


烯烃化合物通常会与含卤化合物(如F、Cl、Br和I)和氮源发生卤化和胺化反应, 这是合成卤胺的直接方法[1-2]. 卤胺是有机化学和药物化学领域公认的多功能构建模块[3], 是后续转化的关键中间体[4]. 在特定条件下, 这些中间体有可能转化为含氮杂环化合物[5-6]. 卤胺化反应可分为分子内卤胺化和分子间卤胺化反应. 值得注意的是, 氨基烯烃的分子内卤胺化反应早在一个多世纪前就已率先进行[7-8]. 卤胺化反应的关键步骤不仅涉及氮鎓离子中间体的形成[9-13], 也可以形成以氮进攻的卤鎓离子中间体. 卤胺化反应一直是现代有机化学和药物化学的重大挑战[14-17], 其产物邻卤胺是各种化合物(包括偶氮吡啶[18-20]和脱氢氨基酸)的重要前体[21].
立体选择性卤胺化反应的实现主要依赖于催化体系, 催化体系大致可分为两类: 有机催化剂和过渡金属催化剂. 后者包括铁[22-23]、银[24]、铜[25-26]和钯[27-28]等金属. 立体选择性卤胺化反应的氮源通常来自胺、磺酰胺、氨基甲酸酯和酰胺[29-33], 而主要卤素来源包括对氯苯磺酰亚胺[34]和N-碘代丁二酰亚胺(NIS)等. 随着研究的不断深入, 立体选择性卤胺化反应的底物范围已大大扩展. 在优化的反应条件下, 包括α,β-不饱和酯[35]、酰胺、腈和酮在内的官能化烯烃表现出卓越的对映选择性和非对映选择性. 通过开发各种方法, 立体选择性卤胺化技术取得了长足的进步. 虽然金属催化剂在促进立体选择性卤胺化反应方面表现出相对较高的活性, 但它们通常价格昂贵, 而且会造成严重的环境污染. 例如, 镍和铜催化剂具有高活性, 但必须使用高效配体, 且成本高昂[36]. 相比之下, 非金属催化剂对环境友好, 符合绿色化学的原则; 总的来说, 每种催化方法都有其独特的优点和缺点. 在选择最合适的方法时, 必须考虑底物的性质、反应条件和所需的产物.
目前, 立体选择性卤胺化领域的研究尚未得到系统地梳理, 因此, 迫切需要对其研究成果进行全面而深入地总结. 本文详细综述了立体选择性卤胺化的反应机理以及各个反应的独特优势, 旨在为研究人员提供一份有价值的参考, 助力该反应在有机合成领域的发展, 并为构建更加多样化的反应类型提供支持. 为了更好地理解卤胺化反应的立体选择性, 本文列举了一些非立体选择性的卤胺化反应, 并根据催化模式不同, 将本文分为一般卤胺化、过渡金属催化和非金属催化三大类. 进一步, 依据卤素来源的不同, 过渡金属和非金属催化模式下的反应又可细分为立体选择性氟胺化、立体选择性氯胺化、立体选择性溴胺化和立体选择性碘胺化. 本文将对这些反应的最新研究进展进行全面总结与分析, 探讨其反应机理, 同时指出可能存在的问题, 并对未来的发展方向进行展望. 通过全面梳理立体选择性卤胺化领域的研究进展, 期望为研究人员提供一份有价值的参考, 推动该领域的进一步发展, 助力其在立体选择性卤胺化领域发挥更大的作用.
1 一般卤胺化反应
近年来, 卤胺化反应及其衍生转化在合成化学领域取得了突破性进展, 不仅为复杂含氮化合物的构建提供了高效策略, 更在药物分子和功能材料合成中展现出广阔的应用前景.
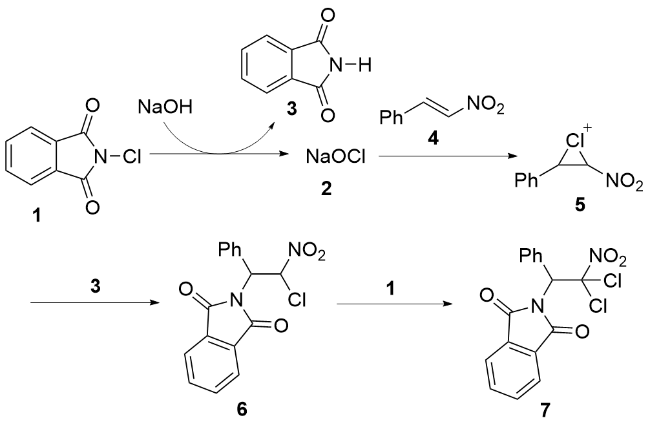
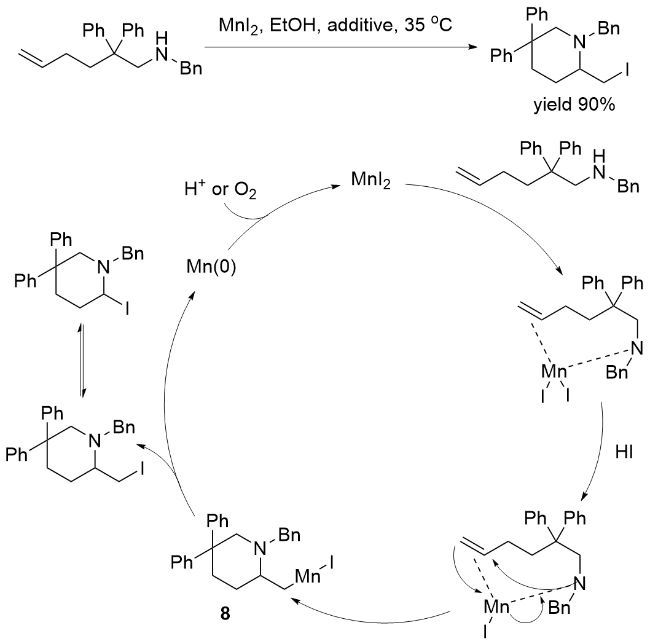
2016年, 孙慧等[39]报道了一种以二碘化锰(MnI2)为催化剂、实现非活化烯烃碘胺化反应的新策略. 研究表明, 在适宜的碘源和酸性添加剂存在下, N-取代的戊-4-烯-1-胺与N-取代的己-5-烯-1-胺可高效发生分子内环化反应, 区域选择性地生成3-碘代哌啶或2-(碘甲基)吡咯烷类化合物, 并取得良好至优异的分离产率(Scheme 3). 该方法的显著优势在于: 反应条件温和、操作简便, 且催化剂与碘源均易于获取. 尤为重要的是, 此项工作首次揭示了基于Mn(II)中心还原消除烷基碘化物的反应路径. 如Scheme 3所示, 其反应机理涉及: MnI2通过与烯烃底物配位活化碳-碳双键, 同时释放碘化氢(HI); 随后, 氮原子对活化的双键进行分子内亲核进攻, 形成关键的氨基金属化中间体8; 最终, 中间体8经历还原消除反应, 生成目标碘胺化产物并释放出金属锰Mn(0), 完成催化循环.
Azzi等[41]开发了一种高效的光催化氯胺化反应体系, 以N-氯代丁二酰亚胺(NCS)作为多功能试剂, 成功实现了2-(1-氯乙烯基)吡咯烷及其相关杂环化合物的合成(Scheme 5). 反应机理研究证实, 反应过程中会生成一类以氮原子为中心的活性自由基中间体, 即氮中心自由基(Nitrogen-Centered Radica, NCR), 该关键中间体可通过两条路径生成: Ru催化的去质子化烯烃光激发态氧化或N-氯烯烃的光解过程, 生成的NCR随后引发分子内环化, 形成高反应活性的吡咯烷乙烯基自由基中间体, 最终通过氯胺化过程得到目标产物. 值得注意的是, NCS在该反应中表现出双重功能: 既作为磺酰胺基团的活化剂促进NCR的形成, 又作为氯化试剂完成最终的氯原子引入.
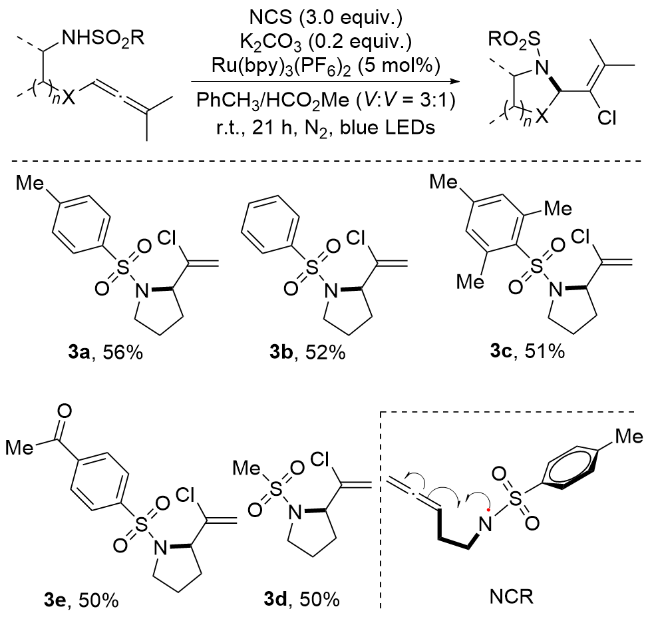
2 过渡金属催化的立体选择性卤胺化反应
2.1 过渡金属催化的立体选择性氯胺化反应
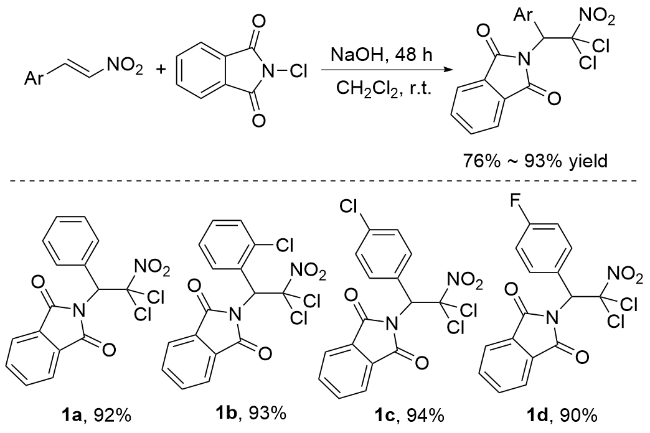
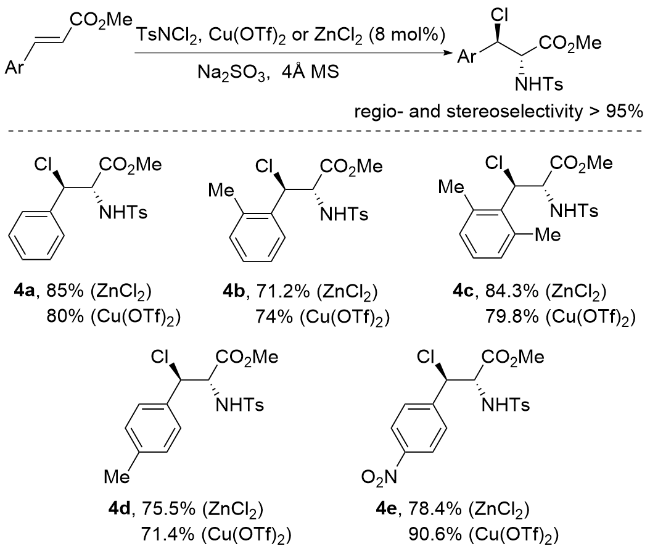
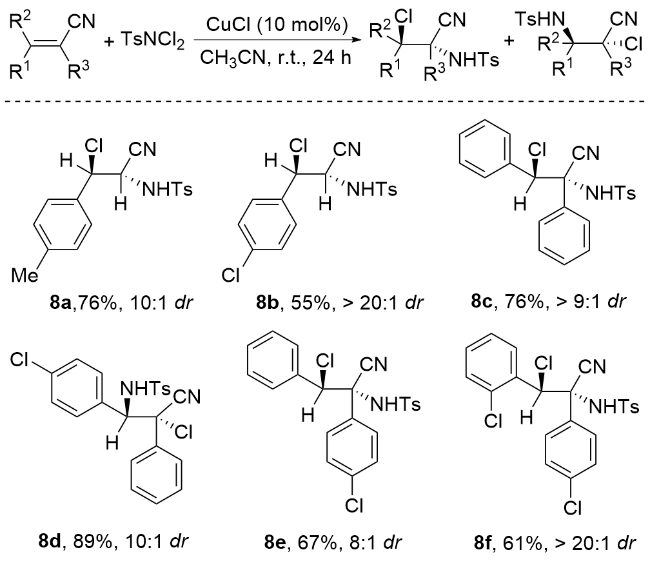
肉桂酸酯被广泛认为是合成化学中通用的底物, 特别是在烯烃的氧化中[42]. 这些底物已被有效地用于各种反应, 包括催化不对称二羟基化[43-44]、环氧化[45]、叠氮化[46-47]和氨基羟基化反应[48-49]. 这种转化为复杂分子结构的合成提供了一种强大而有效的策略. 在肉桂酸酯的基础上, 李桂根等[50]开发了一种合成邻近卤胺衍生物的创新方法(Scheme 6). 该方法以乙腈为溶剂, ZnCl2或Cu(OTf)2为催化剂. N,N-二氯对甲苯磺酰胺既可作为反应的氯源, 又可作为反应的氮源. 实验结果表明, 这种合成方法的产率从良好到优异不等, 同时还表现出良好的区域选择性和立体选择性(>95%).
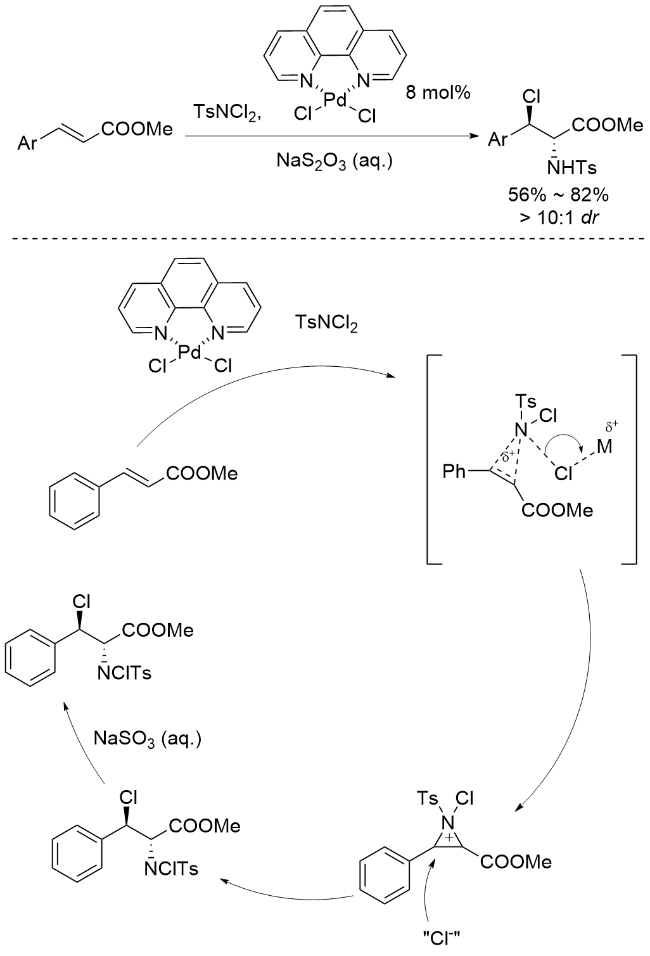
李桂根团队[52]实现了钯催化肉桂酸酯的立体选择性卤胺化反应, 该反应以对甲苯磺酰二氯胺(TsNCl2)作为氮源和氯源, 二氯-(1,10-菲罗啉)钯(II)为催化剂, 以56%~82%的产率获得目标产物, 表现出优异的非对映选择性(dr>10∶1) (Scheme 8). 李桂根等进一步提出了创新反应机制, 认为烯烃首先经历亲电氨基化过程, 生成高活性的N-对甲苯磺酰基-N-氯氮杂环丁烷中间体. 该中间体可与多种亲核试剂反应, 展现出广阔的合成应用前景. 李桂根等通过深入研究发现, 钯催化剂优先与TsNCl2中的N—Cl键配位, 形成关键的“Ns-N+-Cl”活性结构, 随后钯配位的氯离子作为亲核试剂进攻N-对甲苯磺酰基-N-氯氮丙啶鎓离子的三元环, 通过SN2机制实现开环并高效构建最终产物, 这一机理研究为钯催化烯烃官能团化提供了新的理论依据.
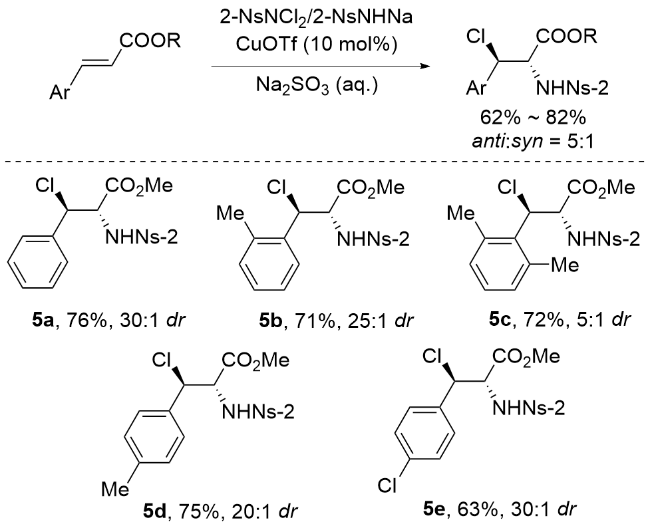
2001年, 李桂根团队[53]在卤胺化反应研究中取得重要突破, 他们发现相较于先前报道的NsNCl2与NsNHNa混合体系, 使用N-氯-N-钠对甲苯磺酰胺作为氮源和氯源能显著简化操作流程(Scheme 9). 值得注意的是, 在铜催化条件下, 该试剂与不饱和烯烃反应时并没有生成预期的氮丙啶产物, 而是出人意料地得到了邻卤酰胺衍生物. 这一反常现象源于反应过程中形成了新型的N-氯- N-铜-2-硝基苯磺酰基氮丙啶中间体, 该中间体的存在使得反应展现出卓越的非对映选择性(dr>95∶1). 反应机理概括如Scheme 9所示: 首先, 化合物9与三氟乙酸铜(I)反应生成关键中间体N-氯-N-铜-2-硝基苯磺酰胺(10). 随后该中间体与烯烃底物通过独特转化, 形成结构新颖的N-氯-N-铜-2-硝基苯磺酰基偶氮嘧啶中间体(11), 其中铜可能同时与氮原子及磺酰氧基形成配位作用. 在反应的关键步骤中, 氯离子(Cl-)通过SN2机制选择性进攻中间体11中β位(因其正电荷密度高于α位)的碳活性中心. 值得注意的是, 为促进中间体13的生成, 需加入过量2-NsNClNa以驱动反应平衡. 最终, N-氯-N-铜-2-硝基苯磺酰胺(10)可从中间体13中再生, 实现高效的催化循环. 该机理揭示了铜配合物在调控反应选择性中的独特作用, 为硝基苯磺酰胺类试剂的催化应用提供了新见解.
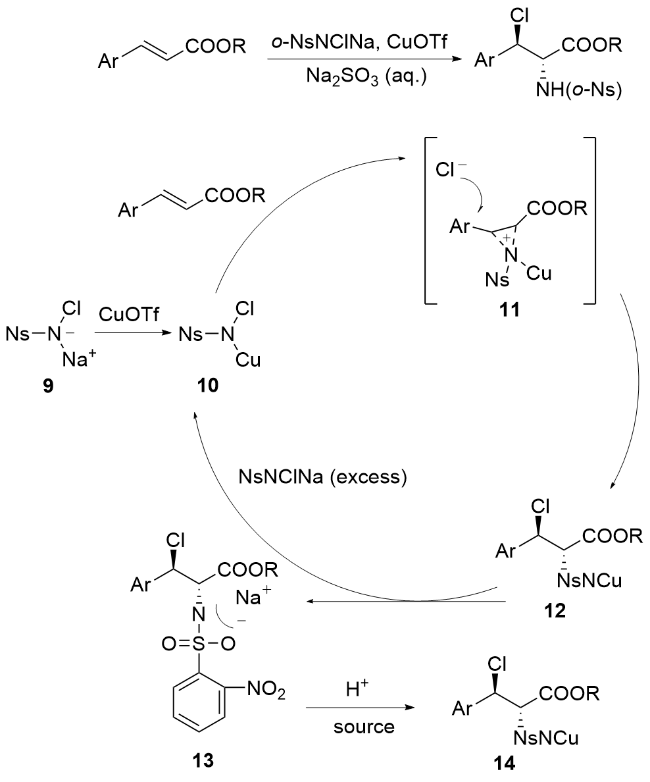
2004年, 李桂根课题组[54]在卤胺化反应领域取得重要突破, 首次将离子液体成功应用于烯烃立体选择性卤胺化反应体系(Scheme 10). 研究发现, 当以α,β-不饱和N-酰基-4-烷基噁唑烷酮为反应底物时, 传统有机溶剂在标准条件下难以有效促进反应进行; 而采用[BMIM][BF4]离子液体作为反应介质后, 不仅显著提高了反应效率, 还能以优异的化学收率和立体选择性获得目标产物. 该离子液体介质展现出多重优势: 兼具低挥发性、高阻燃性等安全特性, 对极性化合物具有出色的溶解能力, 且可通过简单操作实现回收再利用, 在提升反应性能的同时也体现了显著的绿色化学价值. 这一创新性工作为发展环境友好的不对称合成方法提供了重要参考.
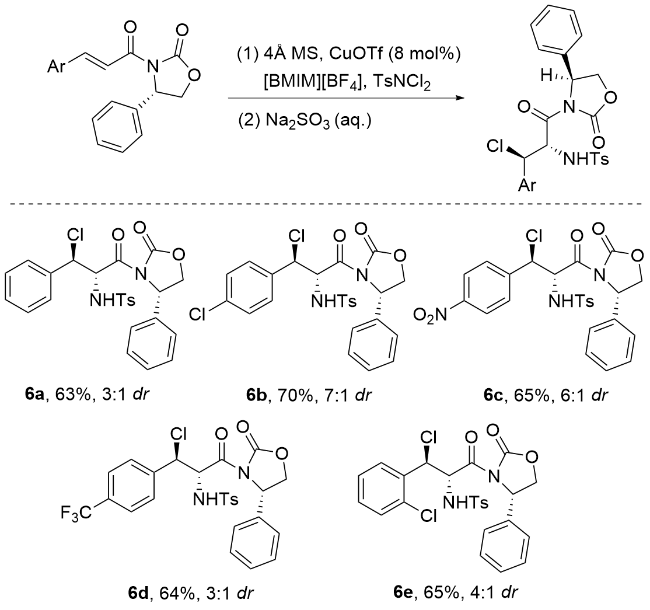
2007年, 李桂根研究团队[55]开发了一种高效的非对映选择性卤胺化反应新策略(Scheme 11). 该反应采用Pd(OAc)2作为催化剂, 以离子液体[BMIM][NTf2]为反应介质[56-59], 乙腈为溶剂, 通过螯合控制实现了优异的非对映选择性. 该方法操作简便, 可在“一锅法”条件下进行且无需惰性气体保护, 反应产率中等到优秀, 并表现出良好的非对映选择性. 反应机理如Scheme 11所示, 涉及亲氮离子选择性地进攻碳碳双键位阻较小的一侧, 同时氯离子(Cl-)进攻双键另一侧, 其中钯金属中心通过形成螯合复合物, 精准调控了反应的立体化学过程. 这一工作为发展绿色高效的立体选择性有机合成方法提供了新思路.
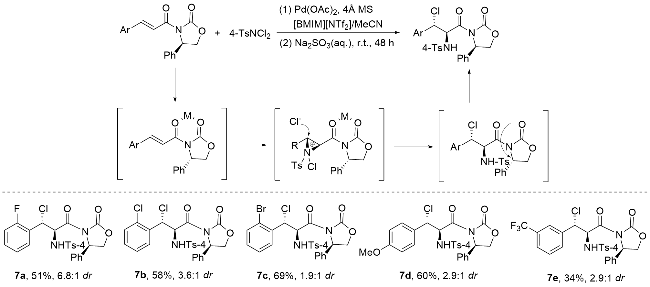
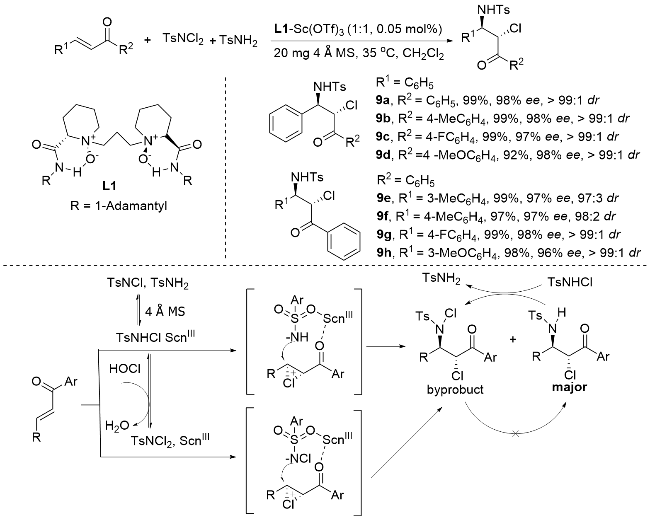
2011年, 蔡云飞研究团队[61]在立体选择性氯胺化反应领域取得重要突破(Scheme 13). 该团队创新性地以α,β-不饱和γ-酮酯和查尔酮为底物, 采用TsNH2/ TsNCl2 (n∶n=1∶1)复合体系作为氯/氮源, 在4Å分子筛存在下, 通过L1-Sc(III)手性络合物催化, 成功实现了高立体选择性的氯胺化反应. 研究表明, 该反应首先在分子筛促进下原位生成高活性TsNHCl中间体, 随后在手性钪络合物的调控下, 通过负氮亲核作用形成关键的手性氯化铵离子中间体, 最终以优异的产率(最高达99%)、出色的对映选择性(>99% ee)和非对映选择性(>20∶1 dr)获得目标产物, 同时仅生成微量副产物. 这一高效催化体系的建立不仅拓展了氯胺化反应的底物范围, 更为手性β-氨基酮酯类化合物的合成提供了新策略.
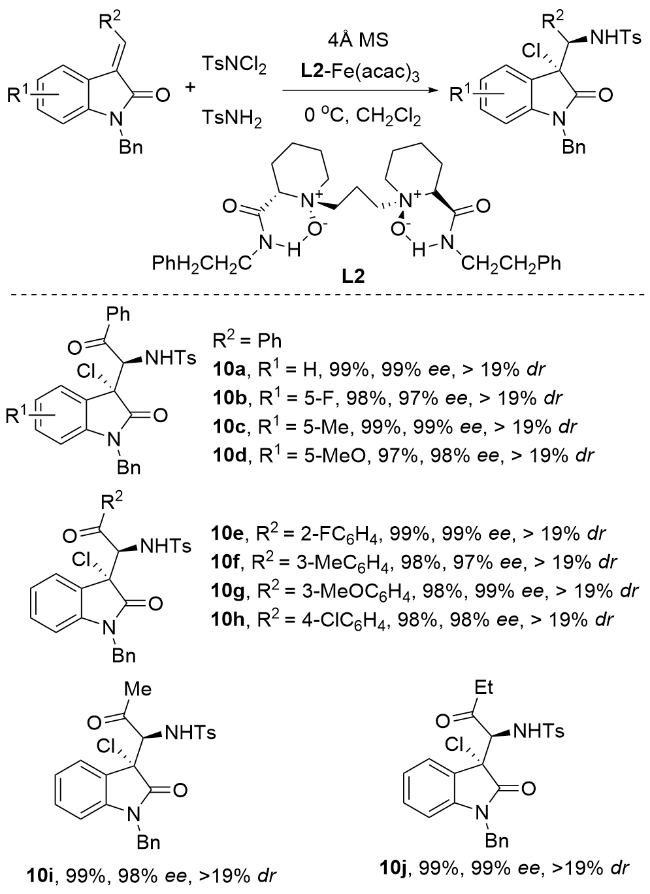
两年后, 蔡云飞课题组[62]在过渡金属催化领域取得重要进展. 他们通过系统研究发现, 相较于其他过渡金属催化剂, 手性铁基催化剂在特定反应体系中展现出卓越的立体控制能力, 能够实现高达99%的对映选择性(Scheme 14). 研究团队创新性地采用铁(III)/手性双氮氧配体催化体系, 成功实现了3-烯基吲哚的分子间氯胺化反应. 该催化体系表现出优异的反应性能, 不仅能以近乎定量的产率(99%)获得目标产物, 同时实现了99%的出色对映选择性和>19∶1的高非对映选择性(dr值). 这一研究成果不仅证实了铁催化剂在立体选择性合成中的独特优势, 也为吲哚类化合物的高效立体选择性修饰提供了新思路.
2.2 过渡金属催化的立体选择性溴胺化反应

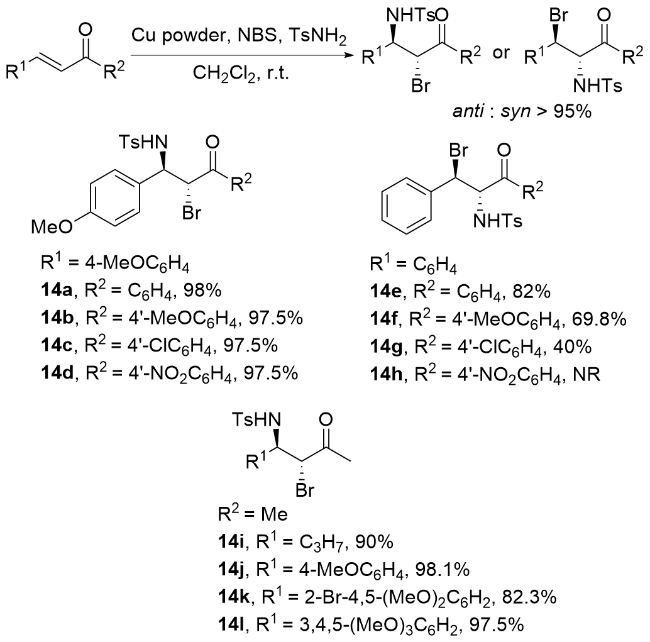
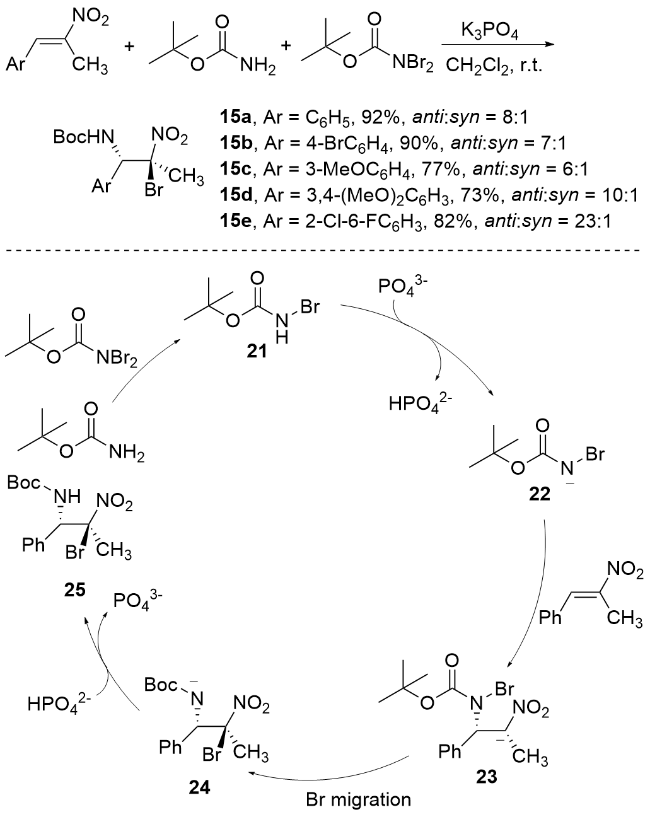
李桂根课题组[67]在非对映选择性溴胺化反应研究中取得重要突破, 成功开发了一种基于α,β-不饱和硝基化合物的高效非对映选择性溴胺化反应体系(Scheme 19). 该团队创新性地采用N,N-二溴氨基甲酸叔丁酯(BocNBr2)和氨基甲酸叔丁酯(BocNH2)分别作为溴源和氮源, 在K3PO4催化下, 实现了对多种β-甲基-β-硝基苯乙烯底物的高效转化, 不仅获得了优异的化学收率, 还展现出卓越的区域和立体选择性. 特别值得关注的是, 该反应条件温和, 产物的N-保护基可便捷脱除, 直接获得游离溴化产物. 反应机理如Scheme 19所示, 揭示了多步协同催化过程: BocNBr2与BocNH2首先生成活性中间体14, 经K3PO4去质子化形成关键中间体15, 随后通过迈克尔加成反应构建中间体16, 经历溴迁移形成中间体17, 最终在 ${\text{HPO}}_{4}^{2-}$ 质子化作用下生成目标产物18并完成催化循环. 这一工作不仅拓展了硝基烯烃的双官能团化策略, 也为含溴氨基化合物的高效合成提供了新思路.
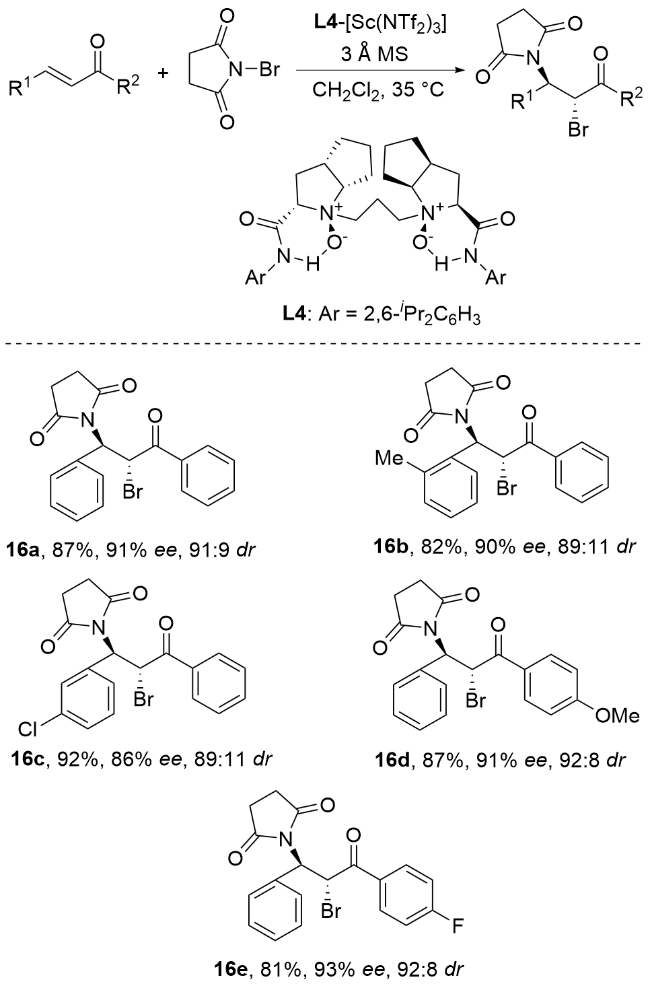
2.3 过渡金属催化的立体选择性碘胺化反应
Bovino团队[70]在铜(II)催化非活性烯烃不对称卤胺化领域取得了系统性突破. 通过精心优化反应条件, 该团队开发出一种具有广泛官能团兼容性的高效催化体系, 能以中等到良好的收率及优异的对映选择性实现烯烃的卤胺化转化(Scheme 21). 机理研究表明, 该催化循环始于三氟乙酸铜(II)与双噁唑啉配体的配位, 随后通过碱辅助的配体交换实现胺与铜中心的结合. 关键步骤涉及顺式氨基通过椅式过渡态选择性引入, 形成C—N键并生成含C—Cu键的有机铜中间体. 该中间体均裂产生铜(I)和伯碳自由基, 其中铜(I)经二氧化锰(IV)氧化再生完成催化循环. 伯碳自由基可进一步与2-碘丙烷反应生成目标卤胺化产物, 或通过分子间氨甲酰化等途径转化为其他衍生物. 该Cu(II)催化的自由基型不对称卤胺化反应通过Cu(II)-双噁唑啉配合物实现了烯烃活化与立体控制, 将自由基反应的高活性与金属催化的精准性相结合. 相较于传统的亲电/亲核反应路径, 该反应具有以下优势: 可高效转化非活化烯烃, 通过手性配体实现优异的对映选择性, 反应条件温和且与敏感官能团兼容. 这种协同机制同时克服了自由基反应的化学计量局限性和亲电路径的选择性难题, 为复杂分子修饰提供了新策略.
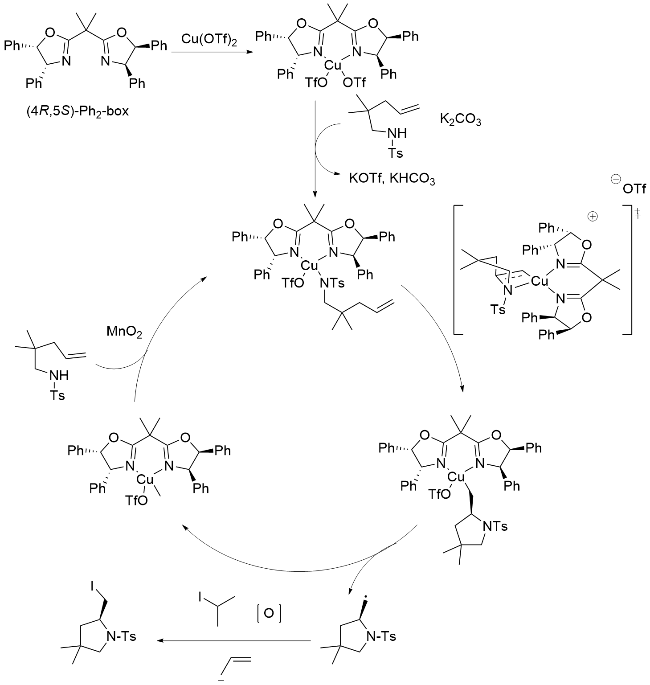
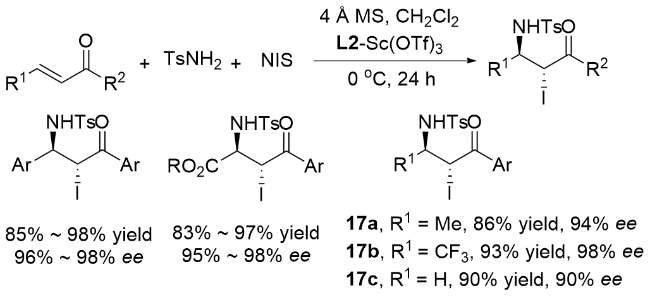
蔡云飞和他的团队[71]对不对称溴胺化进行了进一步的研究. 通过进一步优化反应条件, 包括手性配体、添加剂、温度等, 得到了收率高、对映选择性优异的产物. 他们确定了以查尔酮和4-芳基-4-氧丁基-2-烯酸酯为底物的不对称碘胺化反应, 取得了显著的结果(Scheme 22). 在无水条件下, 使用刚干燥的沸石(4Å MS), 以L2-Sc(OTf)3为催化剂, 以NIS和TsNH2分别为碘源和氮源, 在无光条件下进行了该过程, 得到了高产率和优异对映选择性的产物. 此外, 通过改变底物β位上的取代基, 也进行了类似的实验, 结果同样良好. 值得注意的是, 研究证实卤源和TsNH2(氮源)共同生成了参与卤化反应的活性物种, 这对于形成关键的中间体至关重要. 同时该反应具有典型的卤胺依赖性, 反应活性按以下顺序依次降低: NBS>NIS>>NCS.
2.4 过渡金属既可催化立体选择性氯胺化又可催化立体选择性溴胺化反应
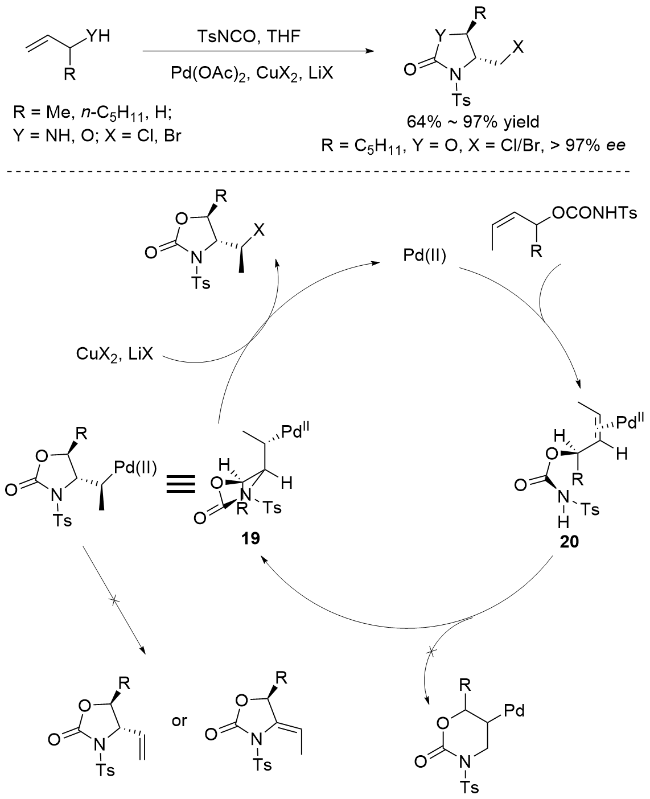
刘国生团队[72]成功开发了一种基于二价钯催化的高区域和立体选择性卤胺化反应新体系(Scheme 23). 该研究创新性地以烯丙基醇和烯丙基胺为底物, 采用Pd(OAc)2作为催化剂, 对甲苯磺酰异氰酸酯(TsNCO)作为氮源, 卤化铜/卤化锂作为卤源, 在四氢呋喃(THF)中实现了高效转化. 该反应的独特之处在于起始阶段底物与TsNCO形成盐或脲中间体, 随后在钯(II)催化下经历关键C—Pd键的形成与断裂过程. 机理研究表明, 反应首先通过Pd(II)催化的烯烃分子亲核反应构建C—Pd键, 随后在卤化铜和卤化锂协同作用下选择性氧化裂解, 专一性地生成中间体19(无取代基时)或20(烯丙位有取代基时). 值得注意的是, 该过程展现出优异的化学选择性, 未观察到其他消除副产物的生成. 这一工作不仅拓展了钯催化烯烃双官能团化的反应类型, 也为烯丙位含杂原子化合物的立体选择性合成提供了新思路.
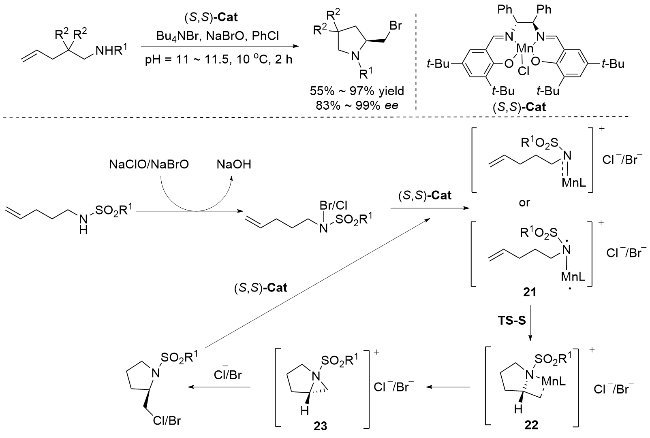
孙慧研究团队[73]在手性催化领域取得重大突破, 首次利用(S,S)-Cat催化剂通过手性氮吡啶鎓离子中间体实现了对映选择性卤胺化反应(Scheme 24). 该团队开发了一种创新的分子内不对称卤胺化策略, 采用(S,S)-Cat作为手性催化剂, 以PhCl为溶剂, Bu4NBr为溴源, 在pH 11~11.5、10 ℃的温和条件下, 高效地构建了光学活性的含氮杂环化合物. 反应机理如Scheme 24所示, 研究揭示了多步立体控制过程: 底物首先与NaClO/NaBrO形成氯胺/溴胺中间体, 随后被催化剂捕获生成三重电子基态的N-Mn中间体21; 在(S,S)-Cat的精准调控下, 成功构筑关键的手性氮丙啶鎓离子中间体23; 最终通过卤素离子的立体选择性进攻, 以优异的对映选择性获得目标产物. (Salen)Mn(III)催化的不对称分子内卤胺化反应通过手性氮丙啶鎓离子的开环路径, 相较于传统的卤胺化自由基机制和亲电卤胺化机制具有显著优势: 氮丙啶鎓中间体的刚性结构结合配体位阻效应, 可实现高达99% ee的对映选择性, 而自由基机制易受底物限制, 亲电卤胺化则难以稳定氯鎓离子. 该反应在温和条件下兼容富电子/缺电子烯烃及杂环化合物等多种底物, 突破了传统氯胺化方法的局限性. 三重态Mn-N中间体通过氮宾转移避免自由基副反应, 且C5—C6键极化作用主导区域选择性(区域比6∶94), 解决了亲电路径中卤正离子选择性差的技术难题, 是一种卤胺化类型的创新机制.
2.5 过渡金属既可催化立体选择性氯胺化又可催化立体选择性碘胺化反应
3 非金属催化立体选择性卤胺化反应
3.1 非金属催化立体选择性氟胺化反应
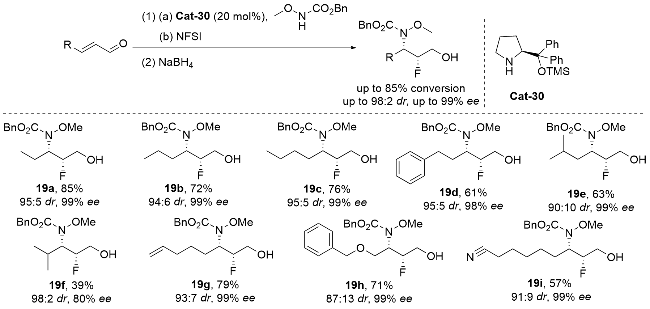
Mennie研究团队[76]在手性氟胺化领域取得重要突破, 成功开发了基于手性芳基碘催化的烯丙胺不对称氟胺化反应(Scheme 27). 该团队创新性地采用HF-吡啶复合物作为亲核氟源, 间氯过氧苯甲酸(mCPBA)作为氧化剂, 实现了对多种功能化烯烃底物的高效转化. 该方法不仅能够立体选择性地构建anti-β-氟吡咯烷骨架, 还可合成多种1,2-氧氟化产物. 特别值得注意的是, 所得高对映选择性β-氟吡咯烷产物中的张力氮杂环结构可与各类亲核试剂发生选择性开环反应, 进一步衍生出多种高产率、高光学纯度的α-氟芳基乙胺类化合物. 这一研究成果为含氟生物活性分子的高效合成提供了新思路, 同时也拓展了手性碘催化在不对称氟化反应中的应用范围.
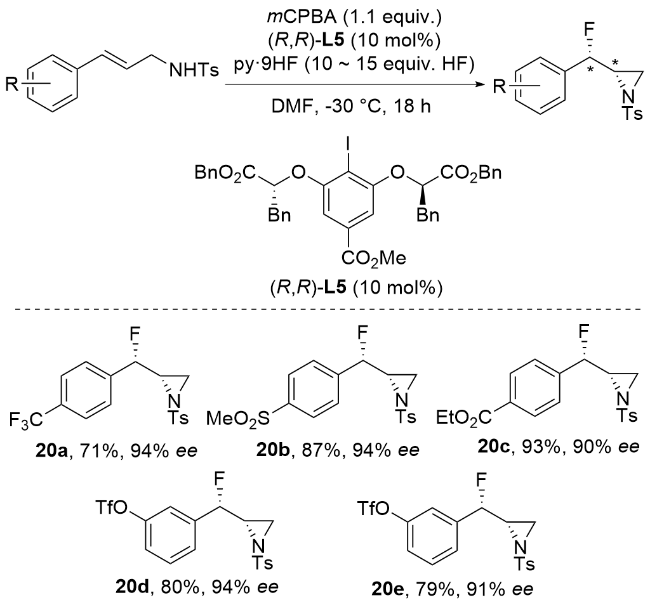
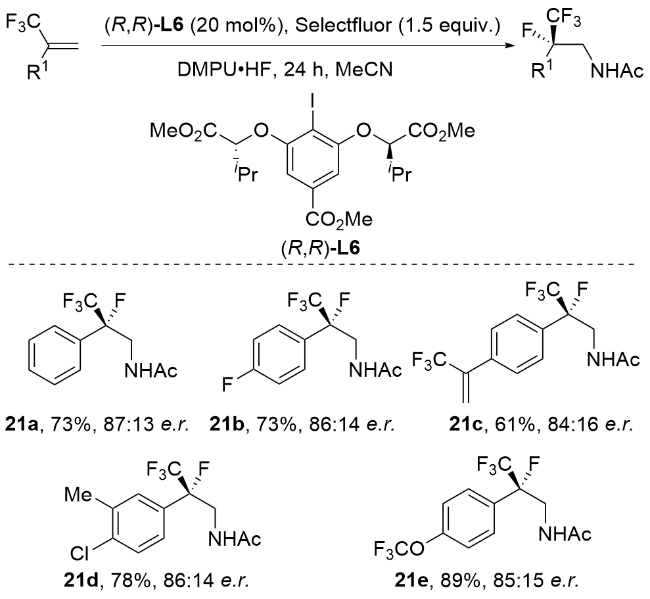
3.2 非金属催化立体选择性氯胺化反应
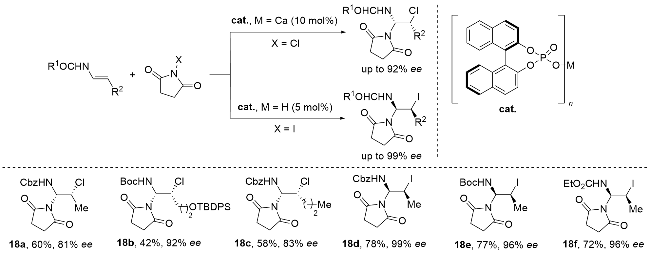
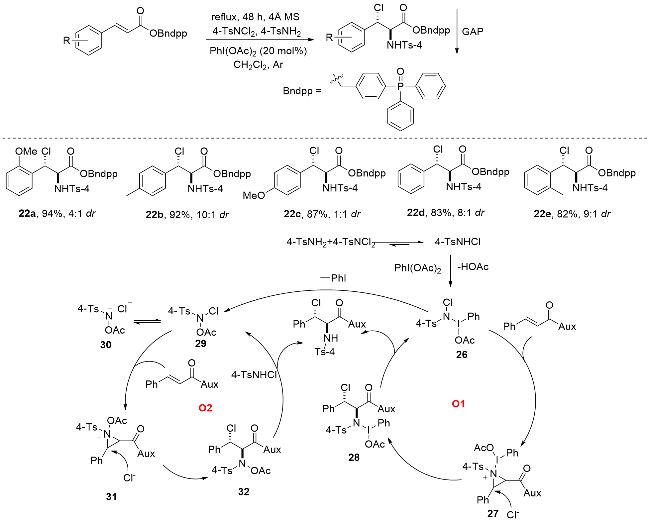
Rahman研究团队[78]创新性地开发了一种基于高价碘(III)介导的基团辅助纯化(GAP)技术, 实现了烯烃非对映选择性氯胺化与产物纯化的高效耦合(Scheme 29). 该方法以含二苯基膦氧基定向基团的dppBnOH/dppBn- NH2 (dpp=Ph2P(O))为底物, 4-TsNH2和4-TsNCl2分别作为氮源与氯源, 在PhI(OAc)2催化、二氯甲烷溶剂体系中, 成功构建了结构多样的邻卤胺类化合物. 该反应不仅表现出优异的区域和立体选择性, 且产率中等到优秀. GAP技术的核心优势在于: 通过高价碘(III)定向官能团的精准引导, 将反应过程与产物纯化无缝整合, 兼具操作简便性、高成本效益和环境友好性, 特别适用于缺电子烯烃及高附加值卤胺分子的绿色合成, 为复杂分子的高效制备提供了创新解决方案. 该反应机理通过双循环路径实现高效转化(Scheme 29): 首先, 4-TsNCl2与4-TsNH2反应生成关键中间体N-氯对甲苯磺酰胺(4-TsNHCl), 随后在PhI(OAc)2氧化作用下形成高价碘(III)中间体24. 该中间体通过O1和O2两条路径推进反应: 在O1循环中, 中间体24与底物结合形成氮-碘配合物25, 经氯离子亲核进攻生成中间体26, 最终与4-TsNHCl结合得到目标产物; 在O2循环中, 中间体24的N—I键断裂生成高活性N-乙酰氧基-N-卤代磺酰胺27(可异构化为28), 28进一步与底物反应形成氮丙啶中间体29, 再经氯离子进攻转化为30, 最终与4-TsNHCl结合完成转化. 这一双循环机制通过碘(III)介导的键合与断裂过程, 实现了反应的高效立体选择性控制.
3.3 非金属催化立体选择性溴胺化反应
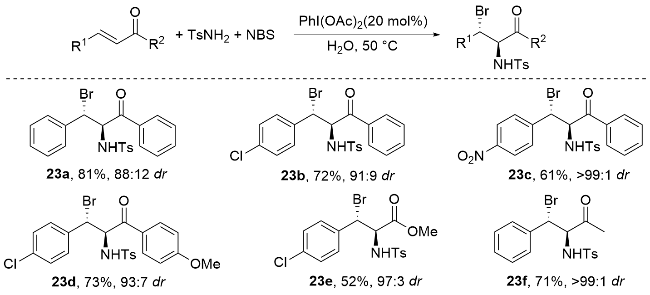
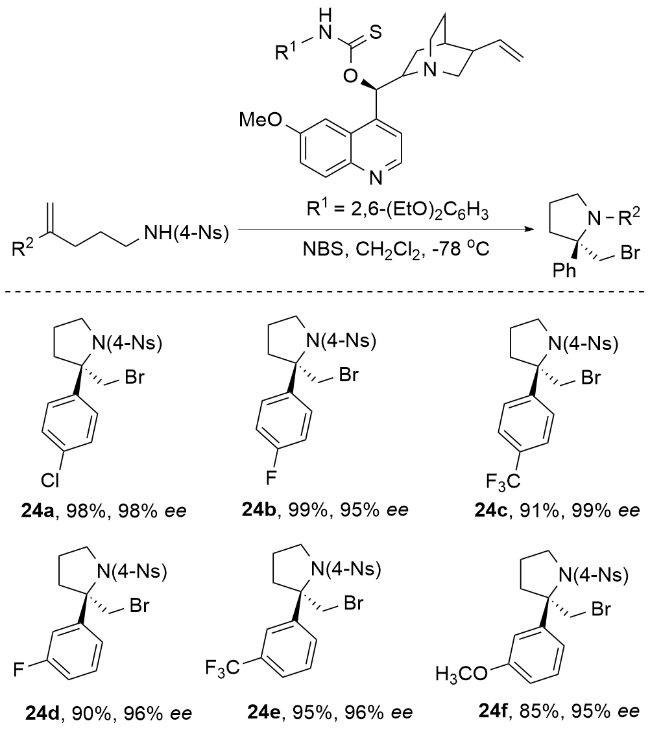
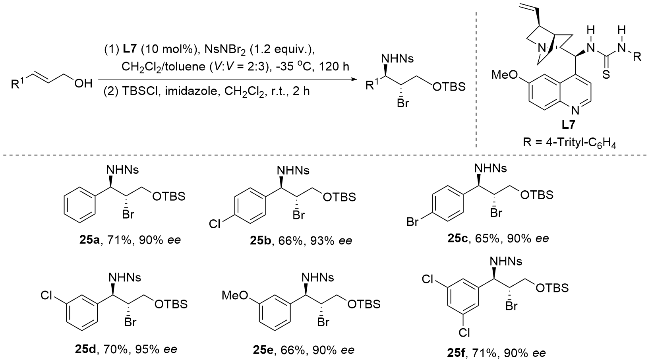
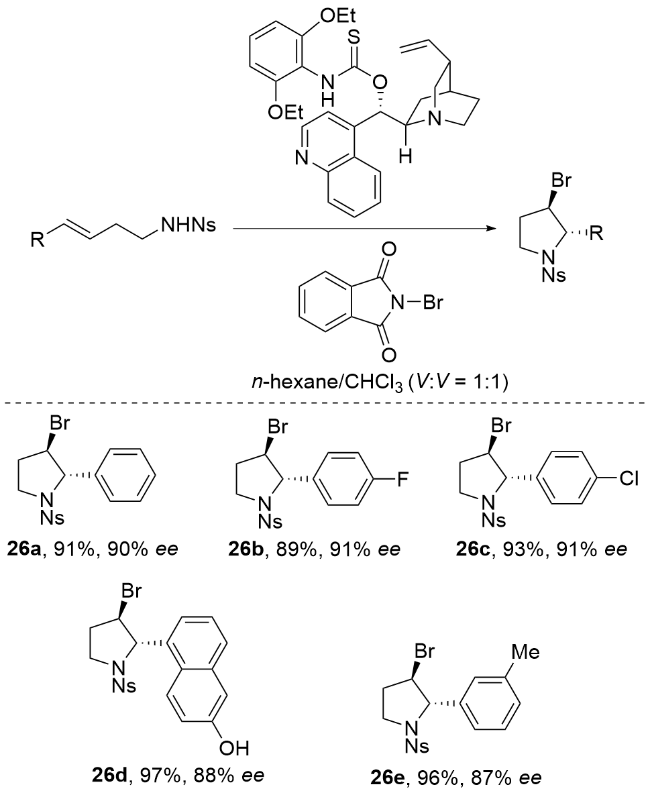
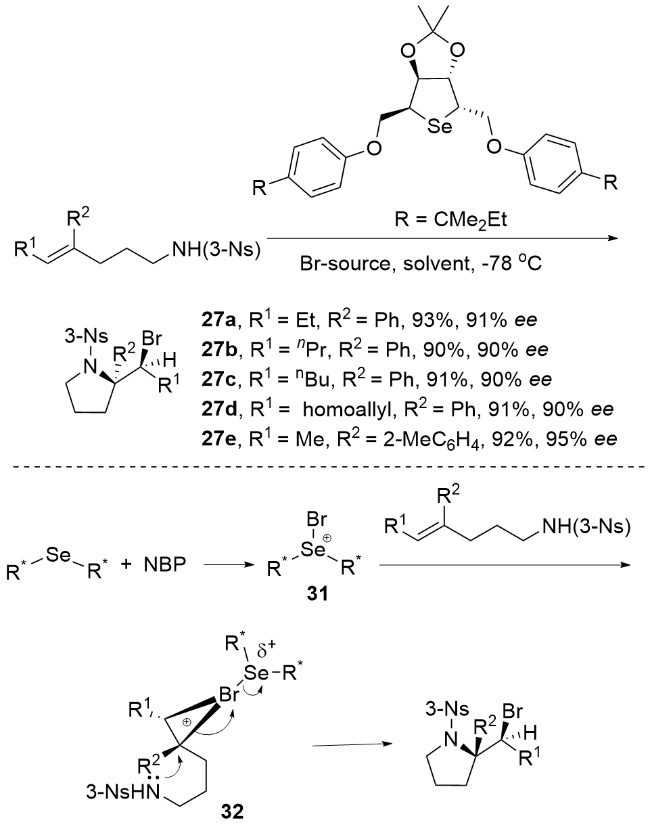
2013年, 杨英洋课题组[83]首次研究了氨基磺酰基氨基酸盐催化剂的硒类似物, 发现氨基甲酸硒表现出优异的对映选择性(Scheme 34). 尽管具有与硫代氨基甲酸酯相当的反应速率, 但氨基甲酸硒表现出不稳定性, 易于分解, 在储存中提出了挑战. 为了解决这一挑战, 杨英洋等将硒原子引入到二烷基取代体系中, 并成功开发了一种C2对称循环硒催化剂, 用于促进N-溴酰亚胺的不对称溴胺化反应. 结果表明, 单功能环硒催化剂在该反应中表现出优于双功能氨基硫代氨基甲酸酯催化剂的活性. 本研究提出了由硒路易斯碱促进的不对称溴胺化环化的第一个实例. 该反应生成的对映体富集吡啶产物具有两个手性中心. 通过随后的重排, 可以得到2,3-二取代哌替啶, 具有优异的对映选择性. 反应机理如Scheme 34所示. 首先, N-溴代邻苯二甲酰亚胺(NBP)配体与Lewis碱式硒结合, 生成亲电溴化合物31. 随后, 31与底物结合生成硒配位溴中间体32. 最后, 32在催化剂存在下被磺胺SN2攻击生成产物. 中间体32的紧密配对结构被假设可能减轻卤素降解和消旋化(无需使用可包封卤鎓中间体的双功能口袋), 这可以解释产物的高对映选择性(高达92% ee). 该C2对称环状硒催化溴胺环化机制属于路易斯碱激活的亲电溴胺化反应机制. 通过硒催化剂与NBP形成紧密的Se-Br配位中间体9, 实现了对溴鎓离子的精确立体控制, 避免了自由基机制中常见的非选择性副反应. 与卤胺化自由基机制相比, 该机制在温和条件下实现(无需光照或高温), 且对三取代烯酰胺底物表现出优异的官能团兼容性. 同时, 硒-溴键动态调控有效抑制了自由基链式反应导致的消旋化问题, 避免了自由基中间体不稳定性对产物收率的影响.
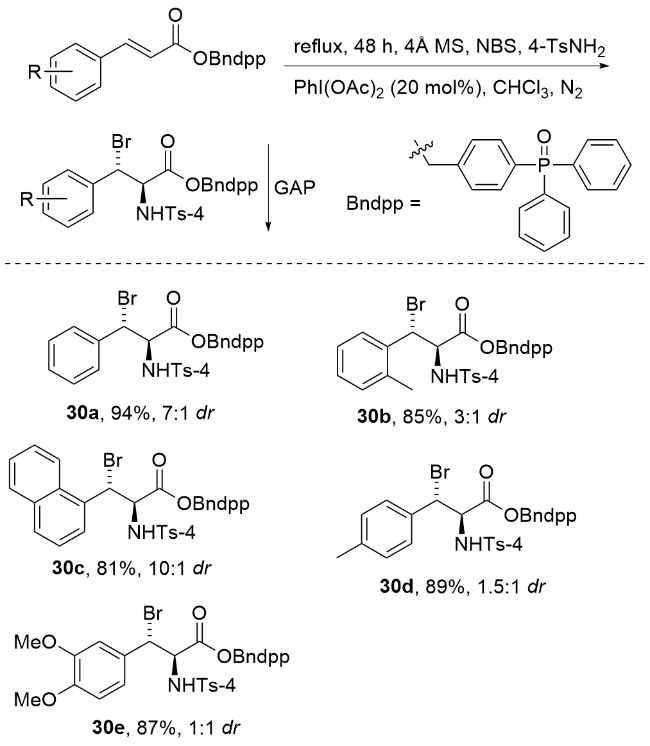
2021年, Rahman研究团队[86]在绿色化学合成领域取得重要进展, 成功将基团辅助纯化(GAP)技术拓展至高价碘(III)介导的缺电子烯烃非对映选择性溴胺化反应(Scheme 37). 该研究创新性地采用PhI(OAc)2作为催化剂, 结合TsNH2-NBS混合试剂与GAP锚定底物, 高效构建了结构多样的邻溴胺类化合物. 该方法具有显著的绿色化学优势: 产物无需传统柱层析或重结晶纯化, 仅通过助溶剂洗涤即可获得高纯度产物, 大幅减少了硅胶、有机溶剂消耗及纯化时间. 更值得注意的是, GAP试剂可回收循环利用, 实现了反应体系的可持续性. 这一工作不仅丰富了溴胺化反应的绿色合成策略, 也为复杂分子的高效制备提供了环保新思路.
3.4 非金属催化立体选择性碘胺化反应
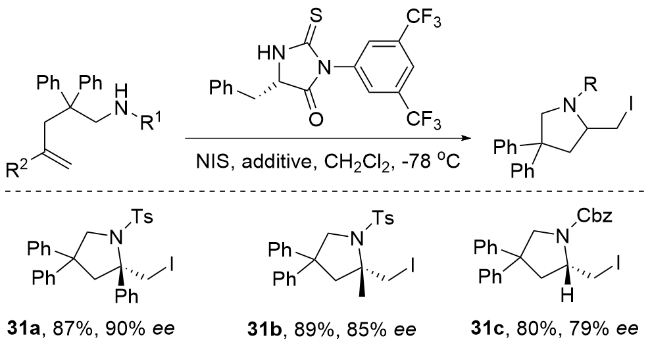
Struble研究团队[88]在手性催化领域取得重要突破, 成功开发了基于C2对称双脒(BAM)催化剂的新型不对称碳氮成环反应(Scheme 39). 该研究通过精准调控双脒催化剂的电子和空间效应, 有效解决了氮原子反应活性不足的难题, 显著提高了邻磺酰亚胺中间体的氮环化选择性. 相较于传统的氧环化路径, 该催化体系实现了对5元和6元环状脲结构的高效构建, 不仅获得优异的产率, 同时展现出卓越的对映选择性(ee值>99%). 所得手性环状脲产物可进一步转化为多种重要结构单元, 包括二元胺、海因类化合物以及完全脱保护的咪唑烷酮等. 特别值得关注的是, 研究人员通过该策略成功实现了NK1拮抗剂中关键咪唑烷酮骨架的全对映选择性合成, 充分证明了该方法在复杂手性分子构筑中的独特价值, 也为烯烃碘胺化反应中的选择性控制提供了新思路.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论与展望
不对称卤胺化作为构建手性C—N和C—X (X=F、Cl、Br、I)键的重要策略, 已在金属催化和有机小分子催化体系方面取得显著进展, 实现了不同卤素的高立体选择性引入. 然而, 该领域仍存在明显不足, 如氟胺化反应的对映选择性控制仍具挑战性, 复杂底物的适用性有限, 苛刻的反应条件限制了工业化应用潜力, 同时反应机理研究尚缺乏深入的原位表征支持, 这些瓶颈问题制约了该反应在药物合成等领域的广泛应用.
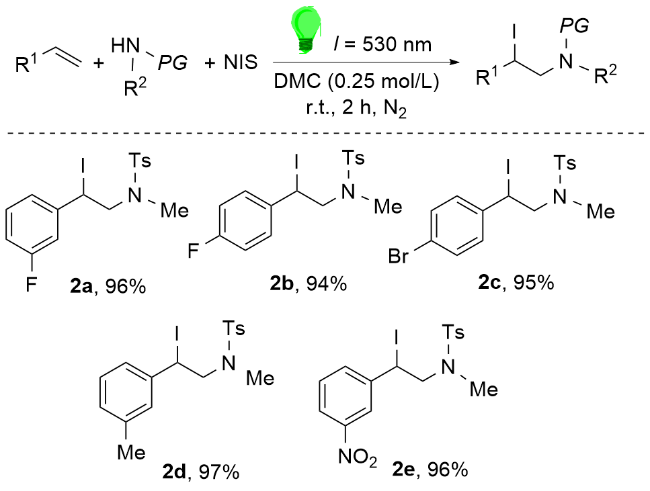
为应对这些挑战, 将电化学合成、连续流技术与人工智能辅助催化剂设计相结合, 有望带来突破性进展. 电化学方法能够原位生成并精准调控活性卤素物种, 可替代传统氧化剂, 如郑柯等[89]成功实现了多种吲哚衍生物与非活化胺的电化学碘胺化反应, 产物收率高达98%. 连续流技术通过微反应器强化传质传热, 显著提升工艺安全性和可放大性, 如Oliver团队[90]开发了无金属、可见光介导的连续流碘胺化体系, 采用商品化NIS和保护胺即可高效完成烯烃转化. 人工智能辅助的催化剂设计则能加速反应优化, 突破选择性瓶颈. 这些前沿技术的协同融合, 或将重塑传统卤胺化反应的研究范式, 为药物及功能材料合成开辟新途径. 尽管在跨学科融合、工艺放大和数据标准化方面仍存在挑战, 这些先进策略必将推动立体选择性卤胺化从基础研究向工业化应用的跨越式发展.
(Cheng, F.)