 2/6-OMe | 3.81 (s) | 56.8 | ||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Compound 2 possessed a molecular formula of C14H19- ClO8, as deduced from HRESIMS (m/z 351.0846 [M+H]+, calcd 351.0841). The 1H NMR data revealed two aromatic protons at δ 7.01 (s, 2H, H-3,5), two methoxyl groups at δ 3.81 (s, 6H, 2/6-OMe), together with many protons arising from sugar moiety. The 13C NMR and 135 distortionless enhancement by polarization transfer (DEPT) data showed 14 resonances, including six aromatic carbons (δ 154.8, 154.8, 137.8, 128.2, 103.3 and 103.3), six O-bearing carbons (δ 106.2, 78.3, 77.9, 75.8, 71.4 and 62.7), and two methoxyl carbons (δ 56.8 and 56.8). These data closely resembled those of known leonuriside A[12] except for the differences of chemical shift at C-4 (δ 128.2 for 2, 154.0 for leonuriside A). These data combined with the molecular formula of C14H19ClO8 of 2 indicated that the OH group at C-4 in leonuriside A was placed by the Cl at C-4 in 2. The 2D structure of 2 was thus constructed. Detailed analysis of 2D NMR (HSQC (heteronuclear single quantum coherence), 1H-1H COSY and HMBC) spectra confirmed that the other part of the molecule was the same as those of leonuriside A. The glycone part of 2, a glucose moiety, was identified and characterized by the presence of six carbon signals (δ 154.8, 154.8, 137.8, 128.2, 103.3 and 103.3) and a signal for one anomeric proton doublet at δ 4.67 (J=7.2 Hz, H-1'), respectively. The large 3JH-1'/2'=7.2 Hz, large 3JH-2'/3'=9.2 Hz, large 3JH-3'/4'=7.6 Hz and large 3JH-4'/5'=8.4 Hz revealed the axial-axial relationship for these protons. These data suggested that the glycosyl moiety was β-glu- cose. Acid hydrolysis of 2 produced glucose as the sole sugar identified on the basis of PMP-labeling HPLC analysis with a ULTRON ES-OVM chiral column. The retention time of the PMP-labeling product of compound 2 (retention time: 44.87 min) was the same as the retention time of the PMP-labeling product of D-glucose standard (retention time: 44.87 min). The optical rotation value of the glucose from compound 2 ($[\alpha]_{\mathrm{D}}^{24}$+51.6, H2O) was also the same as the standard D-glucose ($[\alpha]_{\mathrm{D}}^{24}$+52.0, H2O). Thus, the structure of compound 2 was identified as 4-chloro-2,6- dimethoxylphenol-1-O-β-D-glucopyranoside (Figure 1).
Compound 3 was obtained as white amorphous powder, with the molecular formula C15H18O9 from HRESIMS m/z 341.0875 [M-H]- (C15H17O9, calcd 341.0878) combined with 1H NMR and 13C NMR spectroscopic data. In the 1H NMR spectrum, four aromatic protons at δ 7.59 (d, J=7.6 Hz, 1H, H-6), 7.48 (dd, J=8.4, 7.6 Hz, 1H, H-4), 7.17 (d, J=8.4 Hz, 1H, H-3) and 7.05 (dd, J=7.6, 7.6 Hz, 1H, H-5), one singlet methyl group at δ 1.96 (s, 3H, H-2''), together with many protons arising from sugar moiety were observed. The 13C NMR data showed 15 resonances, including two carbonyl carbons (δ 170.7 and 167.6), six aromatic carbons (δ 155.7, 133.7, 130.8, 122.3, 122.0 and 116.8), six O-bearing carbons, and one methyl carbon (δ 21.1), which are characteristic signals of a phenolic acid glycoside. The 1H NMR and 13C NMR data (Table 1) were very similar to those of known compound 4, except for the presence of one acetyl group signal [δH 1.96 (3H) and δC 167.6/21.1] in 3 and the absence of a methoxyl group signal (δC 52.0) in 4. The location of the acetyl group at C-6' was confirmed by the HMBC correlations from H-6' to C-1'' (Figure 2). The HMBC correlations from H-1' to C-2 indicated that C-1' linked with C-2 by a glycosidic bond. The HMBC correlations from H-6 to C-1/7 and H-3 to C-1 indicated that the carboxyl group located at C-1. Detailed analysis of 2D NMR (HSQC, 1H-1H COSY and HMBC) spectra confirmed that the other part of the molecule was the same as those of 4. The glycone part of 3, a glucose moiety, was identified and characterized by the anomeric proton doublet at δH 4.91 (d, J=6.0 Hz, 1H, H-1')/δc 101.1. These data suggested that the glycosyl moiety was β- glucose. According to the biogenic synthesis pathway and the similar optical rotation value of compounds 3 and 4, the glycosyl moiety was assigned as D-glucose. Furthermore, it was also determined by the PMP-labeling HPLC analysis method. The retention time of the PMP-labeling product of compound 3 (retention time: 44.87 min) was the same as the retention time of the PMP-labeling product of D-glucose standard (retention time: 44.87 min). The optical rotation value of the glucose from compound 3 ($[\alpha]_{\mathrm{D}}^{24}$+51.4, H2O) was also the same as the standard D-glucose ($[\alpha]_{\mathrm{D}}^{24}$+52.0, H2O). This result indicated that the glycosyl of compound 3 was D-glucose (Figure 3). Thus, compound 3 was identified as salicylic acid-2-O-(6'-O-acetyl)-β-D-glucopyranoside, a new phenolic glycoside.
The structures of known compounds 4~7 were identified by comparison of their 1H NMR and 13C NMR spectra and $[\alpha]_{\mathrm{D}}^{24}$ with those in the literature. The four known compounds were identified as 2-O-β-D-gluco- pyranosylbenzoate (4),[13] catechin (5),[14] epicatechin (6)[14] and quercetin (7).[14]
Compounds 1~7 were evaluated their anti-inflammatory activities via testing the inhibitory activities against the NO production induced by lipopolysaccharide (LPS) in mouse macrophage RAW264.7 cells in vitro. As a result, compounds 1 showed significant inhibitory activity with the IC50 value of (12.28±1.25) μmol/L. The positive control, hydrocortisone, showed an inhibitory activity with the IC50 value of (8.16±1.02) μmol/L. Compounds 3 and 4 showed moderate inhibitory activity with the IC50 value of (25.24± 1.58) and (36.65±1.87) μmol/L, respectively. No cytotoxicity was observed in compounds 1~7 treated cells (cell viability>95%). The IC50 values of other compounds higher than 40 μmol/L were regarded as inactive.
3 Conclusions
In this study, a new tetrahydrogenated naphthol syringic acid ester, rubusnolicester (1), two new phenolic glycoside derivatives, 4-chloro-2,6-dimethoxylphenol-1-O-β-D-gluco- pyranoside (2) and salicylic acid-2-O-(6'-O-acetyl)-β-D- glucopyranoside (3), together with one known salicylic acid glycoside derivative (4) and three known flavonoids derivatives (5~7), were isolated from the fruits of R. idaeus. Compound 2 represented the rare example of a chlorine-substituted phenolic glycoside derivative from natural products. The significant inhibitory activities on NO production of compound 1 could be used for the development of new anti-inflammatory agents.
4 Experimental section
4.1 Instruments and reagents
1D NMR and 2D NMR spectra were performed on a Bruker AV spectrometer (400 MHz for 1H and 100 MHz for 13C) with TMS as an internal standard. HRESIMS spectra were recorded on a Q-TOF Ultima Global GAA076 LC mass spectrometer. Optical rotations were acquired on a JASCO P-1020 digital polarimeter. Semi-preparative HPLC was performed on an Agilent 1260 LC series with a DAD detector and a evaporate light scattering detector. Silica gel (Qing Dao Hai Yang Chemical Group Co., 200~300 mesh) was used for column chromatography (CC). Precoated silica gel plates (Yan Tai Zi Fu Chemical Group Co.; G60, F-254) were used for thin-layer chromatography.
The fruits of R. idaeus were collected from Youxian, Hunan Province, China, in May 2023, and identified by Prof. Yu Zhang, Hainan Vocational University of Science and Technology. A voucher specimen (No. FPZ20230508) has been deposited at the Key Laboratory of Medicinal and Edible Plants Resources of Hainan Province, Hainan Vocational University of Science and Technology.
4.2 Extraction and isolation
The air-dried fruit powder (1.5 kg) of R. idaeus was ultrasonic extracted with petroleum ether (5 L×5, 1 h each) at room temperature. After extracting non-polar constituents with petroleum ether, the fruits of R. idaeus were ultrasonic extracted with EtOAc (5 L×5, 1 h each) at room temperature. The EtOAc extract was then filtered and dried using rotary evaporator. The EtOAc extract (12.5 g) was separated using a silica gel column chromatography (CC) (petroleum ether/EtOAc, V∶V=100∶0, 90∶10, 70∶30, 50∶50, 30∶70 and 0∶100, gradient) to generate five fractions (Fr.1~Fr.5). Fr.3 (3.1 g) was purified by using Sephadex LH-20 (100 g) CC eluting with mixtures of CHCl3/MeOH (V∶V=1∶1), and further separated by silica gel CC eluting with petroleum ether/EtOAc (V∶V from 5∶1 to 0∶1) to afford three subfractions (Fr.3a~Fr.3d). Subfraction Fr.3b was further purified by recrystallization to obtain 7 (182 mg). Fr.4 (2.4 g) was purified by using Sephadex LH-20 (100 g) CC eluting with mixtures of CHCl3/MeOH (V∶V=1∶1), and further separated by silica gel CC eluting with CHCl3/MeOH (V∶V from 100∶0 to 1∶1) to afford four subfractions (Fr.4a~Fr.4d). Subfraction Fr.4b was further purified by using Semi-Prepara- tive HPLC (CH3CN/H2O, V∶V=20∶80) to obtain 1 (3.9 mg, retention time: 18.6 min) and 2 (4.2 mg, retention time: 16.3 min). Subfraction Fr.4c further separated by using Semi-Preparative HPLC (CH3CN/H2O, V∶V=15∶85) to obtain 3 (3.5 mg, retention time: 14.9 min), 4 (12.5 mg, retention time: 17.3 min), 5 (18.6 mg, retention time: 16.5 min) and 6 (17.2 mg, retention time: 16.1 min).
Rubusnolicester (1): White amorphous powder. $[\alpha]_{\mathrm{D}}^{24}$+12.8 (c 0.1, MeOH); 1H NMR (CD3OD, 400 MHz) and 13C NMR (100 MHz, CD3OD) data were shown in Table 1. HRESIMS calcd for C19H19O6 [M-H]- 343.1187, found 343.1185.
4-Chloro-2,6-dimethoxylphenol-1-O-β-D-glucopyranoside (2): White amorphous powder. $[\alpha]_{\mathrm{D}}^{24}$+21.16 (c 0.1, MeOH); 1H NMR (CD3OD, 400 MHz) and 13C NMR (100 MHz, CD3OD) data were shown in Table 1. HRESIMS calcd for C14H20ClO8 [M+H]+ 351.0841, found 351.0846.
Salicylic acid-2-O-(6′-O-acetyl)-β-D-glucopyranoside (3): White amorphous powder. $[\alpha]_{\mathrm{D}}^{24}$+20.11 (c 0.1, MeOH); 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (100 MHz, DMSO-d6) data were shown in Table 1. HRESIMS calcd for C15H17O9 341.0878, found 341.0875 [M-H]-.
4.3 Hydrolysis and identification of glycosides
4.3.1 Acid hydrolysis
A solution of 2 or 3 (2 mg) in trifluoroacetate (TFA) (3 mL) and 3 mL of deionized water was heated under 110 ℃ for 3 h in a Ampoule Glass Tube, and then evaporated to dryness, respectively.
4.3.2 PMP derivatization
The hydrolysate was mixed into 400 μL of deionized water. 50 μL of the mixture was took out and added to a 2 mL EP tube, and then 0.6 mol/L NaOH (50 μL), 0.5 mol/L PMP methanol solution (100 μL) were added into the EP tube. The EP tube was heated in a 70 ℃ bath for 2 h. After the reaction, 0.3 mol/L HCl (100 μL) and 600 μL deionized water were added.
4.3.3 Extraction
1 mL of chloroform was added to the EP tube. The mixture was then stirred and allowed to separate into layers. Then, the mixture was separated to obtain a water layer, and the extraction was repeated 4 times. The water layer was filtered by a 0.22 μm of microporous membrane to get the test sample (filtrate).
4.3.4 D-Glucose standards and L-glucose standards
The standard sample was prepared using the same method as described in 4.3.1~4.3.3.
4.3.5 HPLC analysis conditions
Mobile phase: acetonitrile-phosphate buffer (KH2PO4, pH=8.0) (V∶V=15∶85). Column temperature: 30 ℃. Flow rate: 1 mL/min. Sample quantity: 10 μL. Detector: Agilent VWD detector (254 nm). Chiral column: ULTRON ES-OVM column; testing duration was 70 min. In addition, the leftover 350 μL hydrolysate was further separated by using Semi-Preparative HPLC (CH3CN/H2O, V∶V=10∶90) to obtain D-glucose (0.8 mg, retention time: 10.1 min, evaporate light scattering detector).
4.4 Anti-infammatory activity
The inhibitory activities of compounds 1~7 against the NO production induced by LPS in mouse macrophage RAW 264.7 cells in vitro were assessed as reported previously.[15] The experiments were performed in triplicate. Hydrocortisone was used as a positive control. The cytotoxicity assay was performed using the methyl thiazolyl tetrazolium (MTT) method in 96-well microplates.[16]
Supporting Information NMR and HRESIMS spectra of new compounds 1~3, and the HPLC spectra of compounds 2 and 3. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(Zhao, C.)