

吲哚生物碱在自然界中分布广泛, 结构类型多样, 且具有抗炎、镇痛、抗肿瘤、抗氧化和抗菌等一系列的治疗作用[1]. 吲哚生物碱复杂的化学结构以及丰富的药理活性使其全合成研究成为了有机化学和药物化学研究中的重要内容, 同时也推动了新的合成方法学以及高效全合成策略的快速发展[2]. 蕊木属植物为夹竹桃科的重要成员, 在我国以及东南亚、印度等地均有较广泛的分布, 从中分离得到的蕊木属生物碱具有新颖的骨架结构以及明确的药理活性[3], 因此蕊木属生物碱近年来吸引了有机合成领域的广泛关注[4].
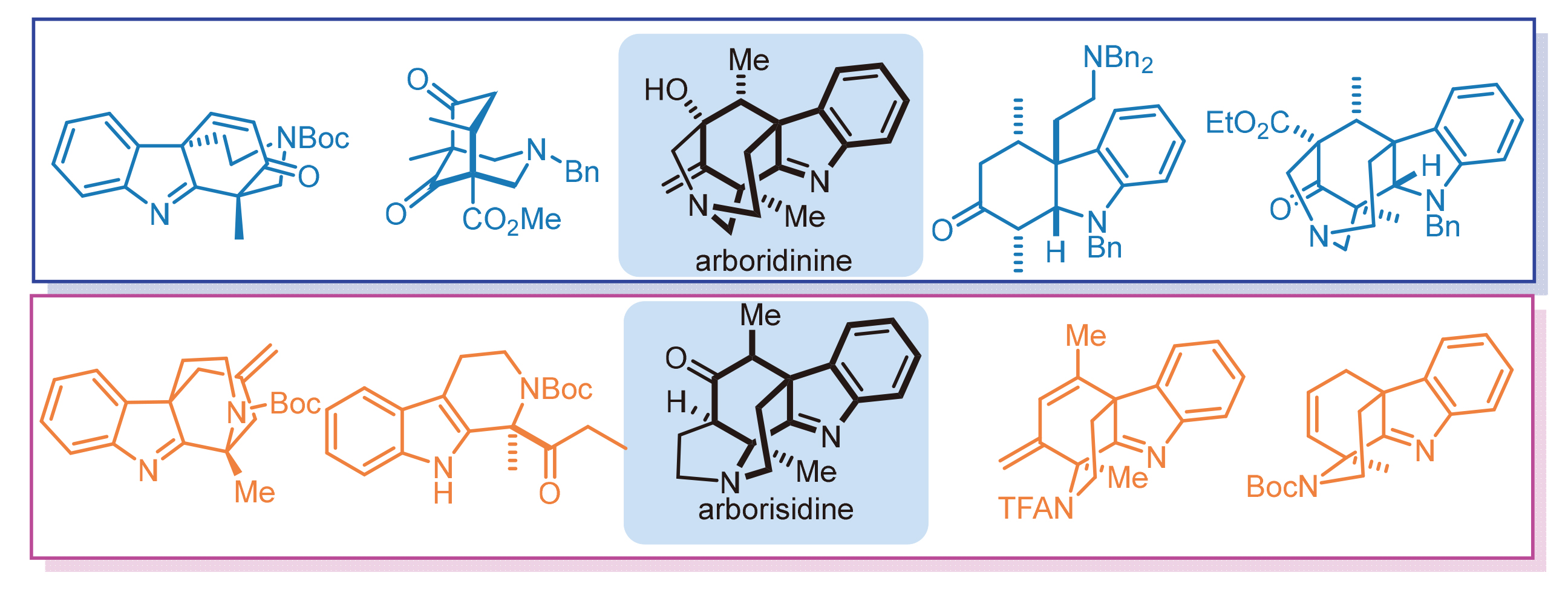
2015年, Kam课题组[5]从马来西亚的蕊木属植物K. arborea中分离得到了一个具有五环结构的吲哚生物碱arboridinine, 随后在2016年, 该课题组[6]又从相同的植物中分离得到了arborisidine. 这两个生物碱均具有较为特殊且复杂的笼状结构, Kam课题组[5-6]推测这两个生物碱可能来源于共同的生源前体pericine. 从结构上看arboridinine和arborisidine的核心骨架(ABCD环系)具有一定的相似性(图1), 但在C环和E环的构造上有所差异. 其中arborisidine的四环骨架在akuammiline家族生物碱(如akuammiline和strictamine)中较为常见, 但arborisidine的E环结构为四氢吡咯环, 这与akuammi-line家族生物碱中常见的桥连哌啶环系有所差异. 这两个生物碱均具有高度复杂的笼状结构以及多个手性中心, arborisidine中还具有一个较为特殊的氮杂季碳手性中心, 为其全合成研究增加了不少难度. 具有复杂笼状结构的生物碱全合成难度大, 这些分子大多是目前有机合成领域的热门分子, 例如灯台生物碱strictamine[7]、钩吻生物碱koumine[8]、蕊木属生物碱kopsine[9]等. Arboridinine和arborisidine由于分离时间较短, 其潜在的药用价值仍有待挖掘, 目前仅有一篇专利报道了arbori-sidine与匹美劳肽联用时具有明确的抗胃癌作用[10]. 由于这两个生物碱具有较为独特而极具合成挑战性的结构特征, 其全合成研究颇受关注, 本文综述了目前arboridinine和arborisidine的全合成研究进展, 希望可以为相关天然产物的合成提供有益的参考.
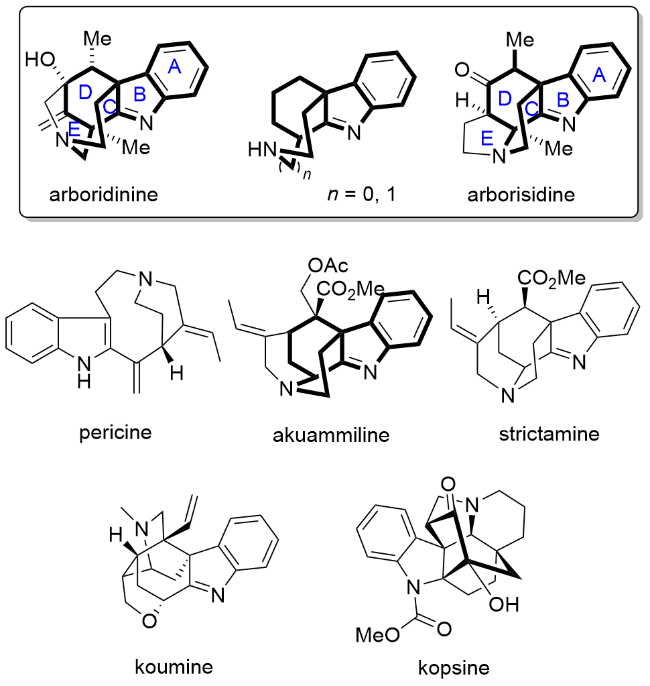
1 Arboridinine和arborisidine的全合成研究进展
1.1 Snyder课题组对arboridinine和arborisidine的全合成研究
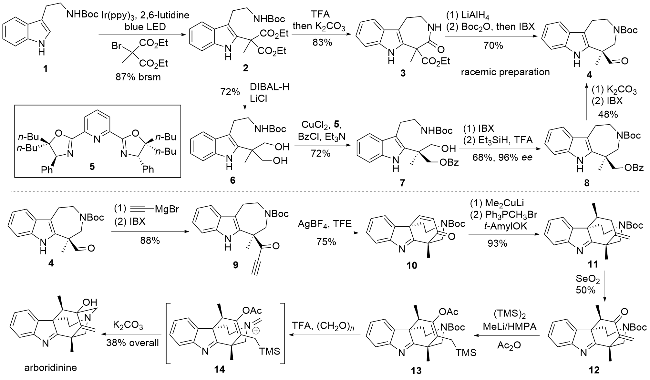
该路线从叔丁氧羰基(Boc)保护的色胺1出发, 利用光自由基反应完成吲哚C2位的官能团化得到化合物2, 随后在三氟乙酸作用下脱除N-Boc保护并实现内酰胺化得到化合物3. 接着利用四氢铝锂还原酰胺和酯基, 并利用Boc酸酐重新实现N-Boc保护, 同时利用2-碘酰基苯甲酸(IBX)将上一步还原得到的伯醇氧化为醛基, 即可实现消旋中间体4的合成. 为了实现目标天然产物的不对称合成, 作者还探索了光学纯中间体4的合成方法. 从化合物2出发, 利用还原条件将两个酯基同时还原得化合物6, 作者尝试利用酯酶实现双醇底物的不对称单酯化, 但未能成功. 最终利用Kang课题组[13]发展的不对称单苯酰反应成功得到了化合物7, 随后利用IBX将羟基氧化为醛, 并利用还原胺化反应完成七元环构建, 经过脱苯酰基保护以及IBX氧化即可完成关键中间体4的不对称合成.
完成关键中间体4的合成之后, 利用炔基格氏试剂引入炔基侧链, 并利用IBX将生成的仲醇氧化为酮得到化合物9, 紧接着利用银催化的分子内6-endo-dig环化反应实现了炔基端位与吲哚C3位的连接得到关环产物10. 通过分子本身位阻利用甲基铜锂实现不对称1,4-加成, 随后通过Wittig反应成功构建环外双键, 并利用SeO2重新构建α,β-不饱和酮得到化合物12. 在最后阶段, 作者希望利用氮杂Prins环化反应完成最后环系的构建, 具体为通过1,4加成在双键端位引入三甲基硅烷, 同时利用乙酸酐原位捕获生成的烯醇得到乙酰化关键关环前体13, 用三氟乙酸将N-Boc脱除, 暴露的氨基与多聚甲醛发生缩合得到中间体14, 随即发生氮杂Prins环化完成关环, 最后将乙酰基脱除即可完成arboridinine的全合成.
2019年Snyder课题组[12]又完成了arborisidine的全合成研究, 在该全合成中同样利用了金属介导的环化反应实现了关键环系的构建, 具体路线如Scheme 2所示. 与arboridinine的全合成较为类似, 作者均采用了不同的路线分别实现消旋以及手性底物的制备, 消旋中间体16的制备较为简单, 以色胺为原料与2,3-丁二酮经过Pictet-Spengler反应(P-S反应)即可顺利得到. 而光学纯中间体16制备则相对复杂, 以D-色氨酸甲酯为原料, 与2,3-丁二酮发生P-S反应, 可以1.9∶1的非对映选择性, 即以62%的收率得到目标构型产物18. 由于直接的脱酯基操作未能成功, 只能通过一锅法的氨解/脱水步骤将酯基转化为氰基得到化合物19. 经过一系列条件筛选, 最后利用氰基硼氢化钠, 并添加对三氟甲基苯甲醛抑制副产物的生成, 成功得到脱氰基化合物16.
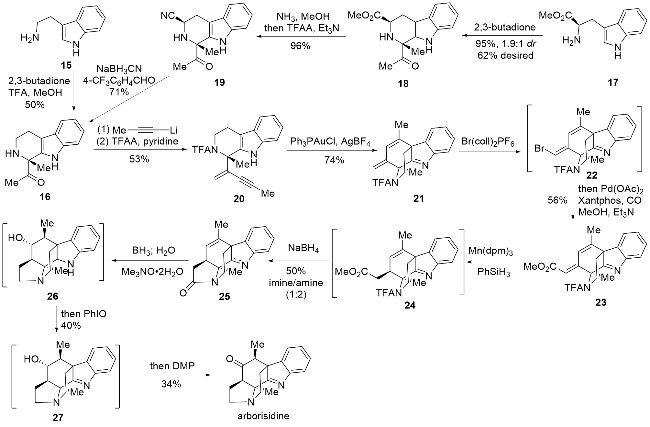
由于arborisidine与strictamine具有较为类似的四环骨架结构, 受到其课题组前期strictamine全合成[14]的启发, 作者希望利用金属催化的6-endo-dig环化实现四环骨架的构建. 在化合物16中引入炔基片段后利用三氟乙酸酐(TFAA)促进生成的羟基发生脱水反应, 同时将氨基进行三氟乙酰化保护得到关环前体20, 通过预期的分子内环化反应即可得到四环化合物21. 随后实现环外双键选择性溴代得到中间体22, 无需纯化即可通过后续的钯催化的羰基化反应得到化合物23. 利用锰催化的氢迁移反应得到1,4-氢化产物24, 该粗品用硼氢化钠处理即可脱除三氟乙酰基保护, 同时促进内酰胺的生成, 此外该过程也会使亚胺部分还原得到化合物25. 接着利用硼氢化氧化反应引入羟基, 该过程也会伴随亚胺和酰胺的完全还原, 之后用亚碘酰苯重新将其氧化为亚胺结构, 随后用戴斯-马丁试剂(DMP)将羟基氧化成酮即可得到arborisidine. 该过程同时也会得到羟基未被氧化的中间体27, 将其分离后仍可用DMP处理得到目标天然产物.
Snyder课题组利用高效的环合策略, 实现了arboridinine和arborisidine中高度复杂笼状骨架的构建, 整体合成路线较为高效, 尤其是关键骨架构建策略对于其他天然产物的合成具有较大的借鉴价值. 主要的不足在于两个天然产物的不对称合成路线相较于消旋路线更加复杂, 尤其是在arborisidine合成中, 利用手性色氨酸甲酯制备手性中间体的策略, 整体转化较为繁杂, 后续如能实现直接利用不对称的P-S反应构建手性中间体, 进一步提升全合成的效率, 因此这也是后续仍值得探究的研究内容.
1.2 翟宏斌、李云课题组对arboridinine的全合成研究
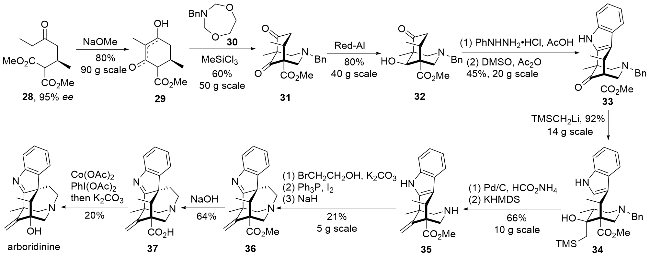
2019年翟宏斌、李云课题组[15]以双Mannich反应高效构建氮杂[3,3,1]双环为关键步骤, 实现了arboridinine的不对称全合成(Scheme 3). 为了实现目标天然产物的不对称合成, 作者首先通过条件筛选, 实现了不饱和酮与丙二酸二甲酯的不对称Michael加成反应, 从而完成了手性原料28的制备. 在碱性条件下发生Dieckmann酯缩合反应得到化合物29, 随后与化合物30反应, 通过双Mannich反应成功构建具有氮杂[3,3,1]双环的关键中间体31. 为了避免后续反应过程中逆Mannich反应的干扰, 作者希望将不参与构建吲哚结构单元的酮羰基先还原, 通过大量的条件筛选, 发现红铝可以实现该酮羰基的选择性还原得到化合物32. 之后利用Fischer吲哚合成法引入吲哚片段, 并利用Swern氧化重新将羟基氧化成酮得到化合物33. 用锂试剂对羰基进行加成得到化合物34, 经过N-Bn脱保护以及Peterson成烯反应生成环外双键得化合物35, 随后通过氮烷基化、羟基碘代以及分子内烷基化反应构建最后一个环系得到化合物36. 在合成的最后阶段, 需要将羧酸甲酯转换成羟基, 首先利用碱性条件将酯基水解为羧酸37, 但是在如此复杂的结构中实现羧基向羟基的转化未能成功. 最终, 作者参考已报道的钴催化脱羧/乙酰氧基化反应[16], 并利用碱性条件水解乙酰基, 成功得到了目标产物arboridinine, 当然该反应的产率不是很理想, 原因是反应过程中还伴随着仅脱羧的副产物.
1.3 祝介平课题组对arborisidine的全合成研究
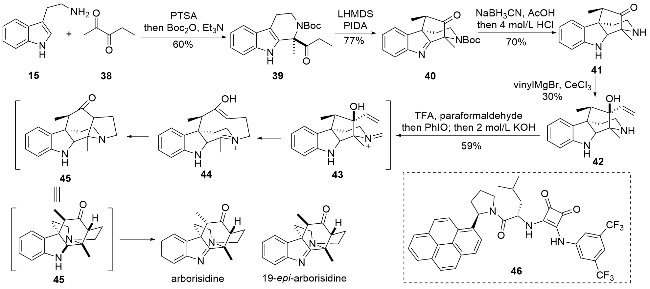
作者首先利用消旋路线进行全合成尝试, 色胺与戊二酮发生P-S反应, 并在氮上引入叔丁氧羰基(Boc)保护基, 得到化合物39, 随后利用分子内的氧化偶联反应关环得到化合物40. 后续利用烯基格氏试剂进行亲核加成时发现存在副反应, 因此先将化合物40中的亚胺结构进行还原, 同时将N-Boc保护脱除. 亚胺还原是为了避免副反应的发生, 而脱除Boc则是为了减少该侧的位阻影响, 使得亲核加成的立体选择性可以符合预期要求. 随后对化合物41进行亲核加成, 顺利得到化合物42, 最终通过氮杂Cope重排串联Mannich反应即可完成所有环系的构建. 该串联反应的具体过程为: 氨基首先缩合形成亚胺盐中间体43, 随即通过氮杂Cope重排反应得到中间体44, 然后发生分子内的Mannich反应重新关环得到中间体45, 为了得到目标天然产物中的亚胺结构, 可以直接加入亚碘酰苯实现该氧化过程, 但是作者发现仅通过上述流程主要得到的是19-epi-arbori- sidine. 为了得到目标天然产物的正确构型, 可以在反应的最后阶段加入KOH促进其异构化, 这样就能顺利完成arborisidine的全合成.
1.4 焦雷课题组对arborisidine的全合成研究
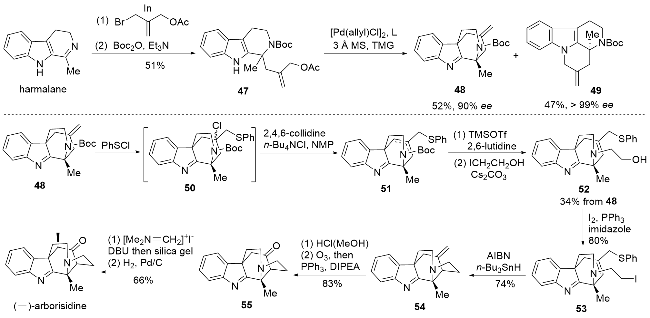
以harmalane为起始原料, 利用铟介导的亲核加成反应以及N-Boc保护即可得到化合物47, 为了进行不对称合成, 作者原本希望利用钯催化的烯丙基化反应实现动力学拆分, 通过一系列条件筛选后发现, 外消旋化合物47的其中一个构型可以在钯催化下实现C3位的烯丙基化得到化合物48, 而另一个构型的底物则在钯催化下实现N1位的烯丙基化得到化合物49, 这一偶然发现的平行动力学拆分[23]为arborisidine的不对称全合成奠定了基础. 随后化合物48与苯硫氯加成得到化合物50, 随即在四丁基氯化铵和2,4,6-collidine的作用下发生消除反应得到化合物51, 这两步反应中的异构体均无需进行分离. 接着将N-Boc保护脱除后, 通过氮烷基化反应引入羟乙基片段得化合物52, 并利用常规的碘代条件实现羟基的碘代. 最后环系构建利用经典的分子内自由基环化条件即可实现, 得到化合物54后利用臭氧分解将环外双键转化为酮, 最终利用Mannich反应构建酮羰基α位环外双键, 通过催化氢化最终完成arborisidine的全合成.
1.5 马志强课题组对arboridinine和arborisidine的全合成研究
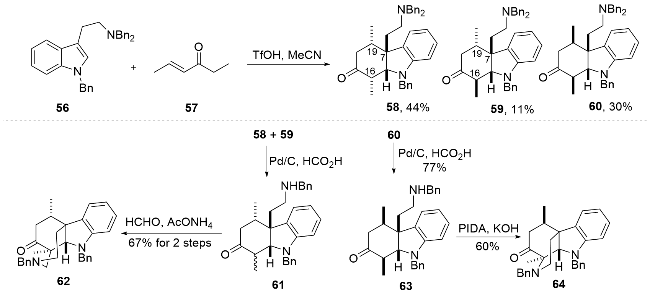

以苄基保护的色胺以及烯酮57为底物, 通过条件筛选发现三氟甲磺酸可以实现预期的串联Michael/ Mannich反应, 得到三个异构体58~60. 基于C7以及C19的相对构型, 化合物58和59符合arboridinine的结构特征, 可用于其全合成, 而化合物60则可用于arborisidine的全合成. 将化合物58与59用催化氢化脱除单个苄基得到化合物61, 随后通过分子内C16位的Mannich反应完成关环, 合成arboridinine四环骨架62. 化合物60通过同样的条件脱除苄基, 随后利用碘苯二乙酸实现了羰基α位的选择性氨基化, 从而得到arbori- sidine四环核心骨架64.
从四环骨架62出发, 首先实现羰基α位的羟基化, 随后将生成的羟基乙酰化, 并利用催化氢化脱除苄基保护得到化合物66. 紧接着通过分子内的Mannich反应完成最后桥环的构筑, 从而完成了目标天然产物核心骨架的构建. 最后阶段主要是完成羰基向环外双键的转化, 该反应看似简单, 但通过条件筛选发现常用的转化条件无法实现该过程. 因此作者引入了一个硅基片段, 该片段可以发生分子内的亲核加成以克服羰基周围较大的立体位阻的影响, 此外硅基的引入也为后续的Peterson成烯反应提供了结构基础. 具体转化过程为利用碱性条件将化合物67中的乙酰基水解, 随后在暴露的羟基中引入硅基片段得到化合物68, 在叔丁基锂的作用下将溴代烷转化为亲核试剂进攻羰基得到化合物69, 最终通过Peterson成烯反应成功构建环外双键, 并利用亚碘酰苯实现向亚胺的转化得到目标天然产物arboridinine.
而从四环中间体64完成arborisidine全合成的过程也相对简单, 首先利用Comins三氟甲磺酸化试剂将羰基转化为烯醇三氟甲磺酸酯, 并通过后续的钯催化偶联反应引入烯基得到化合物70. 随后通过硼氢化氧化反应实现化合物71的制备, 在脱除了一个苄基后, 利用甲磺酰氯将羟基活化, 从而实现分子内的氮烷基化得到化合物72. 最终通过苄基脱保护、氧化为亚胺和羟基氧化成酮三个过程即可完成arborisidine的全合成.
2022年, 马志强课题组[26]在上述工作基础上利用双Mannich反应, 又报道了一条arboridinine的形式全合成路线(Scheme 8). 以前文所述的串联Michael/Mannich反应的产物58为起始, 通过氰基甲酸乙酯在羰基α位引入羧酸乙酯得化合物74, 随后将伯胺上的两个苄基脱除, 紧接着与羰基两侧发生两次Mannich反应, 从而顺利完成五环骨架的构建得到化合物76. 环外双键的构建同样采用的是Peterson成烯反应, 先通过三甲基硅甲基锂对羰基进行亲核加成, 之后在碱性条件下顺利得到环外双键. 经过脱苄基保护、利用亚碘酰苯氧化得到亚胺80, 并利用碱性条件将酯基水解得到了化合物37. 该化合物是翟宏斌、李云课题组[15]合成arboridinine时的关键中间体. 该形式全合成与之前报道的全合成策略相比(包括马志强课题组先前的报道), 主要的优势在于可以一步构建两个环系, 从而完成五环核心骨架的快速构建. 但根据先前的报道, 化合物37向目标天然产物的转化效率并不是很高, 产率只有20%, 这也导致了整个全合成的效率偏低.
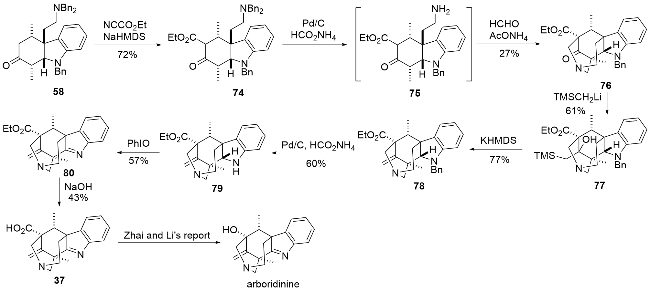
1.6 厍学功、曹小平课题组对arborisidine的形式全合成研究
2024年, 厍学功、曹小平课题组[27]报道了arborisi-dine的形式全合成(Scheme 9), 进一步丰富了此类生物碱的合成策略. 首先利用色胺与化合物81在三氟乙酸作用下发生P-S反应, 并引入N-Boc保护, 将酯基还原为醇, 得到化合物82. 随后将得到的伯醇重新氧化为醛基, 并利用Wittig反应将醛基转化为双键得到化合物84, 而后通过钯催化的烯丙基化实现吲哚C3位的烯丙基化, 这里得到的是非对映异构体的混合物, 无法直接分离. 后续的烯烃复分解反应(RCM)中只有两个双键处于顺式位置的底物才能顺利反应得到化合物86, 而另一个异构体则无法反应, 从而实现了分离. 接着利用烯丙位溴代得到化合物87, 并可在银催化下得到环合产物88, 此外在烯丙位溴代反应中也会部分直接生成环合产物88, 可能是N-Boc中的羰基参与了成环反应. 利用氢氧化钠将化合物88中的噁唑烷酮环水解, 用Boc酸酐对暴露的氨基重新进行保护, 并用IBX将羟基氧化成酮得到化合物89. 用三仲丁基硼氢化锂1,4-还原, 随即用Commins试剂制备烯醇三氟甲磺酸酯得到化合物90. 之后的转化与马志强课题组[24]合成arborisidine时的策略类似, 包括了钯催化的烯基化、硼氢化氧化和分子内的氮烷基化反应, 通过几步转化后可以得到中间体93, 随后用DMP快速处理得到亚胺94, 再利用Swern氧化将羟基氧化成酮, 即可得到焦雷课题组合成arborisidine的中间体55[22]. 整体来说, 该合成路线与之前报道的全合成路线相比并无十分突出的优势, 有些步骤的转化也与之前报道类似, 但是该全合成研究为此类生物碱环系的构建方法提供了新的思路, 也为其他相关生物碱的合成提供了参考.
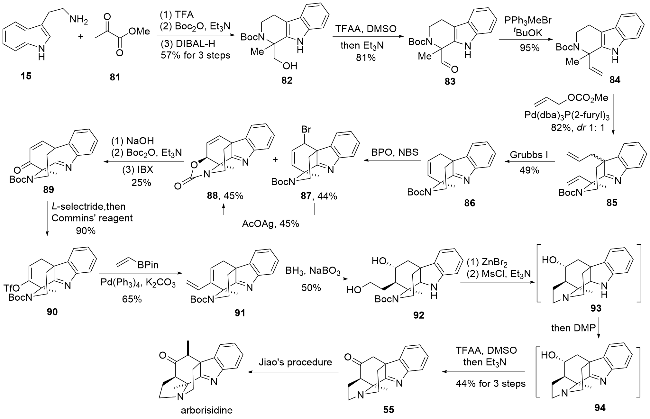
1.7 秦勇、宋颢课题组arborisidine四环骨架的合成研究
秦勇、宋颢课题组[28]关于arborisidine四环骨架的不对称合成研究是该天然产物最早的全合成尝试, 但最终未能完成E环的构建. 该课题组前期发展了铜催化的分子内环丙烷化-开环-亚胺环合串联反应(CRI), 用于构建环己烷四氢吡咯并吲哚啉四环骨架96, 并以此为基础完成了minfiensine[29]以及vincorine[30]消旋体的全合成. 随后, 该课题组又发展了铜催化的Friedel-Crafts环合反应策略, 实现了环己烷四氢吡咯并吲哚啉四环骨架98的不对称合成, 继而完成了strictamine[31]的全合成. 考虑到strictamine和arborisidine的骨架结构具有一定的相似性, 秦勇课题组以手性四环骨架98为基础, 开展arborisidine的全合成研究(Scheme 10).
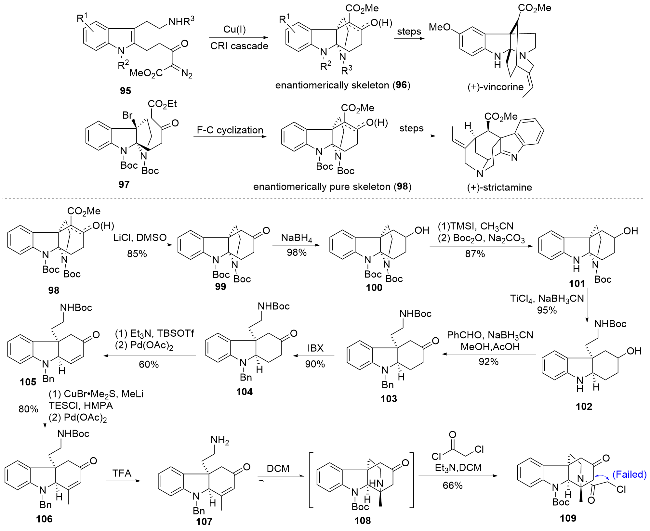
化合物98经Krapcho脱羧反应得到化合物99, 经过硼氢化钠还原得到化合物100. 随后用三甲基碘硅烷(TMSI)将两个Boc保护基同时脱除, 得到的缩醛胺中间体在碳酸钠温和的碱性条件下可以实现选择性的Boc保护, 得到化合物101. 在TiCl4/NaBH3CN条件下实现分子内的还原开环, 并通过还原胺化反应引入苄基保护, 进而利用2-碘酰基苯甲酸(IBX)重新氧化仲醇得到开环产物104. 接着利用叔丁基二甲硅基三氟甲磺酸酯(TBSOTf)和三乙胺将酮羰基转化为烯醇硅醚, 经Pd(OAc)2催化的Saegusa氧化反应, 成功构建α,β-不饱和酮105, 随后与甲基铜锂试剂发生Michael加成反应获得烯醇硅醚中间体, 再次利用Saegusa氧化反应得到化合物106. 接下来作者希望通过分子内的氮杂Michael加成反应构建目标分子中的哌啶C环, 通过对反应条件的探究发现, 在无机碱条件下虽然可以得到目标产物, 但转化率不高. 考虑到氮杂Michael加成反应在碱性条件是可逆的, 因此尝试不添加碱进行反应, 结果发现仅用二氯甲烷作为溶剂即可获得环化的中间体108, 该中间体直接与氯乙酰氯发生酰化反应, 即可得到稳定的四环化合物109. 最后, 作者希望利用分子内的亲核取代反应构建最后的E环, 但该策略经过细致地探索后仍未能获得成功.
1.8 构建arborisidine的四环核心骨架的其他策略
Blakey课题组[32]利用N-酰基-O-TMS氨基醇类底物的串联环化反应可以实现两类重要的吲哚生物碱骨架的构建. 使用给电子能力较强的苄基(Bn)作为取代基的底物110, 可以促进吲哚C3位进攻生成的亚胺离子中间体, 从而得到Malagasy类四环骨架111, 而底物112中的对甲苯磺酰基(Ts)保护基会抑制C3位的亲核性, 从而得到吲哚C2位亲核进攻的产物113, 该产物具有arborisidine的核心四环骨架. Waser课题组[33]利用硅正离子催化的[4+2]环化反应为四环核心骨架的构建提供了新的思路, 利用温度控制吲哚C2和C3位亲核进攻的选择性, 在低温下实现吲哚C2位进攻即可获得arbori-sidine的核心四环骨架115 (Scheme 11). 天然产物arborisidine与strictamine等生物碱具有相似的四环骨架, 因此strictamine等生物碱的全合成中较为成熟的四环骨架构建策略[7]对于arborisidine的全合成也有很好的借鉴意义.
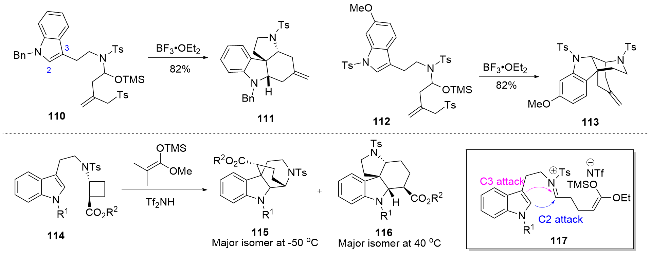
1.9 Arboridinine和arborisidine的合成策略总览
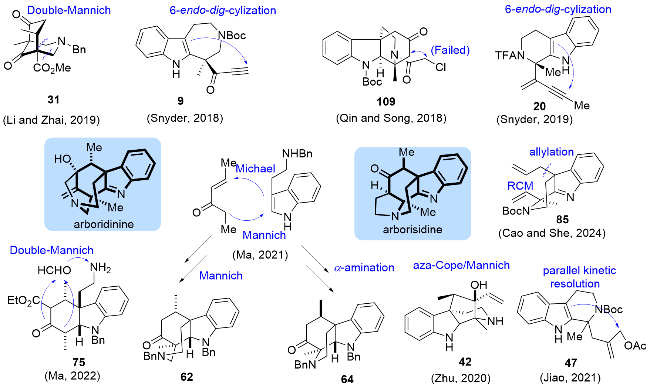
Arboridinine和arborisidine复杂的笼状结构使其全合成研究颇具挑战性, 而自然来源的匮乏以及有待进一步研究的药理活性更是增加了其全合成研究的吸引力, 近年来多个课题组致力于这两个天然产物的全合成研究(图2). 秦勇、宋颢课题组[28]在2018年就报道了arborisidine核心骨架的构建, 但最终利用分子内亲核取代反应实现关环的策略未能成功. Snyder课题组[11-12]最早完成了这两个天然产物的全合成研究, 两条合成路线均是以金属催化的6-endo-dig-环化反应为关键步骤完成了核心四环骨架的构建. 2019年, 翟宏斌、李云课题组[15]利用双Mannich反应为关键步骤, 构建氮杂[3,3,1]双环骨架, 以此为基础完成了arboridinine的全合成. 该策略采用了后期引入吲哚结构的策略, 也是目前已报道中唯一未采用吲哚类底物为起始原料的合成方法. 路线中大部分合成步骤都可以实现克级(甚至是数十克级)的制备, 这是该合成路线的主要优势之一. 2020年, 祝介平课题组[17]利用不对称P-S反应以及氮杂Cope重排串联Mannich反应等关键步骤, 实现了arborisidine的全合成, 该路线总共只有5步反应, 大大提升了目标天然产物的合成效率.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2021年, 马志强课题组[24]利用串联Michael/Man- nich反应构建三环骨架, 以此为基础分别利用Mannich反应以及选择性α-烷基化反应, 可以分别实现这两个天然产物不同的四环骨架的制备, 这种发散性合成策略大大提高了不同类型天然产物的合成效率. 2022年, 马志强课题组[26]又进一步利用双Mannich反应构建了五环笼状骨架, 从而完成了arboridinine的又一条形式全合成路线. 2021年, 焦雷课题组利用独特的平行动力学拆分策略, 实现吲哚C3位的不对称烯丙基化, 并以此为基础完成了arborisidine的全合成[22]. 2024年, 厍学功、曹小平课题组[27]利用P-S反应、吲哚C3位烯丙基化和RCM反应(Ring-closing metathesis)等步骤完成了arbori- sidine的形式全合成研究, 与已有报道相比, 该合成策略在合成效率上并没有突出的优势, 但其不同以往的环系构建思路也为其他吲哚生物碱的全合成提供了有益的参考.
2 结语
综述了目前蕊木属生物碱arboridinine和arborisi-dine的全合成研究进展, 包括形式全合成以及核心骨架的合成. 特殊而复杂的笼状化学结构使得这两个天然产物的全合成研究极具挑战性和吸引力, 因此自这两个天然产物被分离以来, 优秀的全合成案例不断被报道. 各种新颖的合成策略被用于这两个天然产物的全合成, 其骨架的构建效率也不断被提升, 例如祝介平课题组仅用5步反应即可实现arborisidine的全合成. 对于复杂天然产物全合成的探索, 不断推动着有机合成方法学的进步, 而优秀的有机合成方法学研究又进一步提升了天然产物全合成的效率. 相信未来还会有更多的研究小组进行arboridinine和arborisidine的全合成研究, 进一步丰富其合成策略, 为这两个天然产物后续的药学相关研究奠定良好的基础, 同时这些高效的合成策略也为其他天然产物的全合成提供有益的参考和启发.
(Lu, Y.)