


Chiral quaternary phosphonium salts have emerged as effective phase-transfer-catalysts (PTCs) for asymmetric synthesis in 1998, when Manabe[1] reported the asymmetric benzylation of a β-ketoester with promising enantioselectivities. About a decade later, Ooi[2] and Maruoka[3] made significant contributions to the development of phosphonium catalysis and introduced a class of chiral quaternary phosphonium salt catalysts, featuring with tetraaminophosphonium and/or derived from binaphthyl skeletons. They achieved excellent product optical purities through asymmetric Henry, Michael or Mannich addition reactions. In the recent 10 years, chiral quaternary phosphonium salt catalysis has witnessed prosperous development with a diversity of challenging stereogenic centers and axes facilely established through asymmetric addition reactions.[4] Amino acid and peptide structures have been adopted in the design of novel and robust chiral phosphonium catalysts.[5]
Chiral quaternary phosphonium salt catalysis is categorized as ion-pair catalysis. In contrast to traditional organophosphorus catalysts, chiral quaternary phosphonium salt catalysts demonstrate enhanced reactivity and stability in ion-pair interactions with both electron-withdrawing and electron-rich species due to their unique cation-anion structure.[6] In addition, the larger atomic radius of phosphorus relative to nitrogen, allows phosphonium salts to facilitate reactions beyond the capabilities of quaternary ammonium catalysts. Compared to conventional catalytic systems, such as transition metal catalysis, metal-ligand complex catalysis, and phosphoric acid catalysis, chiral quaternary phosphonium catalysis offers notable advantages, including high efficiency, low catalyst loading, easy availability, and exceptional stereoselectivity.[7] Nevertheless, the application of chiral phosphonium catalysts in industrial production remains challenging when compared to conversion catalysts.
Asymmetric addition reactions have proven versatile in the chiral phosphonium salt-catalyzed multi-step cascade transformations for the construction of stereogenic carbon centers,[8] heteroatom centers,[9] axes[10] and planes.[11] Generally speaking, the static electronic effects provided by the phosphonium salts could help stabilize the ionic intermediates generated from the nucleophiles and the adducts during the addition processes, while the oriented non-covalent interactions provided by the H-bondings and the steric effects could help induce the enantioselectivities of the addition reactions. Various transformations including the Michael additions,[12] Henry reactions,[13] Mannich reactions,[14] aromatic nucleophilic substitutions (SNAr)[14] and cycloaddition reactions[15] could be carried out in a highly enantioselective fashion with the promotion of a chiral phosphonium salt catalyst, which is frequently initiated by an asymmetric addition step. Excellent reviews have been documented on the asymmetric phosphonium salt catalysis, which provided readers with comprehensive overviews on this highly active research field.[5,16] We have recently reviewed the chiral phosphonium-catalyzed nucleophilic addition reactions before the year 2023.[17] We noticed that this area has experienced rapid development in the last couple of years, with more than 10 publications newly appeared reporting fantastic success achieved with chiral phosphonium salt catalysts within such a short time. Therefore, we believe that it is necessary to provide a timely update on this topic and give a systematic review on the recent achievements in the chiral phosphonium-cataly- zed asymmetric addition reactions.
Based on the reaction process involving asymmetric addition steps, this review is divided into 3 sections as (1) chiral phosphonium-catalyzed 1,4-addition reactions, (2) chiral phosphonium-catalyzed 1,2-addition reactions, and (3) other chiral phosphonium-catalyzed asymmetric nucleophilic additions. To improve the readability of this review, all the chiral phosphonium salt catalysts that appeared in the context are summarized in Figure 1.
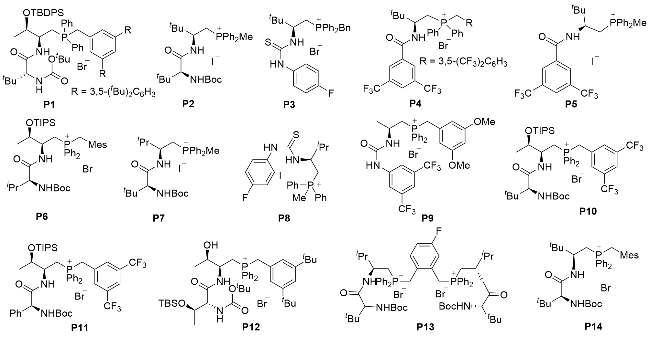
1 Chiral phosphonium-catalyzed 1,4-addi- tion reactions
1.1 Conjugate addition reactions with nitroolefins
In 2023, Wang, Song and co-workers[18] reported a chiral phosphonium-catalyzed cycloaddition reaction bet- ween the nitroolefin 1 and the α-diazophosphonate 2 (Scheme 1). The α-diazophosphonate 2 first reacts as the carbon-nucleophile to attack the electron-deficient nitro- olefin 1 through a conjugate addition process under the direction of the di-peptide based chiral phosphonium catalyst P1. An intramolecular aza-nucleophilic addition reaction occurrs to close the five-membered ring to give the intermediate Int-I bearing three continuous stereogenic centers. Then a nitrous acid is released from the inter- mediate Int-II and the 5-membered ring is aromatized to give the axially chiral pyrazole product 3 through the central-to-axial chirality conversion process. A broad scope of substituents is well tolerated on the nitroolefine substrate 1, providing the axially chiral phosphine compounds with good to excellent yields and optical purities.
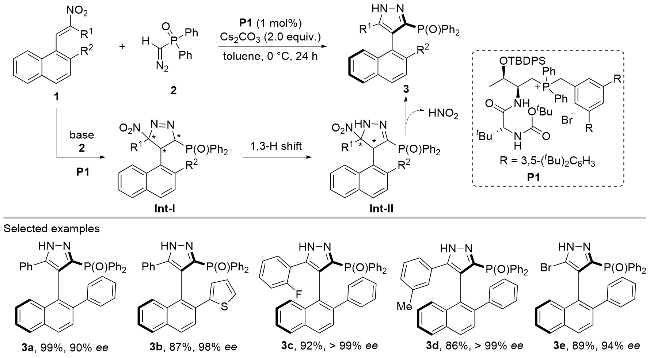
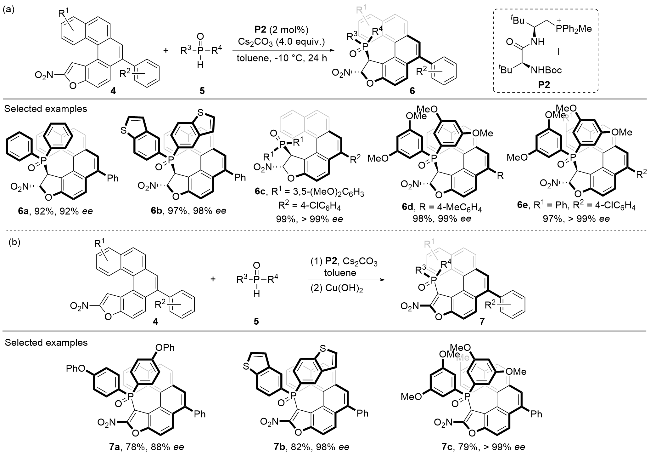
In the same year, Wang and co-workers[19] disclosed a dynamic kinetic resolution (DKR) strategy to convert the rapidly racemized oxa[5]helicene 4 into optically enriched centrally and helically chiral products (Scheme 2). The oxa[5]helicene substrate 4 bears an electron-deficient nitroalkene moiety, which could react with the nucleophilic diarylphosphine oxide 5 through a conjugate addition process in an enantioselective fashion under the promotion of the peptide-derived phosphonium salt catalyst P2. Various substitution patterns were well tolerated on both the oxa[5]helicene substrate 4 and the diarylphosphine oxide substrate 5, with the dearomatized adduct 6 bearing two adjacent stereogenic centers afforded in good to excellent yields and optical purities as single diastereomers. Pleasingly, the optically enriched adduct 6 could be re-aroma- tized under oxidative conditions to provide the helically chiral product 7 in good yields without much erosion on the optical purity. A series of enantio-enriched multi-func- tional oxa[5]helicene products 7 possessing stable stereochemical properties could be facilely achieved through the conjugate addition/oxidation cascade process without isolation of the centrally chiral intermediates.
1.2 1,4-Addition reactions with addition to enone
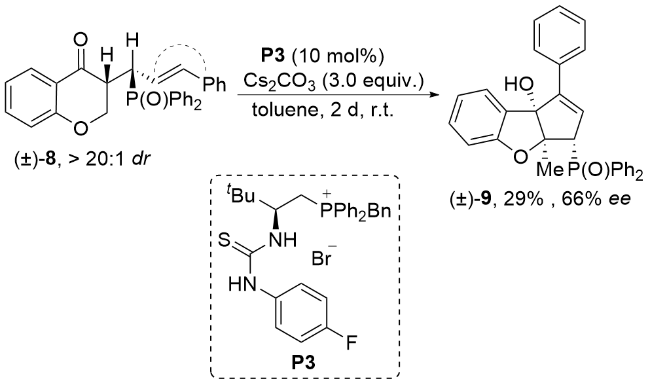
In 2023, Wang, Xu and co-workers[20] disclosed an intramolecular cycloaddition reaction of the benzopyra- none derivatives 8 (Scheme 3). The chiral phosphonium salt P3 was used as the sole catalyst in this transformation. The desired chiral cyclopenta[b]benzofuranol products 9 could be obtained in a moderate yield and moderate enantioselectivity.
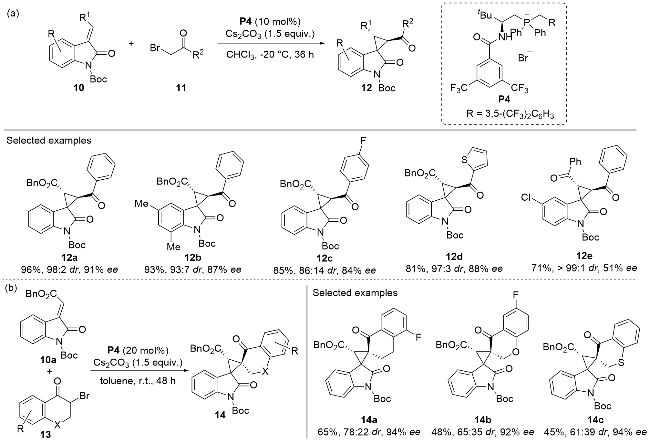
Recently, Wang and co-workers[21] developed a cycloaddition reaction with the 3-alkenyl-oxindoles 10 and α-bromoketones 11 used as substrates (Scheme 4a). The 3-alkenyl-oxindoles 10, containing an electron-deficient alkene moiety, underwent a conjugate addition reaction with the nucleophilic α-bromoketones 11 in an enantioselective fashion, facilitated by the chiral phosphonium salt catalyst P4. This methodology demonstrated excellent substrate tolerance, accommodating a wide range of substitution patterns on both 3-alkenyl-oxindoles 10 andα-bromoketones 11. The resulting spirocyclic compounds 12, which featured three contiguous stereogenic centers, were obtained as single diastereomers with moderate to good yields and high optical purities. Notably, reactions involving 3-alkenyl-oxindoles 10a and heterocyclic compound 13 also afforded chiral products 14 with minimal erosion of optical purity (Scheme 4b). This approach enabled the synthesis of a series of enantio-enriched, multifunctional chiral products 14 with stable stereochemical configurations.
1.3 1,4-Addition reactions with sulfone
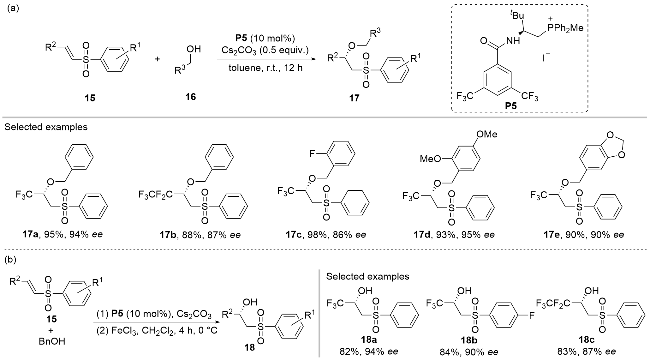
An asymmetric oxa-Michael addition reaction between the vinylsulfone 15 and the phenylmethanol 16 was recently reported by Wang and co-workers,[22] with the L-tert-leucine-derived phosphonium salt P5 adopted as the chiral catalyst (Scheme 5a). The methodology demonstrated broad substrate compatibility, accommodating various substitution patterns on both vinylsulfone 15 and phenylmethanol 16. The resulting adducts 17, containing a stereogenic center, were obtained in good to excellent yields and with high optical purities. Notably, through a cascade operation, the enantioselective oxa-Michael addition products could be efficiently debenzylated under mild conditions by introducing Lewis acid FeCl3 as a promoter (Scheme 5b). This process furnished chiral secondary alcohol products 18 in good yields with minimal loss of optical purity. The method enabled the facile synthesis of a series of enantioenriched multifunctional chiral secondary alcohols 18 with stable stereochemical properties without isolation of the benzyl ether intermediates.
2 Chiral phosphonium-catalyzed asymme- tric 1,2-addition reactions
2.1 1,2-Addition reactions with oxidation
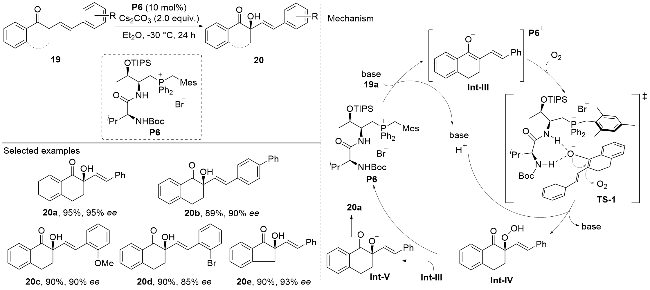
Recently, Wang and co-workers[23] reported a regio- and enantioselective α-hydroxylation of α,β-unsaturated and β,γ-unsaturated compounds 19 catalyzed by peptide-mimic phosphonium salts P6 (Scheme 6). The reaction proceeds via an enantioselective Re-face attacked on O2 through the TS-1 transition state, yielding an α-peroxo intermediate Int-IV that possesses a stereogenic center. This stereo- center is preserved during the subsequent transformation of the protonated intermediate Int-IV into intermediate Int-V via a second attack of the intermediate Int-III. The methodology exhibited broad substrate scope, tolerating diverse substituents on the β,γ-unsaturated ketone 19, and provided the chiral tertiary allylic alcohols 20a in good to excellent yields with high optical purities.
2.2 1,2-Addition reactions with addition to imine
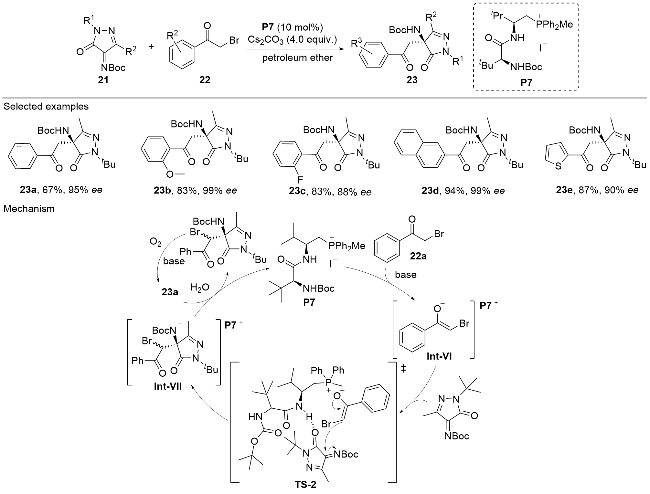
In 2023, Wang and co-workers[24] reported an asymmetric Mannich reaction catalyzed by an L-valine-derived dipeptide chiral phosphonium salt P7 to construct a diversity of chiral β-amino ketone-pyrazolinone scaffolds (Scheme 7). The methodology exhibited broad substrate compatibility, accommodating a wide variety of substitution patterns on both pyrazolinone imines 21 and α-brominated ketones 22. The reaction produced β-amino ketone-pyrazolinones 23 featuring a single stereogenic center, in moderate to good yields with excellent optical purities. Products 23 were formed through an asymmetric Mannich/radical debromination cascade process. Int-VII was initially formed via a Mannich reaction under the enantioselective induction of P7. Subsequently, the Mannich adduct Int- VII underwent an oxygen-promoted radical process to give the debromination product 23a.
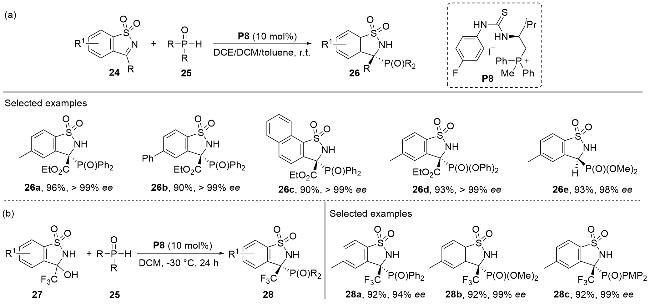
Noteworthily, the same group[25] also completed the first highly enantioselective P-nucleophilic addition to the five-membered cyclic N-sulfonyl imines by a tunable bifunctional ion-pair catalyst P8 (Scheme 8a). The reaction demonstrated broad substrate compatibility, tolerating various substitution patterns on both cyclic N-sulfonyl imines 24 and secondary phosphine oxides 25. The resulting dearomatized adducts 26 were obtained in good to excellent yields with high optical purities. Remarkably, the nucleophilic addition of CF3-substituted cyclic N-sulfonyl imines 27 with P-nucleophiles 25 in the presence of the phosphonium salt catalyst also afforded chiral α-amino- phosphonate products 28 in good yields with minimal loss of optical purity (Scheme 8b). This methodology provided access to a series of enantioenriched, multifunctional chiral cyclic α-aminophosphonates 28 with tetra-substituted stereogenic centers.
3 Other chiral phosphonium-catalyzed asymmetric nucleophilic addition
3.1 Chiral phosphonium-catalyzed asymmetric addition to heteroatoms
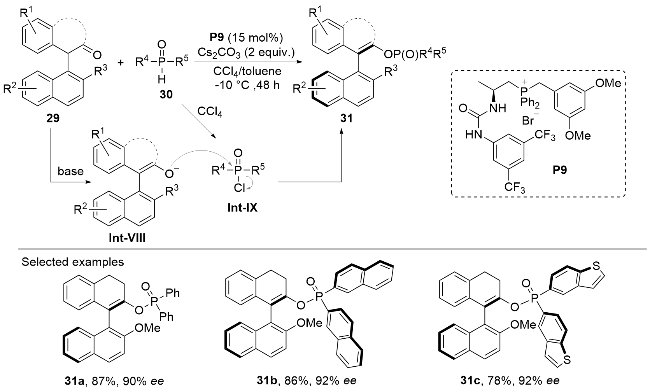
In 2023, Wang, Su, and co-workers[26] developed a facile approach to construct the axially chiral phosphate-con- taining olefin scaffolds by the bifunctional phosphonium salt-promoted Atherton-Todd coupling reaction between cyclic ketones 29 and phosphine oxides 30 (Scheme 9). The nucleophilic diarylphosphine oxides 30 are first chlorinated to give the electrophilic diphenylphosphinyl chloride intermediate Int-XI, which then reacts with the enolate Int-VIII generated from the cyclic ketones 29 through an enantioselective addition/elimination process under the catalysis of the thiourea-based phosphonium salt catalyst P9. This methodology exhibited good substrate tolerance, and various substitution patterns could be accommodated on both cyclic ketones 29 and diarylphosphine oxides 30. The reaction afforded axially chiral olefins with phosphate units 31 in moderate to good yields and excellent optical purities.
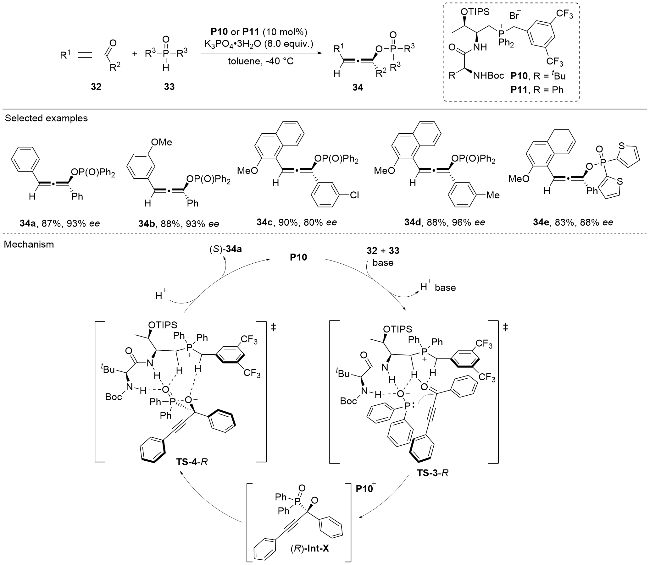
Recently, Wang, Chen, and co-workers[27] disclosed the asymmetric Pudovik addition/phospha-Brook rearrangement reaction between the ethynyl ketones 32 and the phosphine oxides 33 under the catalysis of the chiral phosphonium salt P10 (Scheme 10). Axially chiral allenes 34 bearing a phosphorus functionality were facilely afforded in good to excellent yields and enantioselectivity. Mechanistically, the nucleophilic phosphine oxide 33 could be activated by the phosphonium salt catalyst and attack the carbonyl group of the ethynyl ketone substrate 32 through a nucleophilic addition process to give the oxide anion intermediate Int-X. Then an intramolecular phospha-Brook rearrangement process occurs and the phosphorous unit is migrated to the oxygen atom, with the alkynyl group simultaneously converted to the chiral allene group through a central-to-axial conversion process. The methodology demonstrated excellent substrate compatibility, accommodating various substitution patterns on both ethynyl ketones 32 and phosphine oxides 33. The reaction provided the axially chiral allenyl phosphorus compounds 34 in good to excellent yields with high optical purities.
3.2 Chiral phosphonium-catalyzed asymmetric nucleophilic substitution
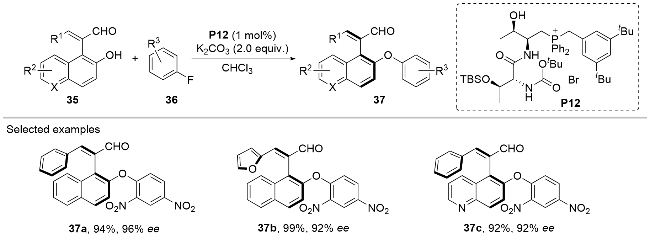
In 2023, Wang, Su and co-workers[14] reported the chiral phosphonium-catalyzed asymmetric nucleophilic aromatic substitution (SNAr) reaction between (o-hydroxy)aryl- alkene aldehydes 35 and fluoroarenes 36 bearing strong electron-withdrawing groups (Scheme 11). The hydroxy group of the (o-hydroxy)aryl-alkene aldehyde substrate 35 could attack the compound 36 at the aromatic carbon attached to the F atom through a nucleophilic addition process in an atroposelective fashion under the promotion of the peptide-mimic phosphonium salt catalyst P12. After elimination of the fluoride anion, the axially chiral ethers 37 were afforded as the final products in generally good to excellent yields and optical purities. The methodology exhibited broad substrate compatibility, tolerating a wide range of substituents on both the aryl-alkene aldehydes 35 and the aromatic compounds 36.
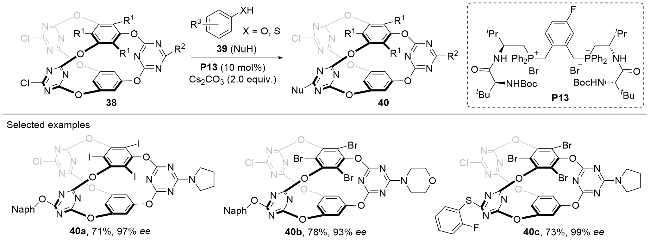
Recently, Wang, Wei and co-workers[28] reported a chiral phosphonium-catalyzed asymmetric SNAr reaction between the prism-like cage molecules 38 and the phenols/thiols 39 (Scheme 12). The two enantioisotopic triazine arms are electron-deficient and could asymmetrically react with the nucleophilic substrates 39 through a nucleophilic addition/elimination process under the catalysis of the peptide-mimic phosphonium salt P13. Various substitution patterns were well tolerated on both the substrates 38 and 39 with the chiral prism-like cage products 40 afforded in moderate to excellent yields and optical purities.
3.3 Chiral phosphonium-catalyzed asymmetric intramolecular ring expansion
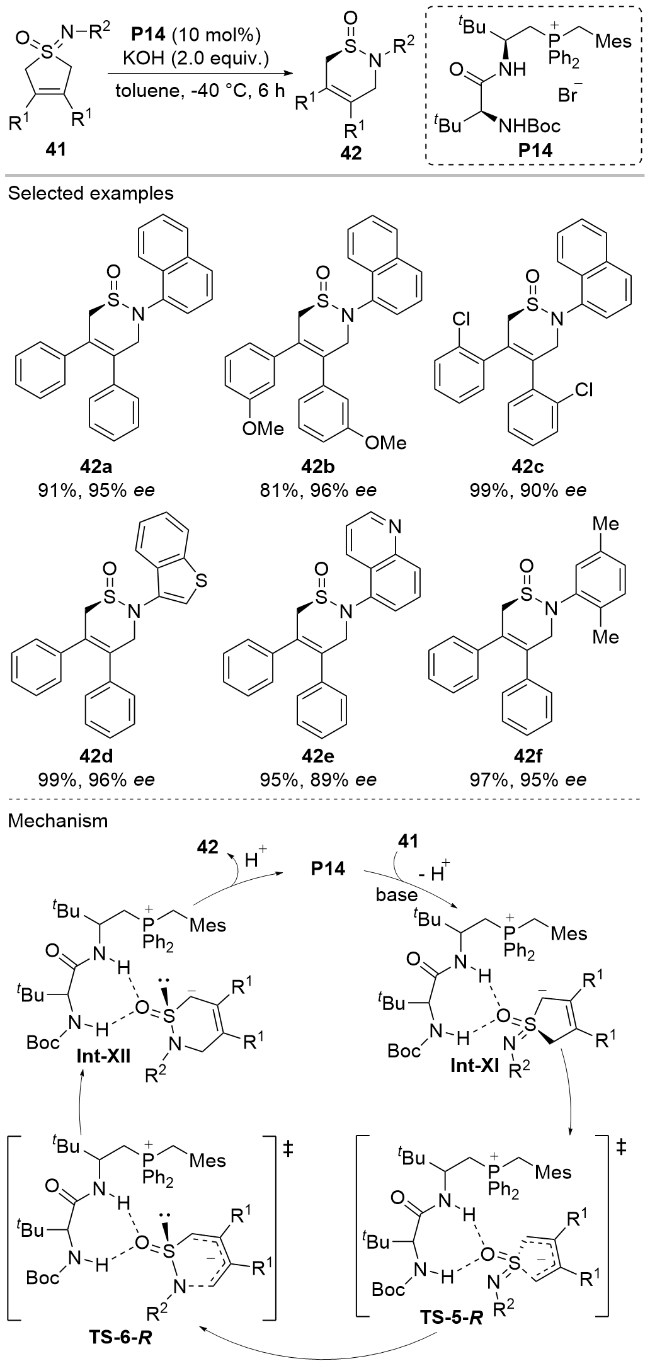
Very recently, Wang, Su, and co-workers[29] disclosed a desymmetric ring expansion reaction of the cyclic sulfoximines 41 for facile construction of the chiral cyclic sulfinamides 42 bearing a stereogenic S(IV) center under the catalysis of the chiral peptide-mimic phosphonium salt P14 (Scheme 13). During the reaction process, the cyclic sulfoximines 41 is deprotonated by the base and undergoes a ring opening process to generate the anionic diene TS- 5-R, which could coordinate with the chiral phosphonium catalyst P14 through non-covalent interactions such as static effects and hydrogen bondings. Then an intramo- lecular asymmetric ring-closing step leads to the formation of the six-membered ring products 42. This methodology demonstrated broad substrate tolerance, accommodating a wide range of substituents on the cyclic sulfoximines 41, and gave the S-stereogenic cyclic sulfinamide compounds 42 in good to excellent yields and optical purities.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 Conclusions and outlook
Chiral phosphonium salts have received considerable attention as robust organic catalysts for asymmetric synthesis in the recent decade. The recent couple of years have witnessed substantial development in the reaction scopes and the challenging chiral structures that could be achieved with phosphonium salt-catalytic strategies. A diversity of asymmetric transformations including 1,4- and 1,6-conju- gate additions, Aldol reactions, Mannich reactions, SNAr reactions and re-arrangement reactions have been effe- ctively promoted by chiral phosphonium salts and chiral molecules bearing multi-functionalities have been facilely produced in highly enantioselective fashion. Chiral functional molecules possessing challenging stereogenic carbon centers, heteroatom centers, axes and planes have been efficiently afforded in good to excellent optical purity through these protocols. A number of novel tetra-substi- tuted phosphonium ion pairs derived from non-natural peptides have been rationally designed and developed as powerful chiral catalysts in recent years for more efficient control on the reaction stereoselectivity.
Nonetheless, challenges still exist in the development of chiral phosphonium salt catalysis. For instance, the application of the chiral phosphonium catalysts in photo- and electro-induced chemical transformations has been limited. The combination of phosphonium catalytic reactions with flow chemistry has not been developed. The application of the chiral phosphonium catalysts in industrial production is still facing obstacles in the improvement on the catalytic efficiencies.
Therefore, although the quaternary phosphonium ion pair catalysis demonstrates a powerful strategy for the synthesis of challenging chiral skeletons, there are plenty of opportunities for future breakthroughs in this highly active research field. We believe that the quaternary phosphonium ion pair catalysis will serve as a powerful tool for the production of various functional molecules that hold significant merits in both pharmaceutical and fine chemical industries.
(Cheng, F.)