


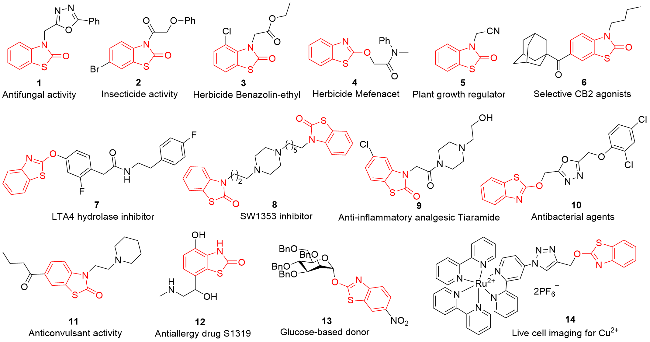
苯并噻唑-2-酮及其衍生物是一类重要的生物活性中间体, 其作为含氮稠杂环类化合物在农业及医药领域有重要应用价值(图1). 在农业领域, 苯并噻唑酮类衍生物不仅可充当植物抗真菌剂(1)[1]、杀虫剂(2)[1]及除草剂(3, 4)[1], 还可用作植物生长调节剂(5)[2]; 在医药领域, 苯并噻唑酮及其衍生物可作为潜在的受体激动剂(6)[3]或抑制剂(7, 8)[4], 起到抗炎镇痛(9)[5]、抗菌(10)[6]、抗惊厥(11)[7]和抗过敏(12)[8]的作用, 还可用于构建糖基供体(13)[9]、活细胞成像检测Cu2+(14)[10]等医疗用途. 鉴于苯并噻唑-2-酮类化合物广泛的应用价值, 其合成方法成为当前科研工作者的研究热点之一.
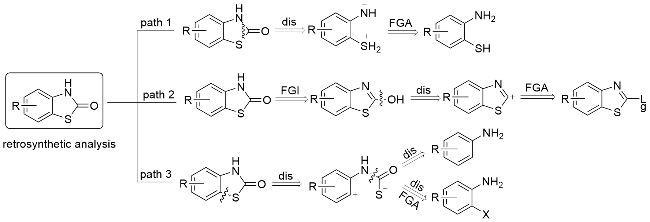
自1910年Besthorn[11]成功合成3-甲基苯并噻唑-2-酮以来, 众多学者不断探索, 开发出多种苯并噻唑-2-酮类化合物的合成方法. 然而, 至今尚未有相关文献对这些合成方法进行全面系统地总结[12]. 为此, 笔者通过调研文献并根据其合成方法对苯并噻唑-2-酮骨架结构进行了逆合成分析(Scheme 1), 发现苯并噻唑-2-酮是由硫酯键、酰胺键及苯环构成的含氮硫杂环羰基化合物, 现有合成路径主要有三种: 当苯并噻唑-2-酮的酰胺键及硫酯键被切断(dis)时, 推出2-氨基苯硫酚是其合成的关键原料(path 1); 当骨架中的羰基互变(FGI)为烯醇式时, 推出2-取代苯并噻唑作为原料可通过官能团转化(FGA)和互变异构为苯并噻唑-2-酮(path 2); 当骨架中与苯环相连的C—S键被切断时, 可推出苯胺或邻卤苯胺是其合成的主要原料(path 3). 然而, 邻卤苯胺中的C—X键与苯胺的C—H键相比, 键能较低, 卤原子更易被取代, 因此在选择底物时, 邻卤苯胺更受青睐. 综上所述, 苯并噻唑-2-酮的反应原料主要有三类: 分别为2-氨基苯硫酚、2位取代的苯并噻唑及邻卤苯胺. 这些原料在不同的反应条件下, 通过引入特定的原子或基团, 并利用底物上的活性位点形成各具特色的活性中间体, 再经过关环或异构化等反应步骤, 最终得到目标产物. 因此, 本文详细总结了以2-氨基苯硫酚、2位取代的苯并噻唑及邻卤苯胺为底物合成苯并噻唑-2-酮的方法与路线, 并深入探讨了其反应机理. 本综述将为苯并噻唑-2-酮的合成方法开发、工艺优化与工业生产提供了重要的理论依据, 也为开发更加高效、经济、环保的合成方法奠定了坚实的基础.
1 以2-氨基苯硫酚为底物合成苯并噻唑-2-酮
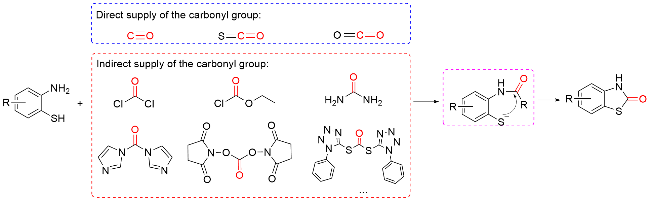
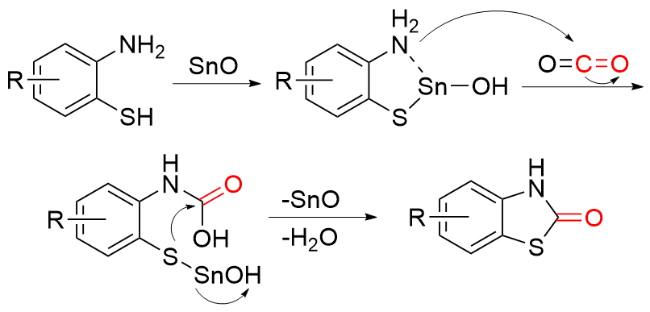
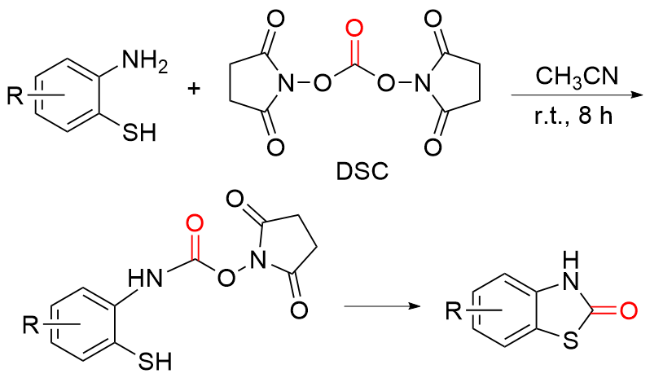
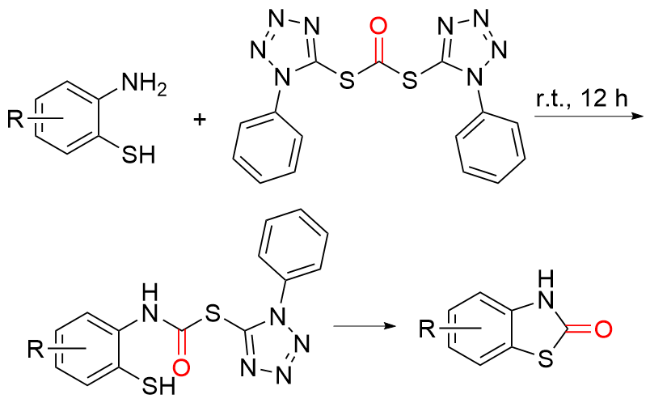
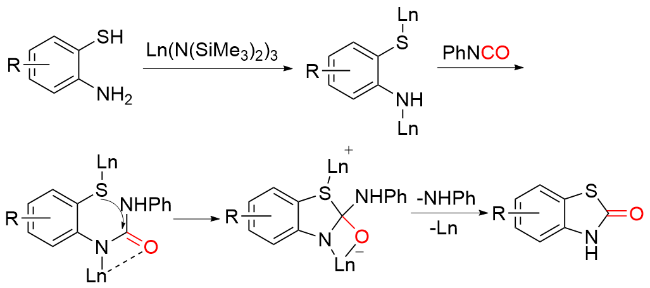
2-氨基苯硫酚与一系列羰基化试剂直接反应是工业合成苯并噻唑-2-酮的重要方法. 底物中的氨基和巯基都具有亲核性, 且氨基还具有弱碱性. 在反应过程中, 氨基亲核进攻羰基化试剂中的羰基碳形成酰胺中间体, 在碱性作用下, 巯基失去氢质子形成硫负离子, 后经分子内亲核进攻酰胺中的羰基碳, 最终环化生成苯并噻 唑-2-酮. 其中羰基化试剂主要分为以下两类: 一类是CO、CO2与COS等直接提供羰基的羰基化试剂, 这类试剂容易与底物中的氮或硫原子发生加成反应形成酰胺或酯. 另一类是光气、三光气、氯甲酸酯类、N,N-羰基二咪唑、N,N'-琥珀酰亚胺基碳酸酯以及尿素等间接提供羰基的羰基化试剂, 这类试剂的羰基两侧连有卤素、氮、氧或硫等基团, 受底物中氮或硫原子的亲核进攻, 易使这些基团离去同时形成酰胺或酯(Scheme 2).
1.1 CO作为羰基化试剂
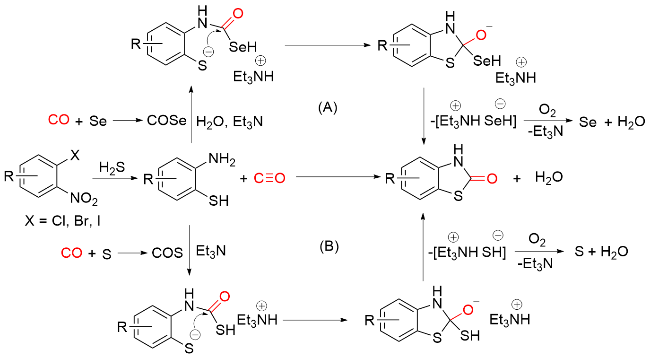
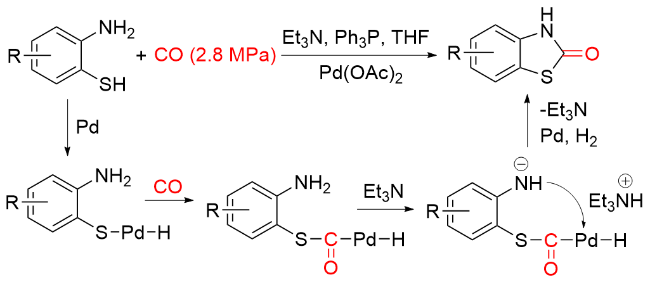
CO作为羰基化试剂具有较好的底物适用性且原子经济性高, 其能与S或Se原位生成具有较强亲电活性的COS或COSe. 1975年, Sonoda等[13]在Se、Et3N和O2存在下, 利用邻氨基苯硫酚与CO在常温常压下反应建立了苯并噻唑-2-酮的合成方法. 机理研究表明, 在Et3N的作用下, Se与CO生成COSe, 随后底物中的氨基氮亲核进攻COSe生成硒代氨基甲酸中间体, 同时Et3N促使巯基亲核进攻羰基发生环化反应生成目标产物. 反应产生的H2Se在O2作用下氧化为硒单质, 使催化剂再生(Scheme 3A). 此外, 与Se亲核性类似的S也能发生类似反应, CO也可与S反应生成COS[14], 与邻卤硝基苯发生类似的反应构建苯并噻唑-2-酮的合成方法[15]. 在反应过程中, CO和S反应生成COS, COS和水生成H2S. 在Et3N作用下, 一部分H2S取代邻卤硝基苯中的卤素生成邻硝基苯硫酚, 然后硝基被H2S还原为氨基, 同时H2S被氧化成单质S[16], 使硫催化剂再生, 最后COS和原位形成的邻氨基苯硫酚反应, 生成苯并噻唑-2-酮(Scheme 3B).
1.2 COY (Y=O/S)作为羰基化试剂
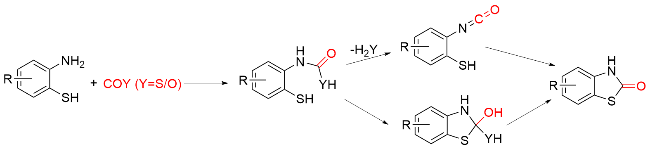
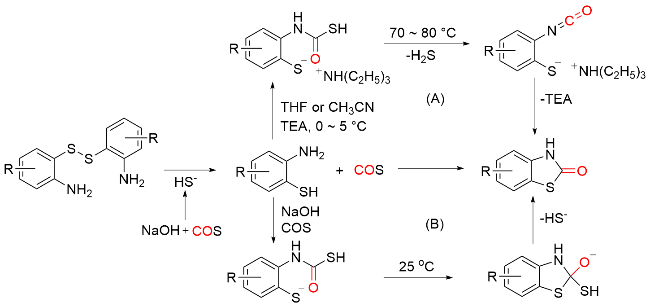
1986年, D'Amico等[20]在三乙胺(TEA)存在下, 利用COS与2-氨基苯硫酚直接反应建立了一种苯并噻唑- 2-酮的合成方法. 首先底物氨基进攻羰基碳形成硫代氨基甲酸中间体, 然后加热脱除H2S形成异氰酸酯中间体, 最后TEA促使底物上的巯基发生分子内环化反应形成目标产物(Scheme 6A). 2018年, 本课题组周博浩[19]利用氢氧化钠开发了一种邻氨基芳香二硫化物与羰基硫在室温下反应合成苯并噻唑-2-酮的新方法. 机理研究表明, 在NaOH存在下部分COS转化成HS-, 然后促使二硫化物中的二硫键断裂生成邻氨基苯硫酚, 最后与剩余的COS结合形成目标产物(Scheme 6B). 该合成方法设计了一个邻氨基芳香二硫化物与COS的偶合反应, 在该反应中, 邻氨基芳香二硫化物与COS反应生成苯并噻唑-2-酮并产生H2S, 而二硫化物中的二硫键又可在H2S的作用下断裂生成邻氨基苯硫酚. 因此, 本课题组将H2S的生成与H2S断裂二硫键两个过程偶合起来, 并利用LC-MS与NMR证实了该反应机理. 此外, 研究还发现碱不仅用于调节反应液的pH, 还用于活化反应中间体以及促进二硫键断裂.
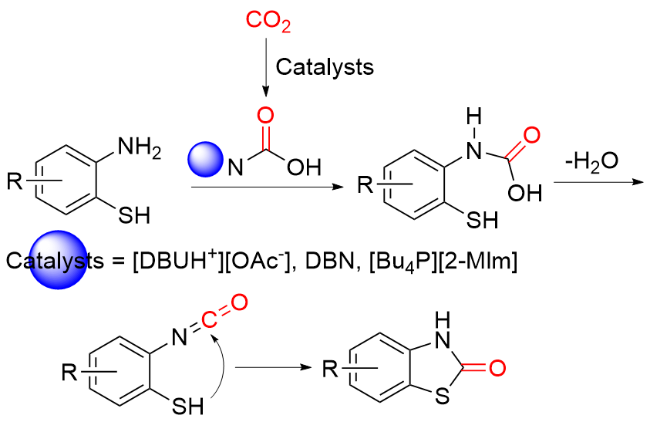
CO2作为一种羰基化试剂, 能够在离子液体、有机强碱或酸性氧化物的催化下, 与2-氨基苯硫酚反应建立苯并噻唑-2-酮的合成方法. 刘志敏课题组利用离子液体[DBUH+][OAc-][21]或[Bu4P][2-Mlm][22]分别催化CO2与2-氨基苯硫酚反应, 建立了合成苯并噻唑-2-酮的方法(Scheme 7). 在此反应中, 离子液体作为双功能催化剂, 首先活化CO2使碳位点更易被亲核进攻, 其次离子液体中的阴离子及其与CO2结合形成的羧酸根阴离子通过与底物氨基形成分子间氢键, 增强了底物氨基氮的亲核性, 使氨基容易与CO2形成氨基甲酸盐, 随后通过分子内环化反应生成苯并噻唑-2-酮. 2018年, 高翔等[23]开发了另一种合成苯并噻唑-2-酮的方法, 该方法利用强碱1,5-二氮杂二环[4.3.0]壬-5-烯(DBN)作催化剂, N-甲基吡咯烷酮(NMP)作溶剂, 在150 ℃下利用5 MPa CO2与2-氨基苯硫酚反应, 可以91%产率合成目标产物. 该反应的具体机制是: CO2首先在催化剂作用下被活化生成CO2加合物, 该加合物中CO2的氧原子与底物的氨基氢形成氢键, 增加了氨基氮对加合物中羰基碳的亲核进攻性. 随后, 受另一分子加合物的作用, 形成不稳定的氨基甲酸盐, 再经过脱水形成异氰酸酯中间体, 最终通过分子内环化生成苯并噻唑-2-酮. 2018年, 曹宪婷 等[24]开发了一种利用1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)/S催化CO2与2-氨基苯硫酚反应, 高效合成苯并噻唑-2-酮的方法. 理论计算结果表明, 在硫和DBU碱的共同作用下可以促进CO2与2-氨基苯硫酚反应形成苯并噻唑-2-酮, 反应过程中硫能够降低反应过渡态的能量, 但硫的具体作用仍不明确.
1.3 光气作为羰基化试剂
光气是最简单的酰氯化合物, 其结构中的C—Cl键易断裂给出羰基, 因此在反应中常被用作羰基化试 剂[26]. 在碱性条件下, 2-氨基苯硫酚的氨基和巯基均具有亲核性, 能够进攻光气中的羰基基团高效合成苯并噻唑-2-酮. 然而, 由于光气是气态反应物, 具有剧毒且难以控制, 为了解决这一问题, 翁建全等[1]利用固体三光气代替光气作为羰基源, 与2-氨基苯硫酚在TEA存在下反应, 以71.4%的产率得到苯并噻唑-2-酮. 反应过程中, 2-氨基苯硫酚的氨基首先亲核进攻三光气分子中的羰基碳, 形成氨基甲酸酯中间体. 三光气分子中的羰基受三氯甲氧基的吸电子诱导效应影响, 使酯基氧与羰基的共轭程度降低, 其碳氧键易断裂, 从而脱除三氯甲氧基, 该中间体再经环化脱除三氯甲氧基生成目标产物苯并噻唑-2-酮. 与此同时, 不稳定的三氯甲氧基负离子中间体离去Cl-再次原位生成光气, 然后继续与2-氨基苯硫酚反应, 经过亲核进攻环化得到苯并噻唑-2-酮(Scheme 9).
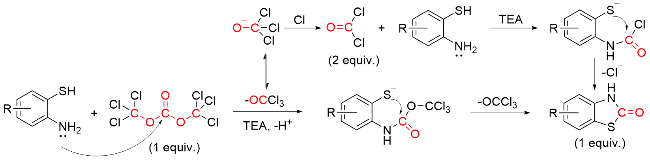
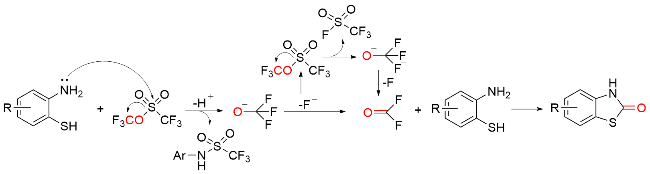
与氯光气类似, 氟光气也能够与底物反应建立苯并噻唑-2-酮的合成方法[27], 其反应机理同光气法一致. 而氟光气还可通过CF3SO3CF3(三氟甲烷磺酸三氟甲酯, 沸点21 ℃)原位生成, 该试剂能够在反应过程中提供高纯度的氟光气, 这不仅简化了氟光气的纯化步骤, 还提高了氟光气与底物的反应速率. 2019年, 宋海霞等[28]利用CF3SO3CF3原位生成的氟光气作为羰基化试剂, 与2-氨基苯硫酚反应制得苯并噻唑-2-酮, 产率为66%. 在该反应过程中, CF3SO3CF3中的三氟甲磺酰基与底物的氨基作用产生三氟甲氧基负离子, 该中间体不稳定, 离去F-生成氟光气, 然后与底物2-氨基苯硫酚反应形成苯并噻唑-2-酮(Scheme 10). 同时, 离去的F-也可亲核进攻CF3SO3CF3中的三氟甲磺酰基, 促进氟光气的生成. 该反应为一种新型Click反应[29], 底物和CF3SO3CF3在CH3CN溶液中室温反应1 h即可得到目标产物.
1.4 氯甲酸酯作为羰基化试剂
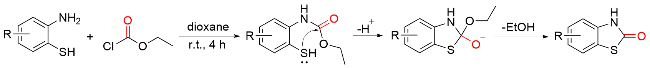
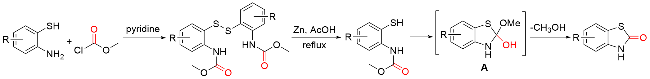
1.5 尿素作为羰基化试剂
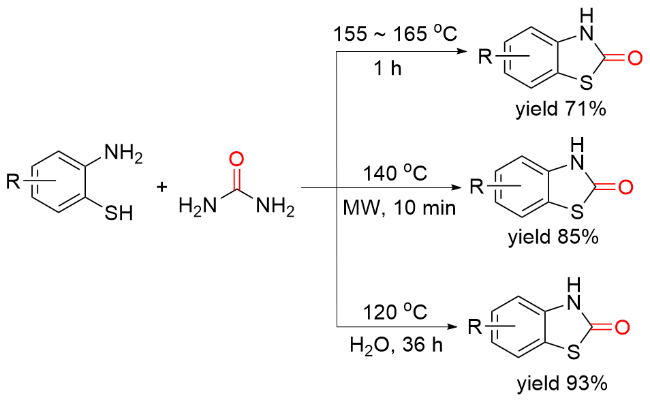
尿素作为工业生产过程中常用的绿色廉价羰基源, 可与底物2-氨基苯硫酚反应建立高效合成苯并噻唑-2-酮的方法. 到目前为止, 科研工作者利用2-氨基苯硫酚和尿素反应, 开发了三种合成方法. 1975年, Fife等[32]在155~165 ℃温度下, 利用2-氨基苯硫酚和尿素直接反应1 h, 以71%的产率生成苯并噻唑-2-酮; 2012年, Önkol等[33]利用微波条件促进反应进行, 在140 ℃下使二者发生反应, 10 min即可获得目标产物, 产率高达85%, 该方法既降低了反应温度又提高了反应效率. 2015年, Bala等[34]以水作溶剂, 在120 ℃下反应36 h, 得到目标产物, 产率为93% (Scheme 13). 该方法又进一步降低了反应温度, 同时提高了目标化合物的产率. 利用尿素作羰基化试剂合成苯并噻唑-2-酮的方法简单高效, 但在反应过程中会产生副产物氨气, 在操作过程中应设置氨气的吸收和回收装置.
1.6 含羰基杂环化合物作为羰基化试剂
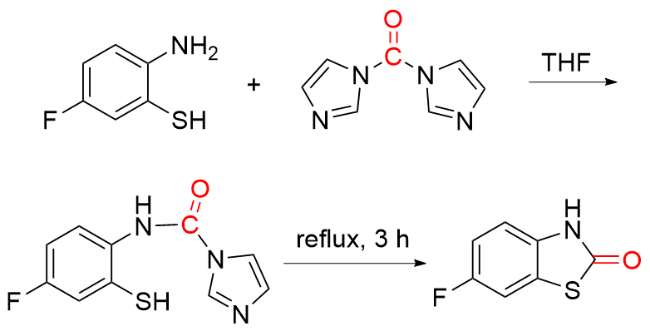
尿素结构中的C—N键共轭程度大, 难以断裂, 在其用作羰基化试剂的过程中常需要高温条件. 要使含羰基部分易于脱落, 需要降低C—N键的共轭程度. 因咪唑环具有芳香性, 当尿素的氨基变为咪唑环形成N,N'-羰基二咪唑时, 与羰基相连的N原子在咪唑环内形成共轭结构, 从而显著降低羰基碳与氮原子之间的C—N键共轭程度, 使其比尿素的酰胺键更易断裂. 2012年, Seminerio等[35]利用N,N'-羰基二咪唑与5-氟-2-氨基苯硫酚在四氢呋喃(THF)中反应3 h, 建立6-氟苯并噻唑-2-酮的合成方法. 该合成方法也是通过底物氨基氮亲核进攻羰基化试剂形成不对称脲类中间体, 随后巯基亲核环化得到目标产物(Scheme 14).
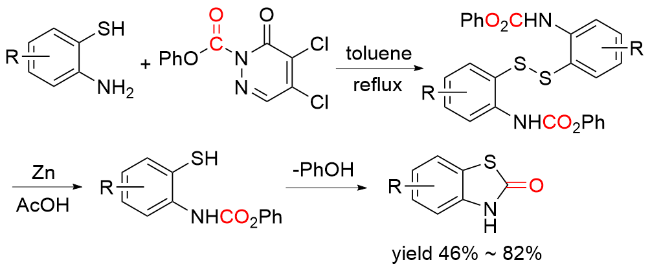
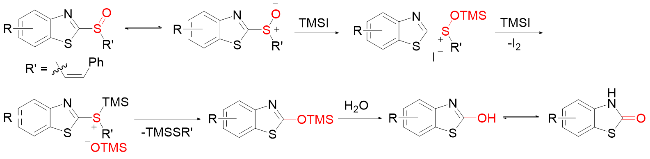
含羰基的杂环类化合物作为羰基化试剂, 因其杂环内部杂原子或芳酰基等基团的影响, 改变了邻近原子或基团的电子云密度, 使得C—N键或C—O键易断裂, 给出羰基直接参与反应. S原子比O原子半径大, 故硫酯键比酯键键能小, 更易断裂. 由于二硫代碳酸二酯结构中四氮唑杂环的吸电子效应, 进一步减弱了硫原子对羰基的共轭效应, 使C—S键键能降低, 易断裂给出羰基参与反应. 1989年, Takeda等[38]利用二硫代碳酸二酯作羰基试剂与2-氨基苯硫酚在室温下反应, 建立了苯并噻唑-2-酮的合成方法. 反应过程中, 底物氨基易亲核进攻羰基碳形成硫代氨基甲酸酯中间体, 然后发生分子内亲核环化反应生成苯并噻唑-2-酮(Scheme 17). 该合成方法的产率较低, 笔者推测其原因可能是2-氨基苯硫酚的巯基被氧化二聚为二硫化物, 限制其发生环化反应形成目标产物[39], 若在反应中加入还原剂断裂二硫键, 就可以有效提高目标化合物的产率.
1.7 其他羰基化试剂
综上所述, 选择合适的羰基化试剂是2-氨基苯硫酚绿色高效转化为苯并噻唑-2-酮的关键控制因素. CO、COS和CO2等羰基化试剂可以直接为反应底物提供羰基, 且反应中脱除的副产物在羰基化试剂中的占比低, 原子利用率高. 当羰基碳与Cl、F、N、O或S相连时, 由于杂原子的直接吸电子效应以及这些杂原子相连基团的间接吸电子诱导或共轭效应, 降低了羰基与其相邻基团之间的共轭程度, 使这些基团更容易从羰基上脱除, 从而促进目标羰基化合物的形成. 在实际应用中, 选择羰基化试剂不仅要考虑其反应活性, 还需关注其原子经济性, 即羰基在试剂中的比例(表1). 较高的羰基占比意味着更好的原子利用率, 减少了废弃物和副产物的生成, 符合绿色化学的原则. 上述总结旨在为科研工作者开发高效环保的羰基化试剂提供参考依据, 从而推动绿色可持续化学工艺的创新发展.
表1 多种羰基化试剂的特点及其性质Table 1 Carbonylation reagent for the preparation of benzothiazole-2-one using 2-aminothiophenol |
| Structure | Molecular weight | State of matter at room temperature | Carbonylation mechanism | Mass ratio of carbonyl groups in the reagent |
|---|---|---|---|---|
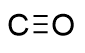 | 27.99 | Gas | CO undergoes in-situ conversion to COS/Se for participation in subsequent reactions or engages in direct reactions mediated by metal catalysis | 100% |
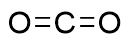 | 43.99 | Gas | The carbonyl group is attacked by a nucleophile to form an amide or ester, which then participates in subsequent reactions | 63.6% |
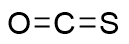 | 59.97 | Gas | The thiocarbonyl group is attacked by an amine nucleophile, forming thiocarbamate intermediates that subsequently engage in further reactions | 46.7% |
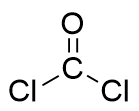 | 97.93 | Gas | The acyl chloride is attacked by an amine nucleophile, forming carbamyl chloride that subsequently engage in further reactions | 28.6% |
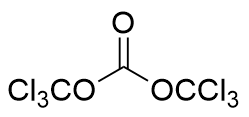 | 293.80 | Solid | Triphosgene cleaves into 3 equiv. of phosgene, which then participate in the reaction | 28.6% |
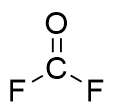 | 65.99 | Gas | The acyl fluoride is attacked by an amine nucleophile, forming carbamyl fluoride that subsequently engage in further reactions | 42.4% |
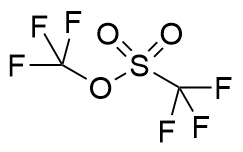 | 217.95 | Gas | The O—S bond cleavage yielded OCF3 and F, generating acyl fluoride, which then participated in the reaction | 12.8% |
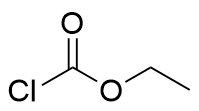 | 108.00 | Liquid | The acyl chloride is attacked by an amine nucleophile, forming carbamate that subsequently engage in further reactions | 25.9% |
 | 60.03 | Solid | The urea is attacked by an amine nucleophile, forming another urea that subsequently engage in further reactions | 46.6% |
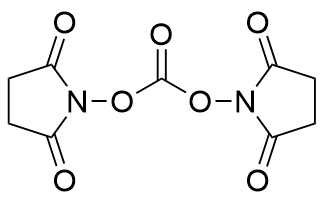 | 256.03 | Solid | The electron-withdrawing inductive effect reduces the conjugation of the ester bond, facilitating the cleavage of the C—O bond to yield the carbonyl group | 11.0% |
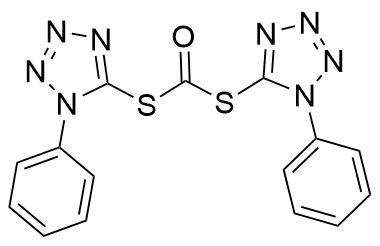 | 382.04 | Solid | The conjugation effect of the aromatic ring reduces the conjugation of the C—S bond in the thioester, promoting the cleavage of the C—S bond to form the carbonyl group | 7.4% |
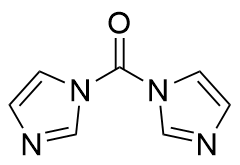 | 162.05 | Solid | The conjugation effect of the aromatic ring reduces the bond energy of the C—N bond in the amide, facilitating the cleavage of the C—N bond to yield the carbonyl group | 17.2% |
 | 283.98 | Liquid | The conjugation effect of the double bond facilitates the cleavage of the C—N bond in the amide, while the conjugation effect of the aromatic ring reduces the conjugation of the ester bond, promoting the cleavage of both the C—N and C—O bonds to yield the carbonyl group | 9.8% |
 | 119.04 | Liquid | The carbon atom in isocyanate, influenced by the electron-withdrawing inductive effects of both the oxygen and nitrogen atoms, is highly susceptible to nucleophilic attack, leading to the formation of urea or carbamate | 23.5% |
2 以2-取代苯并噻唑类衍生物为底物合成苯并噻唑-2-酮类化合物
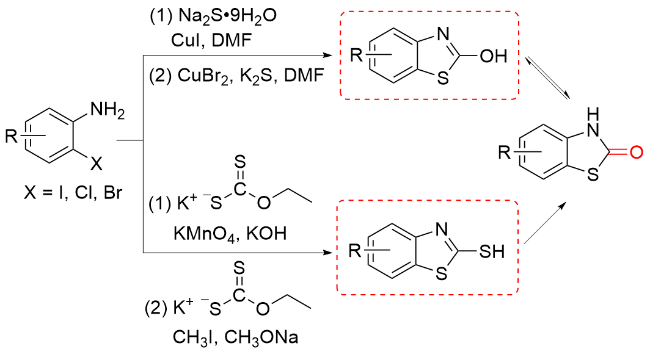
2-氨基苯并噻唑、2-卤代苯并噻唑和2-巯基苯并噻唑等2-取代苯并噻唑类衍生物作为重要的化工中间体, 在多个领域展现出广泛的应用价值. 这些化合物可通过重氮化反应、水解反应、取代反应或亲核加成-消除反应生成2-羟基苯并噻唑, 然后经酮-烯醇互变异构得到目标产物苯并噻唑-2-酮(Scheme 19). 其中, 2-氨基苯并噻唑经重氮化后, 可通过CuCl或CuCl2的催化卤代反应转化为2-卤代苯并噻唑[41], 然后再与硫代硫酸钠反应进一步转化为2-巯基苯并噻唑[42], 2-巯基苯并噻唑还可与磺酰氯反应生成2-氯苯并噻唑[43]. 下面将分别介绍以2-氨基苯并噻唑、2-卤代苯并噻唑、2-巯基苯并噻唑为底物合成苯并噻唑-2-酮的方法.
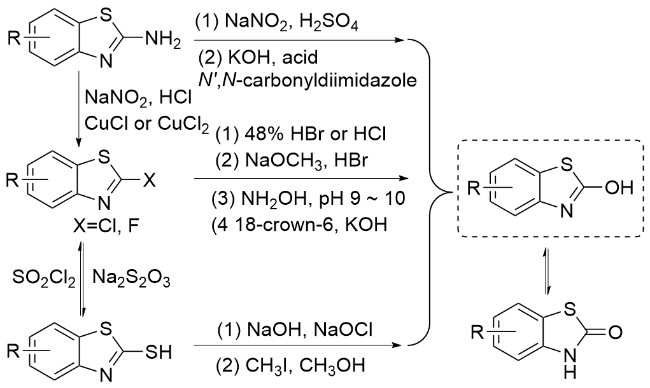
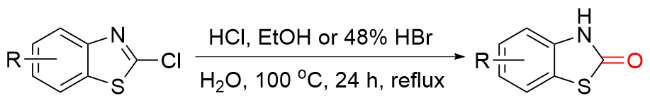
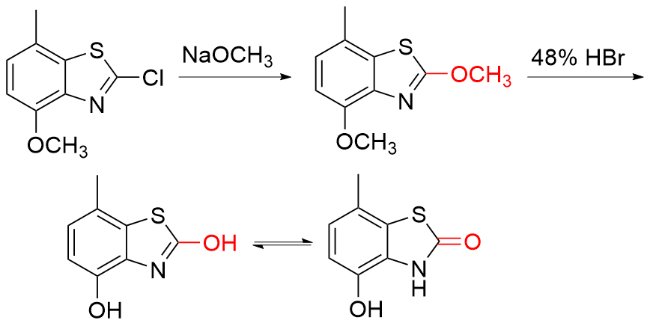
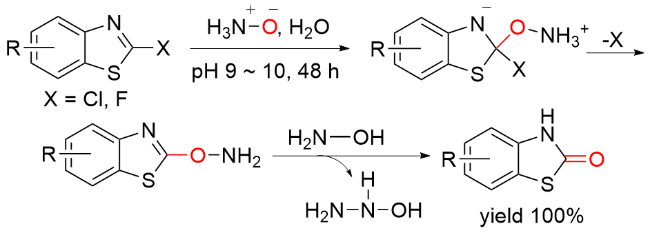
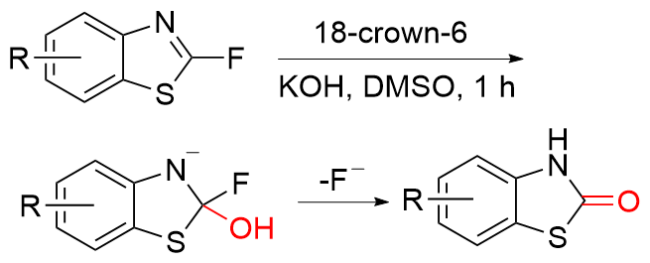
2.1 以2-卤代苯并噻唑为底物
2.2 以2-氨基苯并噻唑类衍生物为底物


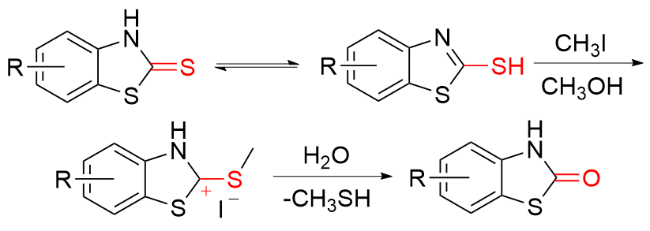
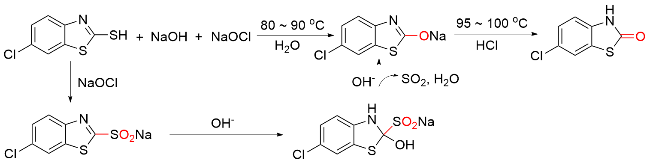
2.3 以2-巯基苯并噻唑类衍生物为底物
2.4 以2-H苯并噻唑为底物
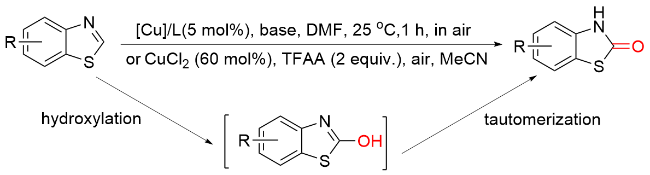
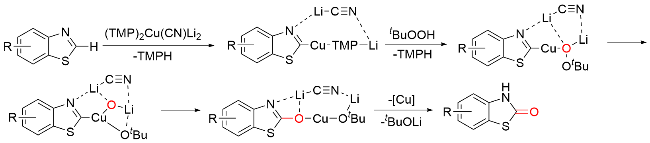
拟加氧酶是一类能够模拟天然加氧酶催化性能的化合物或配合物. 这类物质能够以空气中O2作为氧源, 通过活化作用将其氧原子高效转移至目标底物分子上, 主要用于催化分子氧与底物的氧化反应. 基于这一原理, 科研工作者开发了一种铜催化芳烃和杂芳烃的“拟加氧酶型”氧化反应. 这种铜催化剂具有与酶类似的催化性能, 能使2-H苯并噻唑发生氧化反应. 2012年, 刘强等[51]利用叔丁醇钠和二价铜盐代替酶, 开发了一种“拟加氧酶型”氧化反应, 并建立了一种合成苯并噻 唑-2-酮的新方法. 该方法以N,N-二甲基甲酰胺(DMF)为溶剂, 在CuCl2、NaOtBu和空气的存在下, 底物苯并噻唑先与铜形成配合物, 然后通过单电子转移和过渡金属催化的双重机制, 将O2中的氧原子直接转移到底物. 最终, 经水解反应以70%的产率得到目标产物苯并噻唑- 2-酮. 2024年, 陈丽等[52]利用CuCl2和三氟乙酸酐(TFAA)将2H-苯并噻唑氧化为苯并噻唑-2-酮类衍生物. 在反应过程中, 苯并噻唑、TFAA与CuCl2形成铜配合物, 该配合物再与O2反应生成Cu(III)-过氧配合物, 然后通过自由基环化释放三氟乙酸基得到2-羟基苯并噻唑, 最终经异构化得到苯并噻唑-2-酮(Scheme 28).
2.5 以其它2-取代苯并噻唑为底物
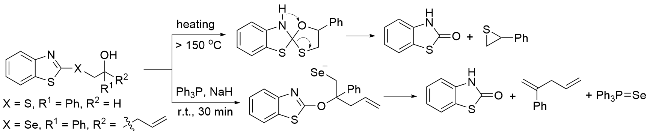
当2-取代苯并噻唑的2位取代的基团中含有羟基时, 其能够发生分子内重排生成苯并噻唑-2-酮. 2007年, Morita等[54]在密闭体系中通过对2-(2-苯并噻唑硫代)-1-苯乙烷-1-醇进行热解(温度>150 ℃), 成功地合成了苯并噻唑-2-酮, 同时有副产物苯乙烯生成. 当2-位碳与Se原子相连时, Se原子半径大于S, 其形成的C—Se键键能较弱, 故重排反应在室温下就能发生. 1992年, Mitsunobu等[55]以1-(2-苯并噻唑硒代)-2-苯基戊-4-烯-2-醇为底物, 在NaH和Ph3P作用下, 于THF中室温反应30 min, 以94%的产率生成苯并噻唑-2-酮. 在反应过程中, 底物在NaH作用下去质子化, 并进行分子内重排生成目标产物(Scheme 30).
当2-取代苯并噻唑的2位取代的基团中含有亚砜基时, 在一定条件下能够发生分子内重排生成目标产物苯并噻唑-2-酮. 1996年, Morita等[56]以1,4-二氧六环作溶剂, 利用2-(2-亚砜基苯并噻唑)-1-苯乙酮与过量的2,3-二甲基-1,3-丁二烯在100 ℃下反应24 h, 以83%的产率生成目标产物苯并噻唑-2-酮. 反应过程中, 受羰基邻位活泼α-H影响, 亚砜基能够异构为烯醇式结构, 增强了烯醇式羟基中氧的亲核性, 使其易亲核进攻邻近的正电性碳原子, 发生分子内重排生成O-取代物, 该取代物中的C=S键与2,3-二甲基-1,3-丁二烯发生加成反应, 生成目标产物(Scheme 31).

总结上述合成方法时发现, 2-取代苯并噻唑经一步反应即可构建苯并噻唑-2-酮的合成方法, 该合成方法相对简单, 但2-取代苯并噻唑通常先由邻氨基苯硫酚或邻卤苯胺合成, 增加了反应过程的成本和复杂性. 因此, 笔者试图寻找直接利用邻卤苯胺合成苯并噻唑-2-酮的方法, 为后续科研工作者提供更简洁的合成策略.
3 以邻卤苯胺衍生物为底物合成苯并噻唑-2-酮
邻卤苯胺类衍生物也是一种常用的医药及染料中间体. 在反应过程中, 氨基的供电子效应使卤素相连的碳原子的电子云密度增大, 进而削弱了卤素的离去能力, 增加了卤素位点的取代难度. 为了解决这一问题, 常采用过渡金属盐作为催化剂, 通过过渡金属催化的 C—S偶联反应, 实现脱卤并引入硫原子构建C—S键, 最终合成目标产物(Scheme 33).
3.1 碱金属硫化物合成法
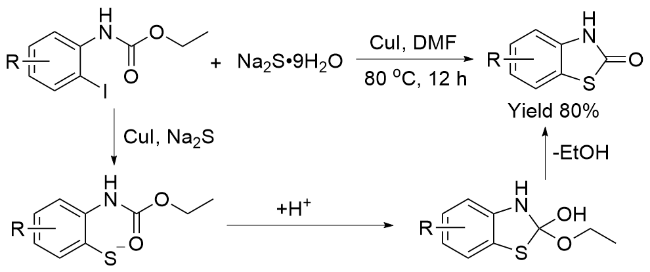
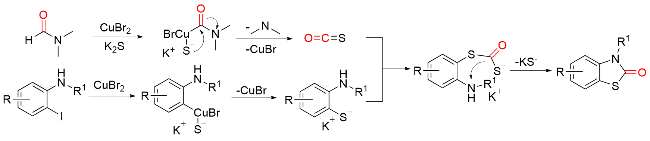
碱金属硫化物(如Na2S、K2S)相对于其它金属硫化物而言, 能在极性溶剂中具有优异的溶解性和离子解离度, 能够解离出大量S2-, 并在铜催化剂存在下, 这些硫化物通过氧化加成-还原消除机制与有机卤化物发生亲核取代反应, 实现C—S键的高效构建, 最终完成卤素取代并生成硫代目标产物. 2012年, 马大为等[58]以CuI为催化剂, 底物(2-碘苯基)氨基甲酸乙酯与Na2S•9H2O在80 ℃下反应12 h, 构建了苯并噻唑-2-酮的合成方法. 在反应过程中, 2-碘苯基氨基甲酸乙酯首先与CuI发生氧化加成反应, 形成铜-碘配合物. 然后, 溶剂中的S2-与Cu配合物发生配体取代反应形成铜-硫配合物中间体, 再通过还原消除反应使S-与芳环偶联, 并发生分子内环化反应得到目标产物(Scheme 34).
3.2 黄原酸钾合成法
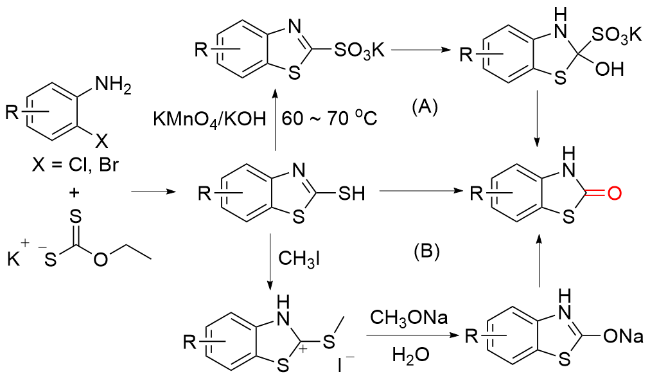
黄原酸钾是二硫化碳与乙醇钾反应的产物, 在有机反应中可作为固体二硫化碳源. 它与邻卤苯胺反应, 通常先生成2-巯基苯并噻唑, 然后通过KMnO4氧化或碘甲烷取代及水解, 最终生成苯并噻唑-2-酮. 2009年, Tzanova等[60]利用邻氯苯胺衍生物与乙基黄原酸钾在DMF中回流生成2-巯基苯并噻唑, 随后在60~70 ℃下, 利用KOH和KMnO4氧化水解制得苯并噻唑-2-酮类衍生物(Scheme 36A). Singh等[61]利用邻溴芳香胺与乙基黄原酸钾反应生成的2-巯基苯并噻唑, 再与碘甲烷反应得到2-甲硫基苯并噻唑碘化盐, 然后水解脱除甲硫基得到目标产物(Scheme 36B).
3.3 其它底物合成法
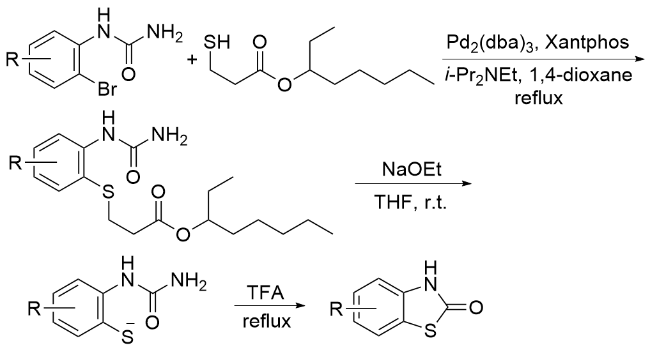
邻卤苯胺衍生物与巯基丙酸酯中的巯基可在过渡金属催化剂的作用下发生偶联反应, 再通过逆Michael加成转化为邻氨基苯硫酚负离子, 继而环化建立了苯并噻唑-2-酮的合成方法. 2007年, Itoh等[62]利用3-巯基丙酸-3-辛醇酯与邻溴苯基脲在钯催化剂的作用下, 经三步反应得到目标产物苯并噻唑-2-酮. 在反应过程中, Pd2(dba)3作催化剂, 双苯基磷衍生物作配体, 邻溴苯基脲与巯基丙酸酯发生偶联反应生成中间体, 然后在EtONa碱的作用下发生逆Michael加成反应形成S-并释放出丙烯酸酯, 最终硫负离子发生分子内亲核环化反应以58%的产率得到苯并噻唑-2-酮(Scheme 37).
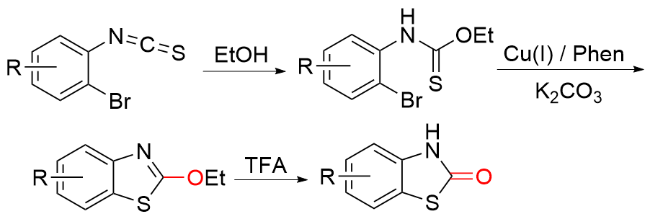
4 其它底物合成苯并噻唑-2-酮
芳胺、CO2和硫(S8)可在强碱叔丁醇盐的存在下, 通过低温纳米金(Au@Cellulose/DFNS)催化[64]或高温非催化[65]的方式建立了苯并噻唑-2-酮类衍生物的合成方法. 在反应过程中, 芳胺在叔丁醇盐的存在下与CO2反应形成脲或氨基甲酸酯, 然后转化为异氰酸酯中间体, 该中间体与S8发生分子间硫化反应生成中间体A, 该中间体通过去质子化和分子内亲核环化反应生成目标化合物苯并噻唑-2-酮(Path A). 此外, 八元环硫在强碱的作用下开环生成B, 随后对异氰酸酯亲核进攻形成C, 该中间体进一步发生分子内环化反应得到D, 最终去质子芳构化生成目标产物(Path B) (Scheme 39).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
苯并噻唑-2-酮类化合物在农业和生物医药等领域的广泛应用, 推动了众多化学工作者对其合成方法的深入研究. 笔者通过对苯并噻唑-2-酮骨架结构的合成方法进行分析, 发现主要包括以下三种: (1)以2-氨基苯硫酚为底物与多种羰基化试剂反应, 合成苯并噻唑-2-酮类衍生物. 该合成方法简单且易操作. 然而, 该底物在空气中稳定性差, 易氧化二聚成邻氨基二硫化物, 使含有多种取代基的底物难以制备, 因此苯并噻唑-2-酮类衍生物的合成受到了限制. (2)以2-取代苯并噻唑为底物合成苯并噻唑-2-酮, 这种方法反应条件温和, 但存在原料2-取代苯并噻唑的合成步骤较复杂, 且原子利用率低等缺点. (3)以邻卤苯胺为底物的合成方法, 原料成本低廉, 但合成路线复杂, 易产生副产物. 这些方法在合成路线、反应机理以及操作过程上各有优缺点, 为未来合成技术的创新提供了极为宝贵的研究方法. 本文在总结各类羰基化试剂的应用时, 发现了其与底物2-氨基苯硫酚的反应规律, 并依据原子经济性及羰基化机理对其展开分析, 为后续化学工作者利用现有羰基化试剂以及开发更多绿色高效的羰基化试剂奠定了坚实基础.
未来, 苯并噻唑-2-酮类化合物的研究将聚焦于绿色化学、高效催化、新型底物设计以及先进合成技术的整合, 致力于开发更可持续、经济和高效的合成路径. 首先, 绿色化学理念将贯穿合成过程, 通过开发环保型催化剂(如金属有机框架或生物酶)和绿色溶剂(如超临界流体或离子液体), 减少反应过程对环境的影响, 并提高原子经济性. 其次, 新型羰基化试剂的开发将成为重点, 例如利用CO2作为羰基源, 不仅降低成本, 还能实现碳资源的循环利用. 此外, 通过计算化学和人工智能技术, 深入理解反应机理并优化反应条件, 有望加速新型合成方法的开发进程. 在底物设计方面, 将开发更稳定的多功能底物, 以解决现有底物易氧化的问题, 并通过引入多样化的取代基, 拓展苯并噻唑-2-酮类化合物的结构多样性, 从而发掘更多具有优异生物活性的衍生物. 先进合成技术如光催化、电催化和流动化学的应用, 也将为温和条件下的高效合成提供新思路, 同时实现连续化生产, 提高工业化效率. 最后, 结合多功能衍生物的设计和工业化生产的优化, 苯并噻唑-2-酮类化合物的合成将更加高效、环保和经济, 充分满足医药、农药等领域对高性能化合物的需求. 总之, 未来的研究将融合多学科前沿技术, 推动苯并噻唑-2-酮类化合物的合成方法向绿色、高效和可持续方向发展, 为其在更广泛领域的应用奠定坚实基础.
(Cheng, F.)