


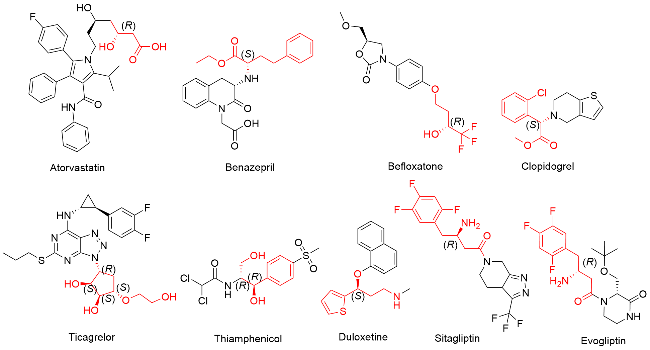
手性化合物是多种药物活性成分和精细化学品的核心骨架, 因其立体化学的独特性和合成挑战性, 在医药、化工和农业等领域具有重要的地位. 其中, 手性醇和手性胺是众多高价值药物中间体的基础砌块, 具有普遍性和典型性[1-2], 例如心脏治疗药阿托伐他汀(Atorva- statin)、降压药苯那普利(Benazepril)、抗抑郁药贝氟沙通(Befloxatone)等[3](图1). 特别是手性醇, 作为一种基础手性砌块, 市场份额占全球药物市场30%以上, 是药物化学领域的重要研究和应用对象[4-5]. 手性化合物不同异构体往往具有不同的生理活性, 甚至是截然不同的功效, 因此单一构型手性醇的高效精准合成仍然是药物化学领域长期需要解决的难题与挑战.
过去几十年来, 科学家们致力于探索不对称合成手性醇的绿色高效方法, 包括化学合成法和生物合成法. 化学合成是生产手性醇的主流方式, 但也不可避免地存在着一些局限, 例如手性金属配体催化剂设计困难、立体选择性不够理想以及生产成本高昂等; 与之相比, 生物催化剂因其立体选择性优、反应条件温和及环境友好等优势, 使得生物催化在手性醇的合成技术中脱颖而出[6]. 其中, 醇脱氢酶凭借其可催化羰基不对称还原或手性醇的对映体选择性脱氢的独特功能, 被广泛地应用于手性醇的不对称合成或动态动力学拆分反应[7].
酶分子由于其本身精巧的结构而具有精准催化的功能; 然而, 酶的催化混杂性也使其能够催化多种底物. 这种特异性与广泛性的共存形成了独特的“双向适配”动态平衡, 而维系该平衡的核心锚点是目前仍不清楚的多酶与多底物间的相互适配机制. 随着绿色可持续发展理念的不断深入以及复杂多样手性药物砌块精准合成的需求日益增长, 传统“大海捞针”式的酶资源挖掘策略日渐乏力, 研究者亟需借助AI深度学习模型实现酶功能的“智能预测”, 而模型预测的准确率离不开对广泛多样的多酶-多底物构效关系数据的深度学习. 本文作者倡议通过深入探究多酶与多底物之间的定量构效关系, 克服以往单酶-单底物适配机制的非定量认知局限. 为推动这一领域的起步和发展, 本文将聚焦醇脱氢酶/羰基还原酶与多底物分子适配性研究的进展, 总结目前对于构效关系关键数据-酶催化动力学参数的高通量实验测定方法, 分析现有ADHs底物谱设计和酶库构建的技术缺陷, 以便为手性药物砌块精准酶促合成技术的进步提供新的底层解决思路, 并助力我国药用醇脱氢酶-多底物构效关系开放数据库及智能设计模型的发展与应用.
1 醇脱氢酶简介
醇脱氢酶(Alcohol dehydrogenases, ADHs, 酶学编号EC 1.1.1.x)也称羰基还原酶(Carbonyl reductases, CRs)或酮还原酶(Ketoreductases, KREDs), 这类酶属于氧化还原酶家族, 依赖烟酰胺腺嘌呤二核苷酸(NAD+/ NADH)或烟酰胺腺嘌呤二核苷酸磷酸酯(NADP+/NADPH)辅酶作为电子供体或受体, 可催化醇羟基的脱氢氧化或其逆反应羰基还原[8-9]. 根据结构与机制的不同, 目前将醇脱氢酶分为三个超家族(图2): (a)短链脱氢酶/还原酶(Short-chain dehydrogenase/reductases, SDRs), 其属于非金属依赖酶, 包括经典型和扩展型, 每条肽链一般含250或350个左右的氨基酸残基[10], 短链脱氢酶通常以单体、二聚体、四聚体、八聚体或十聚体等同源多聚体的形式存在; (b)中链脱氢酶/还原酶(Medium- chain dehydrogenases/reductases, MDRs), 长度为350个左右的氨基酸[11], 大都为锌离子依赖型, 中链脱氢酶一般是单聚体、二聚体或四聚体的形式存在; (c)醛酮还原酶(Aldo-keto reductases, AKRs), 每条链一般含385个左右的氨基酸, 少数含900个左右的氨基酸, 并大多以单体形式存在[12]. NAD(P)+依赖的醇脱氢酶催化醇(羰基)底物的不对称氧化(还原)反应遵循顺序反应机制. 首先, 辅因子与酶结合诱导蛋白由无序的状态形成适合底物分子结合的催化口袋, 成为全酶. 此后, 底物分子结合在活性口袋进一步诱导全酶形成合适的催化构象推动催化进行. 反应生成产物和氧化态或还原态的辅酶, 先释放产物, 最后释放辅酶[11]. 醇脱氢酶催化前手性羰基化合物还原反应的过程简便, 理论收率可达100%, 故以醇脱氢酶催化合成手性醇的方案因应用前景广阔而备受学术界和产业界瞩目.
然而, 现有ADHs酶催化体系的产业化应用受限于酶的底物宽泛性不足问题: ADHs与多样性底物分子的构效关系解析尚浅. 研究表明[13], ADH对不同底物的催化效率存在数量级的差异, 尤其对含杂原子、大位阻或刚性结构的底物适配性普遍较低, 因此在合成高价值的非典型、非天然手性化合物时, 通常难以快速地定制获得具有足够活性的酶催化剂. 为了克服上述限制, 深度探索ADHs与各类手性醇类药物前体的适配关系(特别是定量的构效关系)逐渐显现出其必要性与重要性, 这对酶的智能设计和制药产业的发展均具有重要战略意义.
2 多酶-多底物构效关系矩阵(数据库)的设计、构建与应用
之前酶工程领域的研究大部分遵循“单酶-单底物”的研究模式, 如通过理性或半理性设计, 针对某一手性醇的合成实施ADH酶的定向改造, 或者利用定向进化或定点饱和突变, 经过大量的筛选来寻找催化特定底物的高效ADH突变体. 这种“一对一”的传统研发策略导致酶的资源开发周期长且不确定性高, 无论在经济上还是在时间上, 都难以满足制药工业多样化手性砌块的合成需求. 面对大量前所未见的非天然底物进行单一醇脱氢酶结构改造的传统方法已经日渐乏力. 随着人工智能时代的到来, 涌现出一批蛋白质功能预测模型, 但是目前模型所存在的共性问题是对未见底物的泛化能力严重不足. 因此, 构建多酶-多底物构效关系数据库是突破酶的功能预测模型泛化性瓶颈的关键所在, 其工业价值不亚于蛋白质结构数据库(PDB)对结构生物学的推动作用. 唯有更全面、更规范的构效关系大数据驱动, 才能真正提升酶的底物适配与功能预测模型的精度和泛化能力.
近年来, 已有研究人员逐渐将目光转向ADHs-多底物适配性的研究, 然而目前报道的底物谱设计方法多遵循“单核衍生”扩展模式, 即以一模式底物为母核, 通过局部的基团修饰(如苯环取代基、碳链长度微调)构建衍生底物库, 平均每个ADH只能验证少数几种非天然底物. 当然, 最近也有部分团队开始尝试将底物谱从“单核衍生”向“功能基团分级覆盖”的方向延伸.

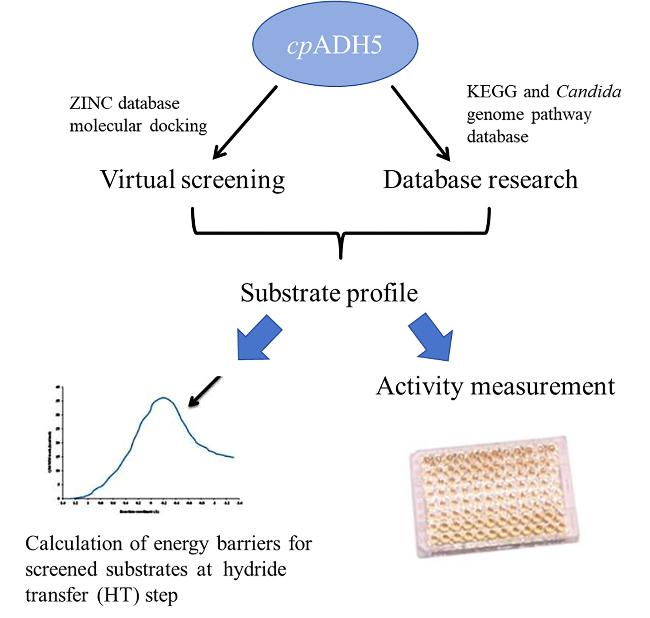
该研究选择了基于底物分子对接计算的虚拟结合数据库搜索方法(图4), 最终确定了52种具有潜在活性构象的底物集合(库). 由此构建的底物谱涵盖范围比较广, 通过从大量化合物中筛选出潜在底物, 挖掘出多种可能与cpADH5结合的化合物, 准确性相对较高, 但仅仅包含部分基础醛/酮和多元醛/酮, 其典型性和覆盖度仍显不足, 且催化活性的表征仍然围绕单一酶展开, 不具有统计学意义.
2018年, 倪晔团队[15]采用多样性统计指标: 香农-威纳指数(Shannon-Wiener index)
$\left.\mathrm{S}-\mathrm{W} \text { index }=-\sum_{i=1}^{n} P \mathrm{si} \times \ln P \mathrm{si}, P \mathrm{si}=A \mathrm{si} / A\right)$
n为所有测试底物的数量, Asi为酶对特定底物i (si)的比活性, A为酶对所有测试的前手性酮的比活性之和, 即酶对某一底物的特异性活力与酶对全部底物活力总和的比值, 来描述醇脱氢酶底物谱的宽泛性. S-W指数的生物学意义在于量化酶对不同底物的催化活力分布均匀性: 指数数值越高, 表明酶能高效催化的底物种类越多, 且对各类底物的活力分配越均衡, 即底物谱越宽泛; 反之, 数值越低则说明酶的催化偏好性越强, 仅能催化少数几种底物, 底物谱较窄. 该研究采用基因组挖掘策略从Candida glabrata中筛选羰基还原酶, 从生物信息学推定的55个羰基还原酶基因编码库中, 利用大肠杆菌BL21 (DE3)成功表达了37种羰基还原酶, 分别进行酶学性质的测定和表征. 所选底物分为4类, 共有32种不同的酮基底物, 确定Cg26酶的活性最高, 且底物谱最广(S-W index=2.82), 显著高于其他酶(图5). 该研究引入了兼具活性高低程度和底物范围宽窄程度的定量化筛选标准, 突破了传统的“单酶-单底物”研究模式, 不仅建立了较为完善的酶库, 底物谱也更加多样化; 同时通过S-W index指数评估了每个ADH所催化的底物多样性, 筛选出了底物谱宽且效率高的羰基还原酶. 该课题组在2022年延续类似的研究思路, 开发了定向复活具有所需特性祖先醇脱氢酶的策略, 并且同样使用S-W index来描述所复活的祖先酶A64的底物谱广度[16]. 综合两次研究结果来看(图6, 7), 该团队综合考虑了底物结构的多样性和酶序列的多样性, 为构建酶的多底物适配模型提供了更加精确的数据, 并且“多酶-多底物”矩阵的构建思想也初步得到体现. 略有遗憾的是, 作者两次选择的底物库仍遵从“母核衍生”策略, 底物类型还较单一, 不能全面反映ADH酶的潜在底物化学空间, 限制了对酶催化能力的认知边界.
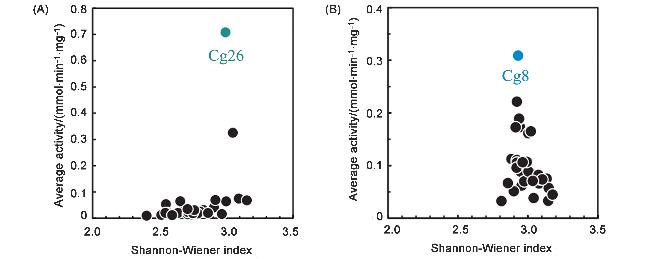
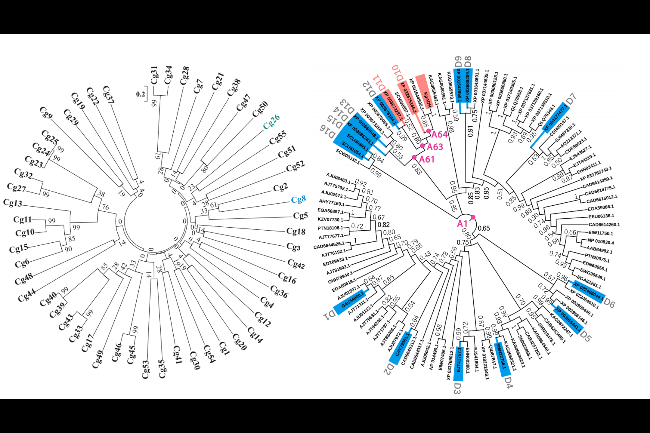
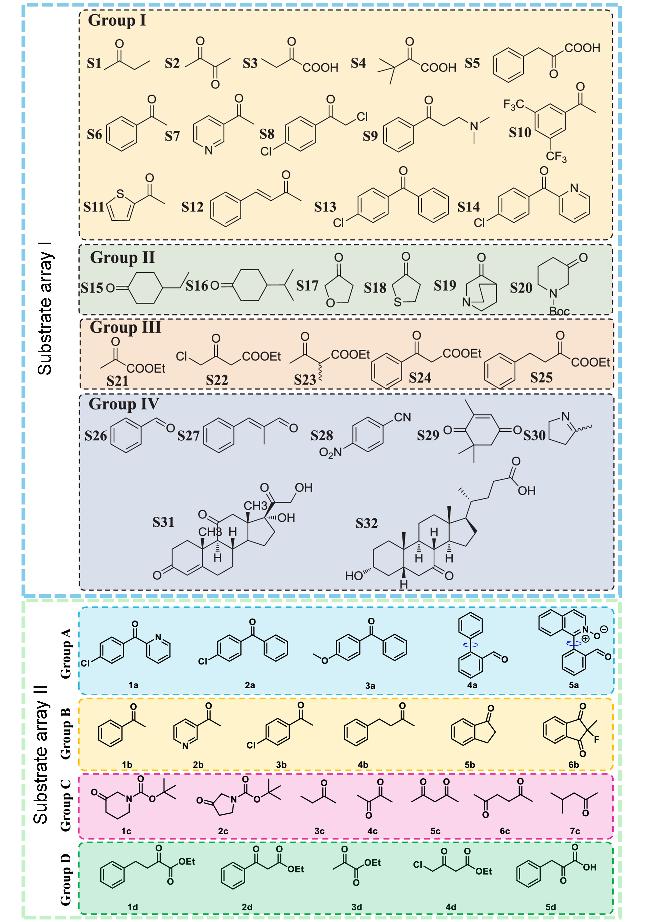
图7 两次酶学表征过程所用的不同羰基底物集合: 底物阵列I和底物阵列II[15-16]Figure 7 Collection of different carbonyl substrates used in the two enzymatic characterization processes: substrate array I and substrate array II[15-16] Substrate libraries are classified into subgroups according to the type of substituent and reducing group of the substrate and are distinguished by different background colors |
目前, 生物催化技术在手性化合物的实际生产中已经得到广泛应用, 但是因其难以快速将不同手性药物砌块同时与酶库相匹配的问题, 使得酶催化剂在早期药物研发阶段的应用较少. 因此, 研究人员不仅尝试挖掘具有底物宽泛性的催化元件, 还有意识地通过多种实验方法全方位探究酶的底物谱范围. 近些年, 随着大数据科学与人工智能技术的推广与应用, 有望建立起更高效、更全面的底物导向型酶序列智能化筛选方法, 甚至是针对新颖结构的“未见”底物具有一定泛化能力的ADH功能预测模型.
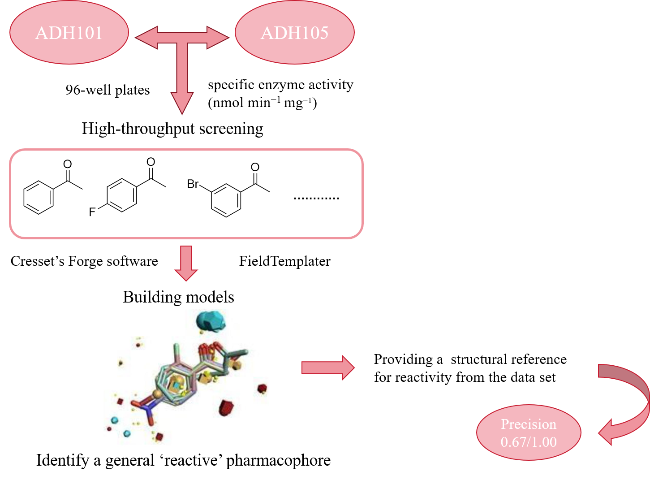
2023年, Reeve课题组[19]以两种商用醇脱氢酶(R)- ADH (Johnson Matthey, ADH101)和(S)-ADH (Johnson Matthey, ADH105)为研究对象, 对40多种药物基序的酮基底物进行了比活力测定, 运用Cresset的Forge软件, 采用基于药效团(从部分结构多样的底物中识别出一个通用结构参照标准)的方法对ADH的反应性进行建模, 开发了一种预测底物反应性的筛选工具, 在无需了解酶序列的情况下能够预测给定底物的反应可能性, 对测试集化合物反应性的定性分类精度达67/100(图10). 该研究证明了醇脱氢酶在多种手性醇合成方面表现出色, 研究成果为生物催化技术早期应用于药物化学合成提供了新途径, 未来可为进一步用于酶促合成手性药物砌块探索更多的催化空间.
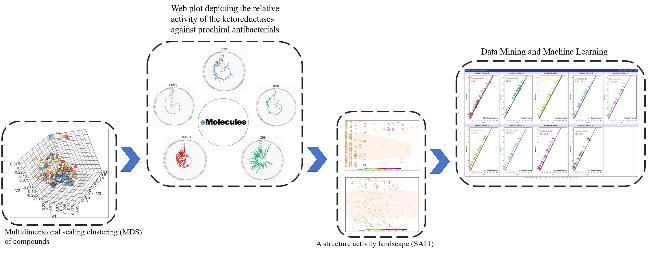
2024年, Santanu研究团队[20]报道了一种利用机器学习算法所建立的不同ADHs与其理想底物的关联性模型来解析二者匹配能力的研究. 作者选取4种短链脱氢酶(分别是经典型短链脱氢酶FabG、SCR和扩展型短链脱氢酶DHK、ZRK)和一种中链脱氢酶(LKADH), 对多达284种具有抗菌活性的潜手性酮依次进行了反应活性测定, 随后利用机器学习算法对化合物进行聚类分析. 比Reeve课题组基于药效团的研究更进一步的是, 作者发现仅依靠底物的化学结构分类, 不足以确定不同ADHs对底物的偏好性, 因此作者利用偏最小二乘(PLS)回归、K最近邻(KNN)回归和支持向量机(SVM)回归等算法, 结合33个特征参数和4个描述符进行了回归分析, 在酮基化合物的分类上取得较好的决定系数(R-square=0.8). 在此基础上, 作者深度挖掘数据的内在区分模式和方差, 增强了对化合物类别的区分和分离能力, 使其分类精度和可靠性显著提升, 最终将化合物分为9类, 更精确地确定了不同ADHs可催化的化合物类别(图11). 这种方法的优势在于能够更高效地对化合物进行可催化性分类, 有助于预测新的活性底物-酶适配对子, 减少对高通量筛选方法和复杂酶工程技术的依赖, 但是当前模型的训练集规模(酶<10种, 底物<10³种)仍不能满足深度机器学习的数据需求(>106), 应考虑设计构建“百万级”多酶-多底物适配关系数据库, 以提升模型预测的精度.
以上研究揭示了明显的发展趋势, 即手性醇合成酶催化剂的开发正逐渐趋向多酶与多底物之间适配关系的学习, 而不再是单个蛋白质如何与感兴趣的化合物相互作用. 与传统的酶筛选和工程化方法相比, 机器学习对ADH的精准设计及手性醇类药物的开发与应用具有变革性创新意义. 建模方法更加侧重于多酶与多底物之间适配关系的机器深度学习, 而不仅是基于单一靶点的分子对接.
3 分子适配度的动力学实验表征方法
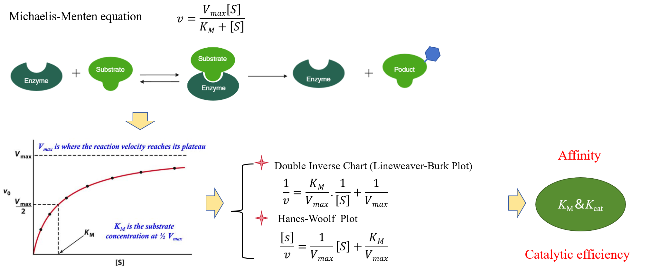
醇脱氢酶催化氧化或还原反应时总是伴随着辅因子的生成或消耗, 因此可以通过检测辅因子浓度的变化来反映底物的消耗量. 例如, Schwaneberg课题组[14]在测定cpADH5活力时, 在微孔板中放置了不同浓度的底物溶液, 并加入0.25 mmol/L的NADH或NAD+辅酶溶液, 最后加入含有cpADH5的细胞裂解液上清启动反应. 在30 ℃进行保温过程中, 使酶与底物充分接触并发生反应, NADH或NAD+参与反应, 其浓度会随反应进程发生变化, 最后在340 nm紫外光的照射下, 按照一定的时间间隔记录微孔板各孔穴的吸光度, 收集吸光度随时间变化的数据, 选取数据变化呈线性的部分进行分析. 基于米氏方程拟合出cpADH5对各底物的KM和Vmax值. 由于细胞裂解物上清液对紫外光也有较强的背景吸收, 这种方法一定程度上存在较大误差.
为了减少背景噪声, 考虑到细胞裂解液对可见光的吸收明显弱于紫外线, 研究人员提出了一种将氧化还原反应与染料生成反应相结合的可见光比色方法. 目前使用最广泛的一种比色方法是硝基蓝四氮唑/甲基硫酸吩嗪(nitroblue tetrazolium/phenazine methosulfate, 简称NBT/PMS)测定法, 该法在1993年由Fibla和Gonzàlez-Duarte提出, 并用于测定ADH的活性[21]. 在PMS存在下, ADH催化的醇脱氢反应所产生的NAD(P)H会使NBT还原, 生成水不溶性的蓝紫色甲瓒产物(Formazan)[22], 据此可通过测定580 nm处吸光度的变化来反映底物的消耗量, 进而测定反应进程曲线, 计算出ADH活性. 这种方法在显著增强低浓度NAD(P)H响应信号的同时, 原位移除了NAD(P)H, 使其不会干扰ADH所催化的醇羟基脱氢反应.
2021年, 周康课题组[23]开发了一种基于分泌物的双荧光检测法SDFA (Secretion‐based dual fluorescence assay), 并应用于ADHs的高通量筛选. 该方法将ADH与突变的超级折叠绿色荧光蛋白(Monomeric Superfolder Green Fluorescent Protein, MsfGFP)进行基因融合表达, 并使融合蛋白分泌到培养液中, 通过产物的红色荧光信号测定ADH活性, 同时利用MsfGFP的绿色荧光信号定量测定ADH的浓度, 大大提高了筛选效率. 研究人员利用该法对来自Pichia finlandica的ADH成功进行了定向进化, 筛选出的变体对(S)-2-辛醇的底物特异性常数(kcat/KM值)比野生型提高了197倍[23], 但操作流程稍显复杂, 普遍推广使用还面临挑战.
2021年, Hollfelder团队[24]报道了一种基于微流控液滴荧光的检测方法, 合成了新型吡喃型荧光底物用于检测醇脱氢酶的活性. 该底物经ADH催化反应释放吡喃型荧光团, 荧光物质在液滴中保留时间长达6周以上, 稳定性远高于荧光素. 该方法的检测限低至100 nmol/ L, 能检测到单个酶分子催化≤1次的反应. 通过模型筛选实验, 实现了对活性ADH的高效富集, 富集倍数达800以上, 为快速筛选高活力的ADH提供了新手段, 实现了液滴微流控技术在ADH检测中的应用, 但相较于传统表征方法, 必须购置昂贵的实验设备.
从米氏方程驱动的系列底物浓度的传统催化参数测定到融合荧光技术与微流控平台的创新检测方法开发, 酶的多底物适配度表征方法正经历从定性分析到定量测定、从单一酶对单一底物的测定到高通量集成方法的深刻变革(图13), 从而为ADHs与多底物之间构效关系的定量快速解析提供了更敏锐、更高效的“观测窗口”.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 挑战与展望
尽管醇脱氢酶的多底物适配度定量研究取得一定进展, 但该领域仍面临诸多挑战: 底物多样性不足、覆盖度有限, 且大多限于天然底物的衍生结构, 而酶对杂环底物以及空间大位阻底物的适配性研究仍显不足; 现有动力学表征体系仍然存在测量误差或成本约束, 制约了酶动力学参数的高通量测定; 机器学习模型的泛化能力低, 现有的计算模型对非天然底物可用ADH的预测精度严重不足, 缺乏覆盖度更加宽泛的ADHs-多底物适配关系标准数据的强化训练.
酶作为精准催化绿色合成的典型代表, 往往在“一酶专一催化单一分子”和“一酶全能催化多个分子”的矛盾中徘徊. 本文所倡议的多酶-多底物构效关系研究, 不仅为手性药物砌块的酶促合成提供了变革性思路和技术可行路径, 更预示着一个崭新的智能化生物催化时代正在迅猛到来. 通过解码活性酶的编码序列与活性底物的结构编码之间的适配逻辑和定量关系, 有望突破过去传统的“单酶-单底物”匹配机制(锁-钥关系学说)的历史局限, 实现酶催化剂的理性设计与功能预测. 更深远的意义在于, 这一领域或将成为绿色精准化学的新旗帜: 当多酶-多底物的适配逻辑被完全解码, 生物制造将实现更高的原子经济性, 推动制药工业向碳中和和生态文明目标迈进关键的一步. 这既是合成生物学从“格物致知”到“造物致用”的跨越式发展, 更是绿色精准合成化学从理念倡导到技术落地的关键突破口. 期待更多的研究力量投入这一领域, 共同绘制生物催化新时代深刻而宽广的多酶-多底物构效关系“全景图谱”.
(Lu, Y.)