

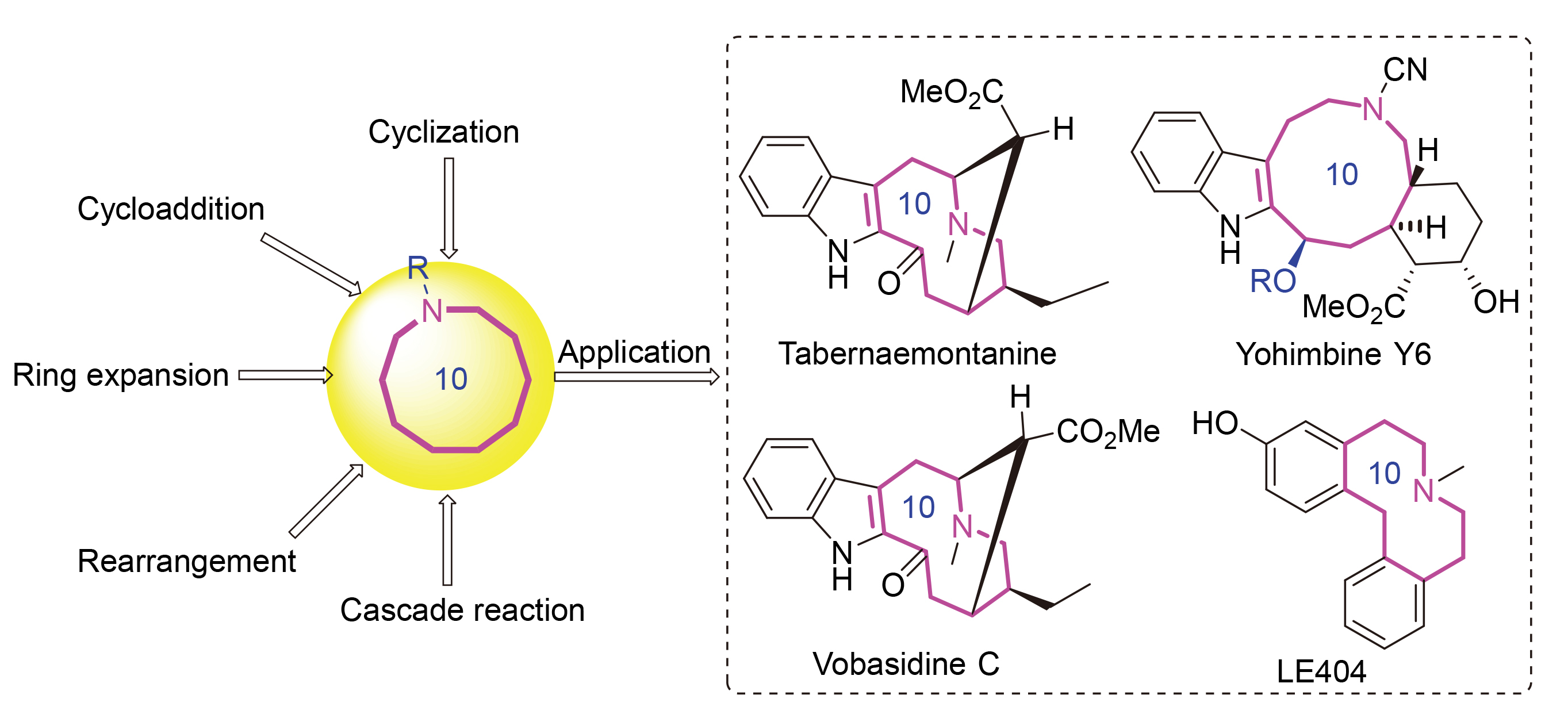
中环杂环骨架(8~12元环), 因其具有独特的刚性空间结构和环熵增的不利因素, 相较于线性分子或平面杂环体系, 在现代药物发现中可作为优势结构单元[1]. 中环含氮杂环化合物作为一类重要含氮原子的环状结构单元, 广泛存在于天然产物和生物活性分子中, 在药物研发领域也展现出巨大潜力[2]. 尤其值得注意的是十元氮杂环化合物, 它们是多种具有重要生物活性的天然产物(如原阿片碱、丹参酮和苦参碱衍生物等)的核心结构(图1)[3]. 这些含氮杂环化合物的骨架已被充分验证为有价值的受体配体和天然产物全合成中的关键中间体[4].
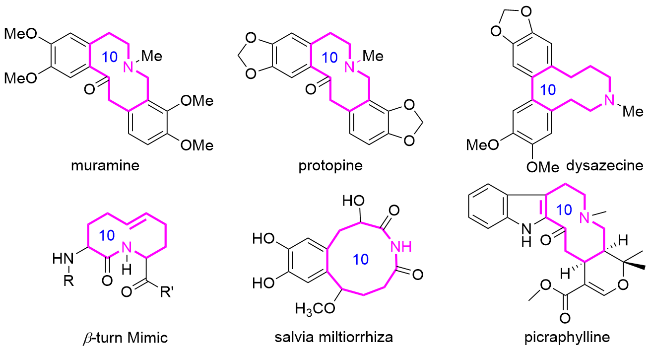
近年来, 十元氮杂环化合物的构建一直吸引着合成化学家的广泛关注, 但其合成仍然面临着巨大的挑战. 这些困难主要源于两个基本因素: (1)中环体系中非理想键角和扭转应变导致的跨环相互作用; (2)环化过渡态显著的熵/焓损失[5]. 这些固有的热力学和动力学障碍使得传统的环化策略对中环骨架的合成效率低, 往往导致产物的产率低或发生竞争性聚合反应. 对于十元氮杂环而言, 其特定的构象要求以及在关键位点引入氮原子的额外挑战, 使得其合成复杂度倍增.
尽管存在这些合成困难, 十元氮杂环化合物作为药物化学和天然产物全合成中的优势骨架, 其价值日益得到认可, 持续推动着合成方法学的创新与发展. 通过发展环扩增级联反应、过渡金属催化环化和光化学转化等新策略, 该领域已取得显著进展. 这些突破不仅拓宽了中环氮杂环化合物合成的应用范围, 更为其构象特征与构效关系研究提供了重要指导. 然而, 十元氮杂环的高效立体可控构建仍是一项极大的挑战, 是未来药物化学中一个活跃且重要的研究方向.
1 分子间反应构建十元氮杂环化合物
1.1 串联反应构建十元氮杂环化合物
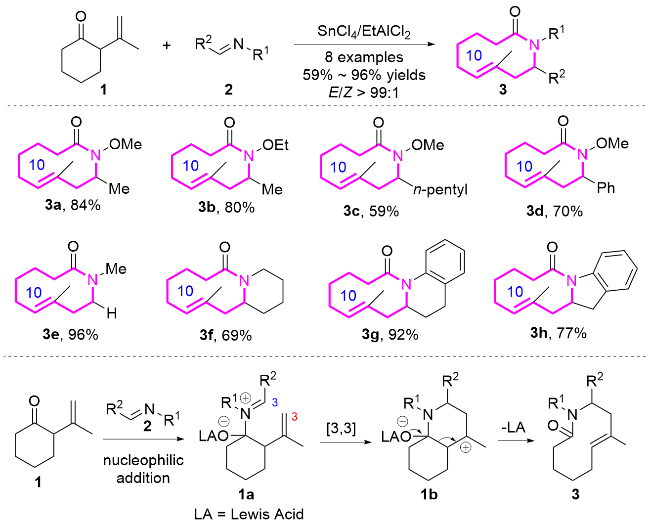
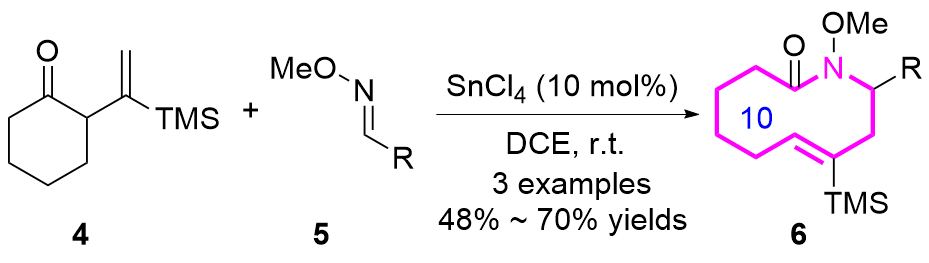
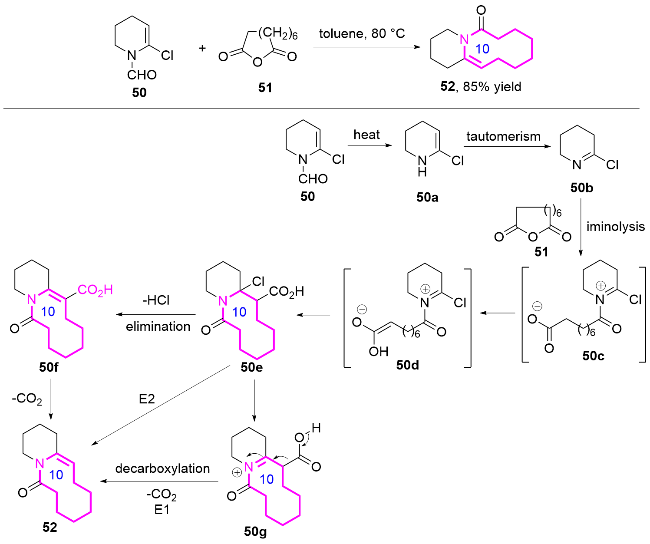
2015年, Goeke课题组[7]报道了Lewis酸催化氧杂- 2-氧鎓Cope重排串联反应的新策略, 实现了十元环内酰胺6的高效构建, 及其在天然大环内酰胺立体选择性全合成中的应用(Eq. 1). 研究表明, 含三甲基硅基(TMS)的β,γ-不饱和酮类底物4, 在SnCl4催化下与醛发生亲核加成, 形成两性离子中间体, 随后通过氧杂-2-氧鎓Cope重排生成十元环内酰胺产物. 该过程的关键在于, TMS基团通过β-硅基效应稳定了反应中的三级碳正离子中间体, 同时其空间位阻有效抑制了双键异构化的副反应, 并通过过渡态构象控制实现了环内双键E构型产物的形成.
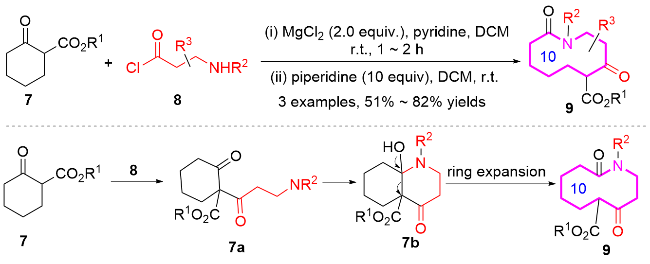
2015年, Unsworth课题组[8]开发了一种新的连续环扩张(SuRE)的串联方法, 用于高效合成结构多样化的大环和中环化合物9 (Scheme 2). 与传统依赖线性前体末端-末端环化的方法不同, SuRE通过将线性片段8(如Fmoc或Cbz保护的酰氯)插入环状β-酮酯底物中, 实现逐步扩环, 且无需高稀释条件或特殊模板辅助. 该反应的机理为: 首先化合物9在酰氯发生C酰化反应生成酰化中间体7a, 然后在哌啶作用下发生亲核加成得到双环中间体7b, 最后扩环得到十元氮杂环化合物9. 此外, 作者还将这种连续扩环串联策略应用到硫代酰胺参与的反应中, 通过酰化、环化和扩环等多步串联反应, 成功获得了十元氮杂环化合物. 该方法操作简便、可实现规模化反应, 为大环/中环药物、功能材料和天然产物合成提供了模块化合成策略.
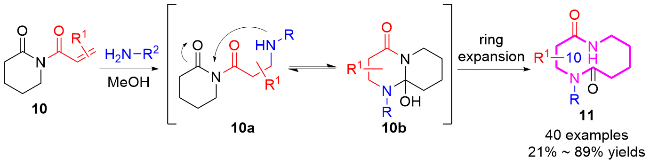
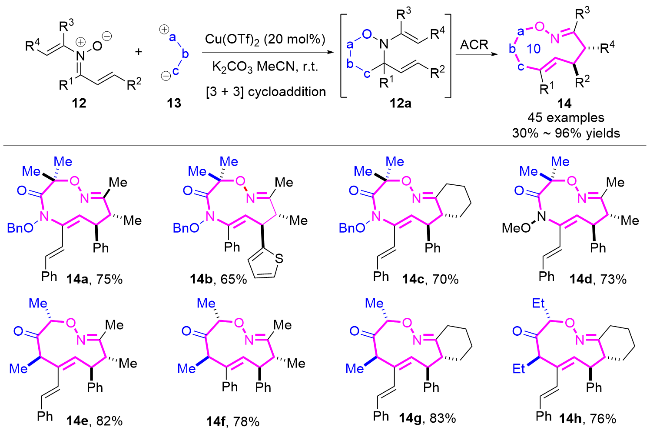
硝酮作为一种重要的1,3偶极子, 在有机合成中展现出了广泛的应用价值, 特别是与各种不饱和键的环加成反应构建不同类型的杂环衍生物[10]. 2025年, 莫冬亮课题组[10e]通过两种1,3-偶极体的高效组装串联策略, 实现十元氮杂环化合物14的立体选择性合成(Scheme 4). 作者开发了铜催化的N-烯基-α,β-不饱和硝酮12与氮杂氧鎓或氧鎓阳离子的[3+3]环加成/氮杂克莱森重排串联反应, 在温和条件下快速构建了含两个或多个立体中心的多取代十元氮杂环化合物14, 最高可达96%产率, 且具有优异的区域选择性和非对映选择性. 值得注意的是, 该合成策略通过巧妙设计1,3-偶极体的[3+3]环加成形成六元环中间体, 随后利用季碳中心的张力驱动氮杂克莱森重排, 成功解决了中环化合物合成中取代基间距过大导致的立体控制难题.
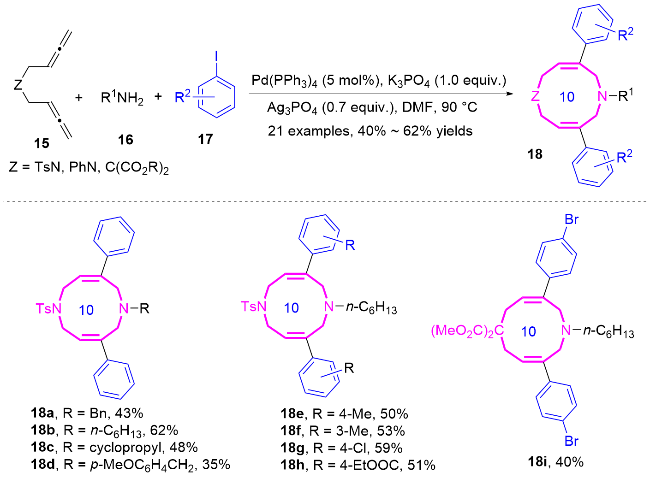
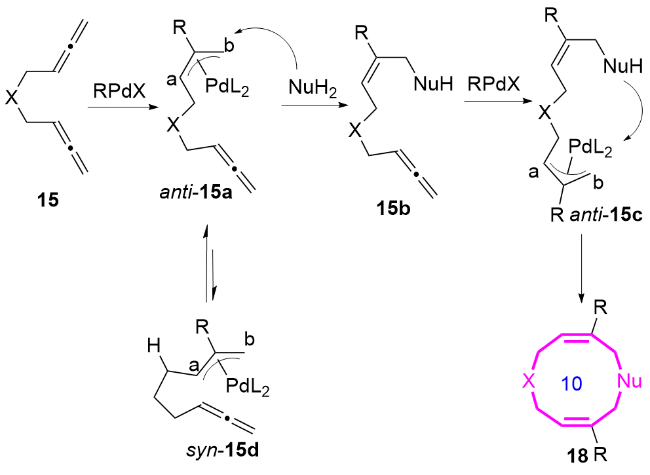
结合相关的控制实验, 作者提出了钯催化1,5-二联烯15合成十元氮杂环化合物18的可能机理(Scheme 6). 底物中两个联烯基中的一个联烯发生碳钯化反应, 优先生成反式的π-烯丙基Pd中间体, 这是由于R基团在与顺式构型中另一个联烯基之间存在空间位阻. 反式中间体anti-15a发生区域选择性分子间烯丙基化反应, 进而生成中间体15b. 随后, 底物中第二个联烯基的碳钯化过程将再次有利于反式π-烯基Pd中间15c的生成. 最终, 通过分子内的区域选择性烯丙基取代反应, 得到十元氮杂环化合物18.
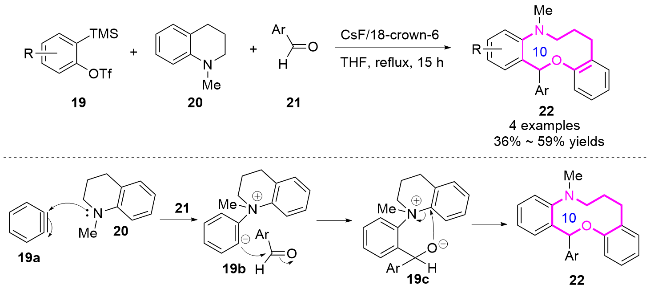
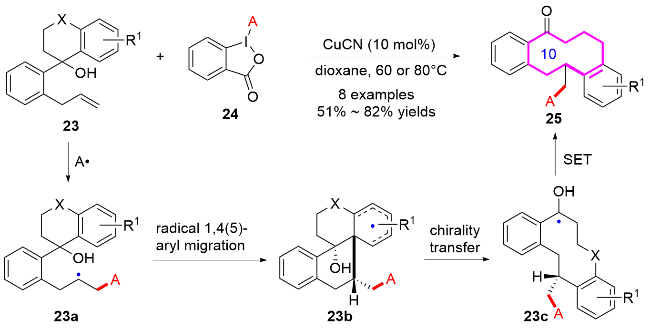
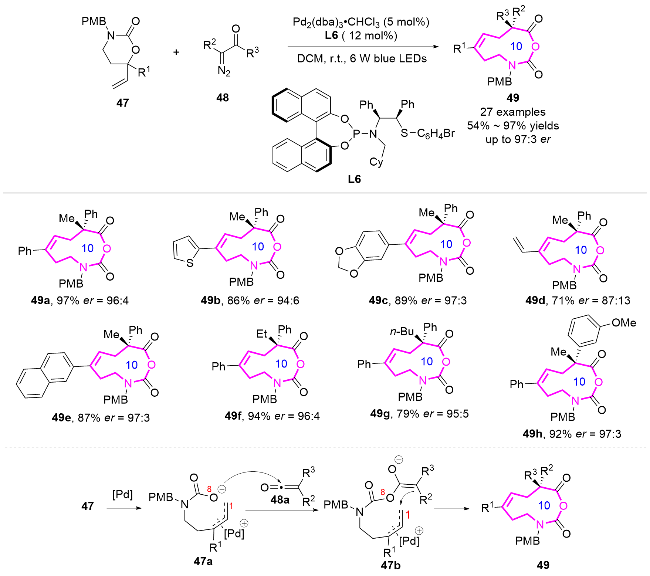
2016年, 刘心元课题组[13]发展了自由基引发的多样性导向合成策略. 通过未活化烯烃23的1,4-或1,5-芳基迁移/环扩增串联反应, 高效构建了苯并十元氮杂环衍生物25 (Scheme 8). 研究团队以含烯丙基的环状苄醇为底物23, 在铜催化条件下, 利用叠氮化、三氟甲基化、膦酰化、磺酰化等自由基反应引发远程芳基迁移, 成功实现了十元环骨架25的立体选择性构建. 这种模块化合成方法突破了传统环化反应中不利的熵/焓因素限制, 为天然产物(如byssochlamic acid类似物)核心骨架的构建提供了新思路. 同时合成的功能化十元氮杂环可作为重要药效团, 用于生物活性分子开发, 初步实验显示部分产物对癌细胞系具有显著的抑制活性. 该工作通过密度泛函理论(DFT)计算, 阐明了自由基迁移/环扩增的协同机制, 为复杂中环体系的高效合成开辟了新途径.
1.2 环加成反应构建十元氮杂环化合物
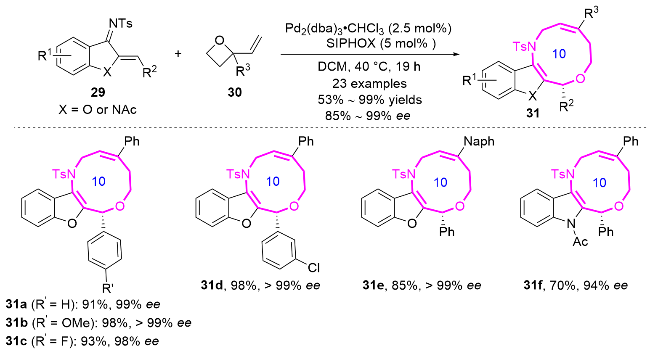
2020年, 陈应春课题组[16]利用简单易得的苯并噻吩氮杂二烯29和2-甲基吲哚不饱和醛32进行形式[6+4]环加成反应, 以99%的产率和95% ee得到苯并噻吩并十元氮杂环化合物33 (Eq. 2). 这些氮杂环化合物的多样性和功能性在药物化学中具有潜在的应用前景.
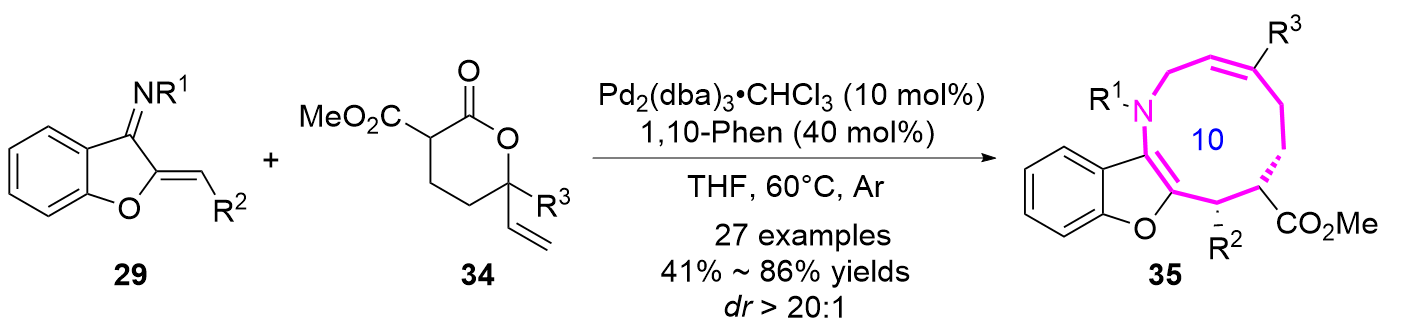
2023年, 赵立明课题组[17]报道了以苯并呋喃氮杂二烯29和乙烯基戊内酯34作为底物, 通过钯催化的 [6+4]环加成反应, 以良好的产率和优秀的非对映选择性合成了苯并呋喃并十元氮杂环化合物35 (Eq. 3).
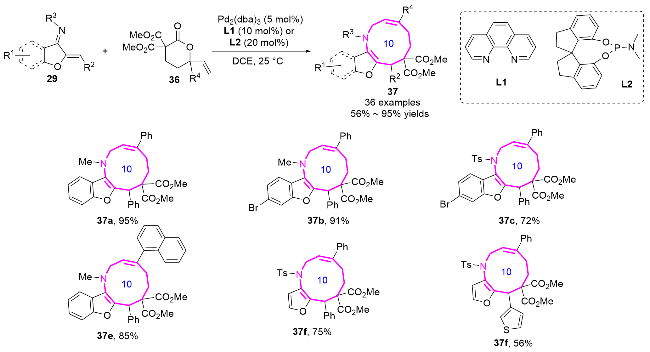
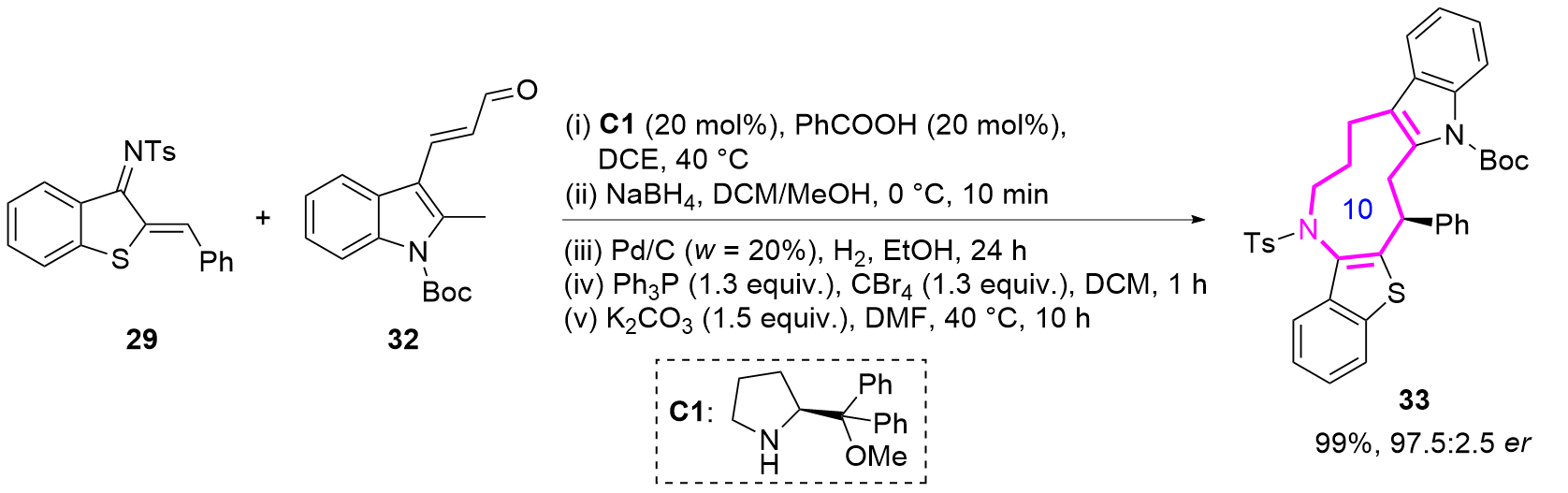
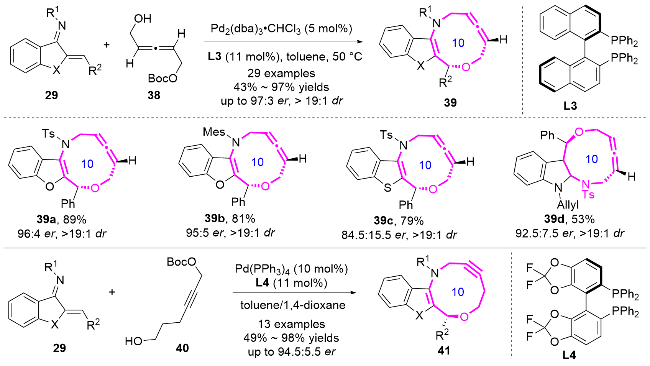
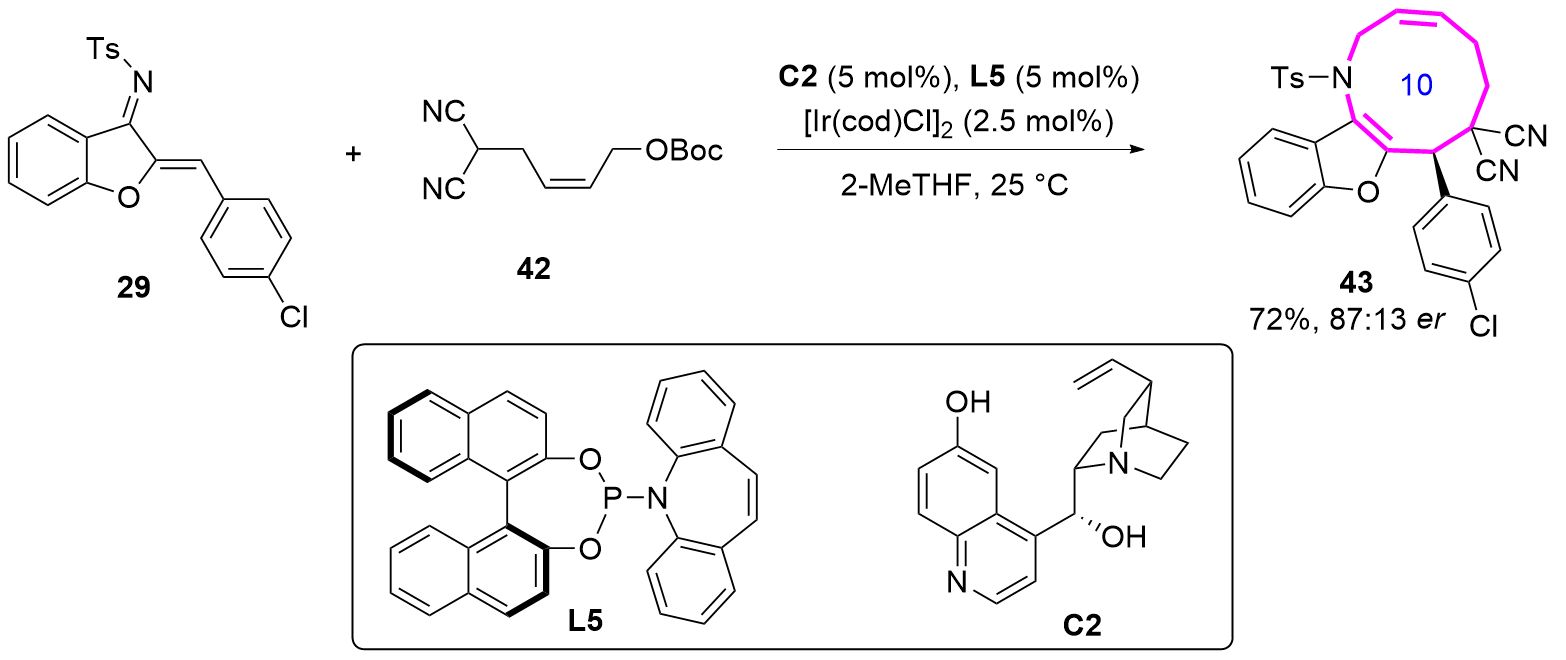
2024年, 游书力课题组[20]通过铱-L5/金鸡纳碱C2双催化体系, 实现了Z-不对称烯丙基取代反应, 成功开发了十元环氮杂化合物43的高对映选择性合成方法(Eq. 4). 该反应利用铱催化剂独特的支链区域选择性特征, 结合金鸡纳碱C2的手性控制作用, 通过捕获热力学不利的anti-π-烯丙基-铱中间体, 实现了分子间[6+4]环加成过程. 本策略不仅为十元中环的立体选择性合成提供了新思路, 其温和的反应条件、良好的官能团耐受性以及克级规模反应的优点, 更凸显了该方法在复杂中环骨架构建中的重要应用价值.
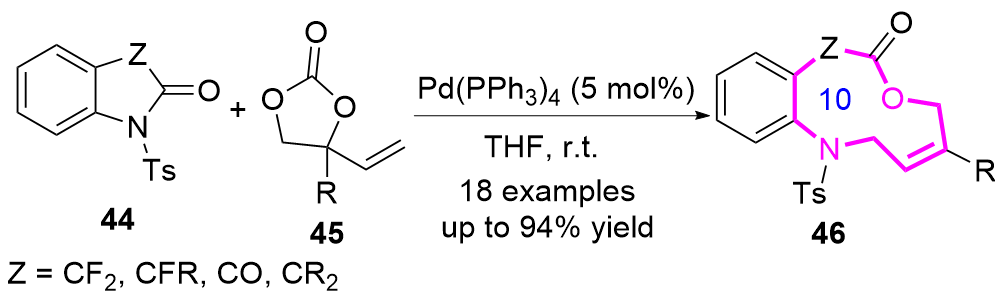
随着过去几十年里金属催化的发展, 高阶偶极环化反应(HODAs)已成为构建中环化合物的一种高效且通用的策略. 例如, 2020年Shibata课题组[21]开发了一种利用碳酸乙烯酯45 (VECs)作为1,5偶极子, 在Pd(PPh3)4催化下与靛红或其类似物44反应, 合成了一系列十元氮杂环化合物46 (Eq. 5). 该反应成功的关键在于酰胺 C—N键的断裂, 该过程是由两个相邻吸电子基团的存在和钯配位的两性离子的亲核进攻共同促进的.
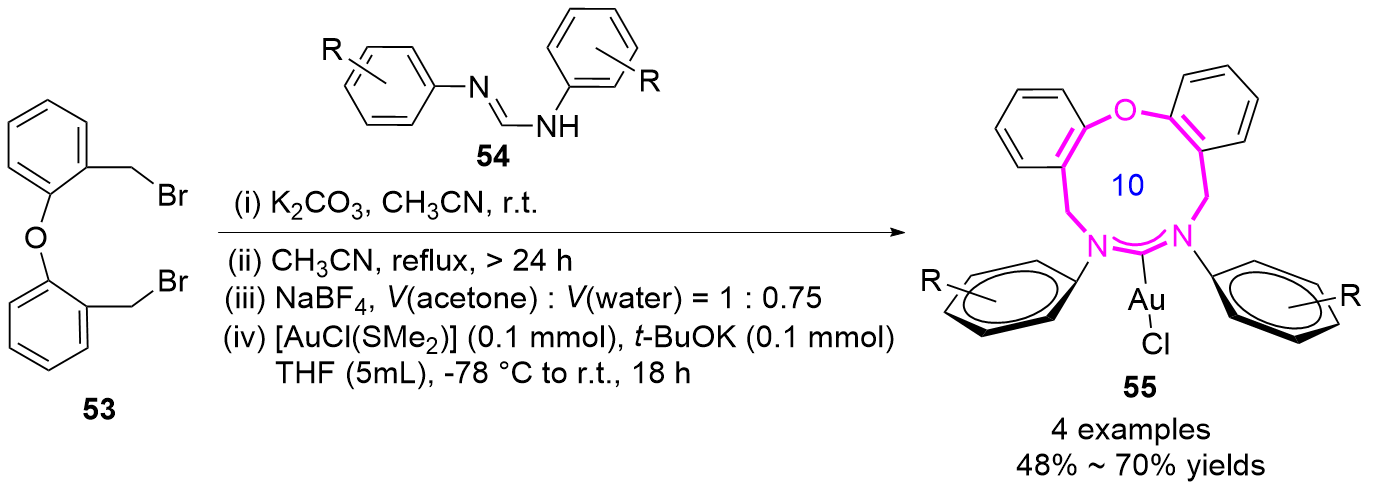
氮杂环卡宾作为一类过渡金属的特殊配体, 具有良好的热稳定性、低毒性和抗氧化性等优点. 这些特性使它们适用于各种金属催化的反应. 到目前为止, 关于八元环以上氮杂环卡宾配体配位的金复合物的报道有限. 2019年, Hashmi课题组[24]报道了十元环N-杂环卡宾(NHC)金(I)配合物55的合成及其催化性能研究(Eq. 6). 作者使用二溴二苯醚53与N,N-二芳基甲脒54反应, 合成了十元氮杂环卡宾配体55. 该工作不仅填补了八元以上大环NHC-金配合物的研究空白, 也为调控金属配合物的立体电子效应开辟了新途径.
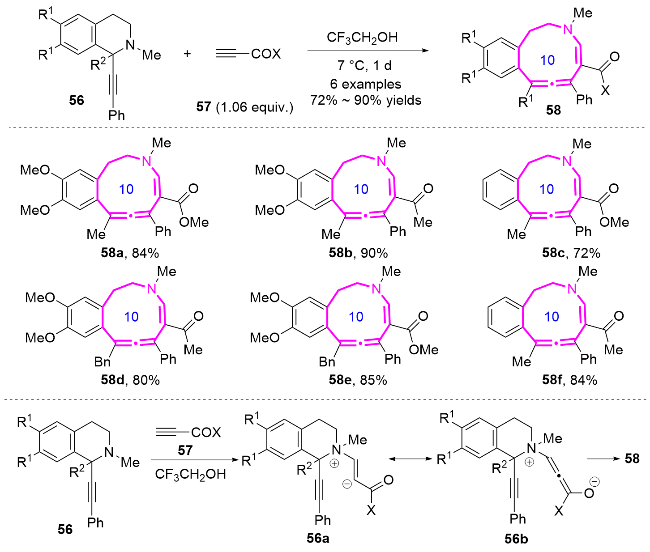
1.3 Michael加成反应构建十元氮杂环化合物
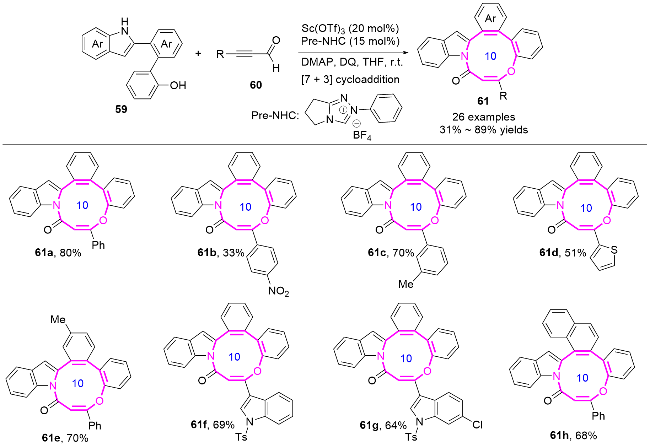
2 分子内反应构建十元氮杂环化合物
2.1 扩环反应构建十元氮杂环化合物
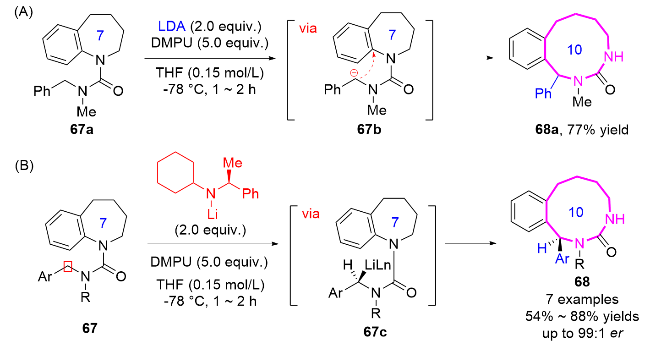
2016年, Clayden课题组[29]发现苯并稠合氮杂环在金属化脲衍生物催化下, 经迁移扩环反应可高效合成十元氮杂环化合物67/68 (Scheme 19A). 研究团队以七元氮杂环为前体, 在二异丙基氨基锂(LDA)和N,N'-二甲基丙烯基脲(DMPU)存在下, 通过选择性去质子化形成苄基阴离子中间体67b, 通过扩环反应成功构建了十元苯并二氮杂环体系. 该策略首次实现了通过金属化脲的迁移扩环策略直接构建十元及以上中环体系, 反应条件温和、底物适用范围广. 此外, 该小组将手性氨基锂引入到反应中, 成功开发了不对称的迁移扩环反应, 合成了具有高对映选择性的十元环脲衍生物68 (Scheme 19B).
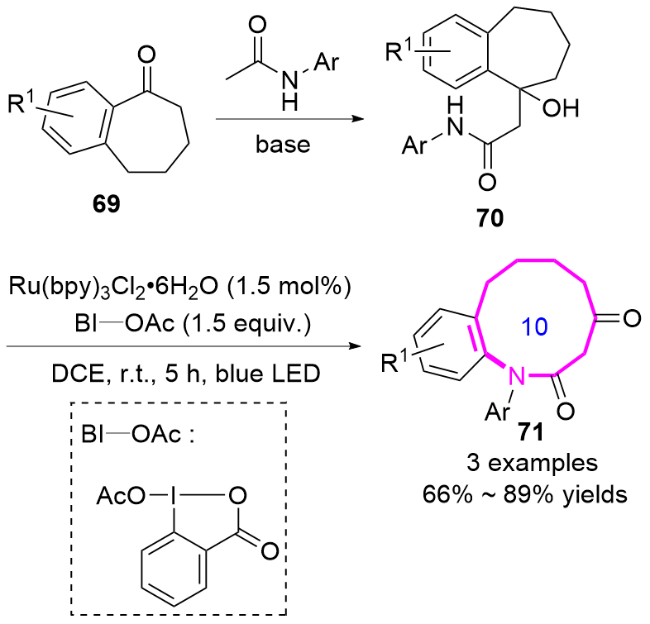
机理研究表明, 该反应可能经历羧酸促进的酰胺基自由基生成过程. 通过荧光猝灭实验和循环伏安法证实, 苯并碘唑自由基(BIC)与羧酸协同氧化底物70生成关键酰胺基自由基中间体70a, 随后发生分子内(杂)芳基迁移和C—C键的选择性断裂, 最终形成扩环产物71 (Scheme 21).
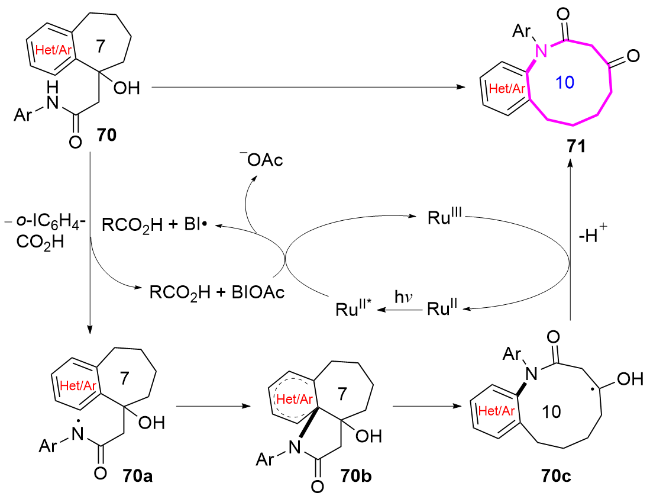
2020年, 阮志雄课题组[31]发展了一种无催化剂、电化学催化的策略, 通过C—C键断裂和酰胺自由基迁移, 实现了十元环内酰胺的高效合成(Scheme 22). 与传统的酰胺基自由基环化不同, 该策略在温和、无金属和无外部氧化剂的条件下, 通过独特的远程酰胺基迁移, 以原子经济和步骤经济的方式构建了具有挑战性的中环骨架. 机理研究表明, 化合物72中的酰胺基N—H键通过单电子转移(SET)过程生成酰胺基自由基72a, 随后经历分子内环化和选择性C—C键断裂实现扩环, 得到十元氮杂环化合物73. 这一绿色电化学合成策略为中环内酰胺的构建提供了高效、可持续的解决方案, 填补了电化学合成中环内酰胺的空白, 为天然产物和药物分子中重要结构单元的制备开辟了新途径.
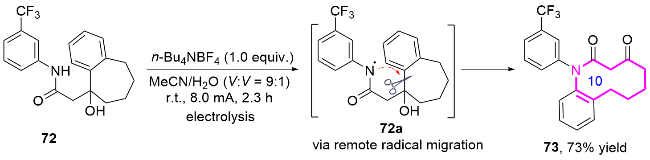
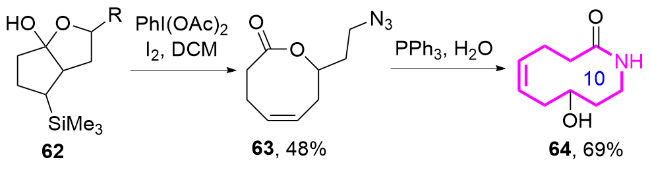
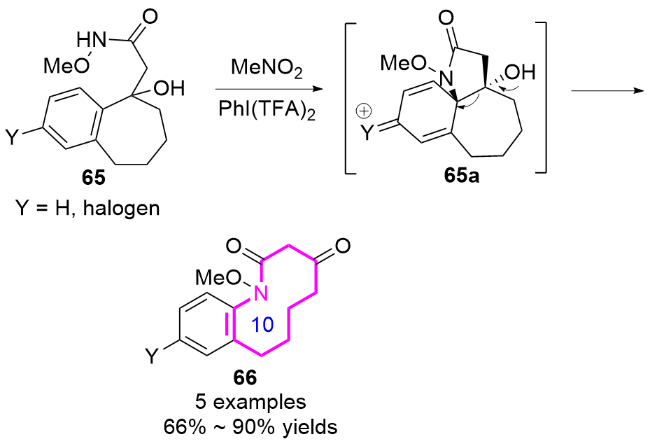
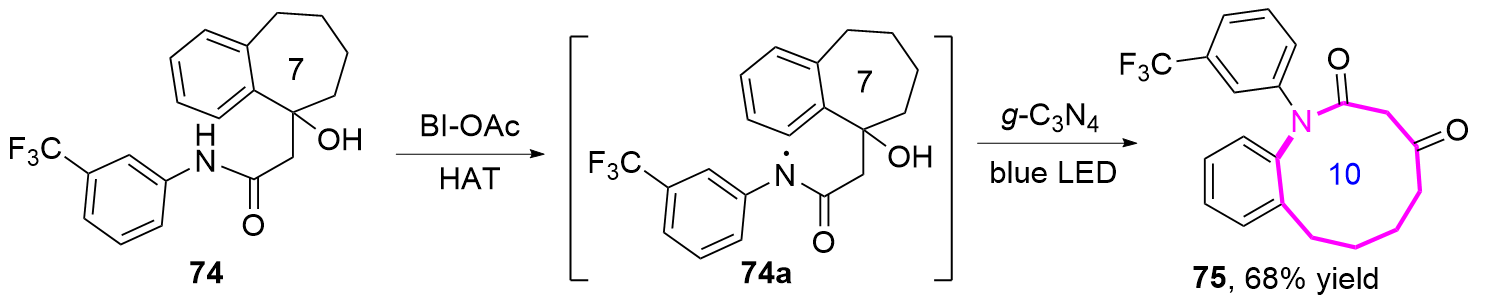
2024年, 郭臻课题组[32]基于类似的反应机理, 报道了一种基于可见光驱动的非均相光催化策略, 通过苯并稠环醇的扩环反应, 高效构建了十元环内酰胺的方法(Eq. 7). 该团队以石墨相氮化碳(g-C3N4)作为非贵金属光催化剂, 化合物74通过氧化性氢原子转移(HAT)过程生成氮自由基中间体74a, 进而扩环生成了十元环内酰胺化合物75. 该反应的关键在于利用双乙酰氧基碘苯(BI-OAc)作为氧化剂, 其光解产生的乙酰氧自由基(AcO•)选择性夺取底物氮原子上的氢原子, 引发氮自由基的分子内亲核进攻, 随后经历选择性C—C键断裂和单电子氧化等步骤, 最终形成热力学稳定的十元环内酰胺.
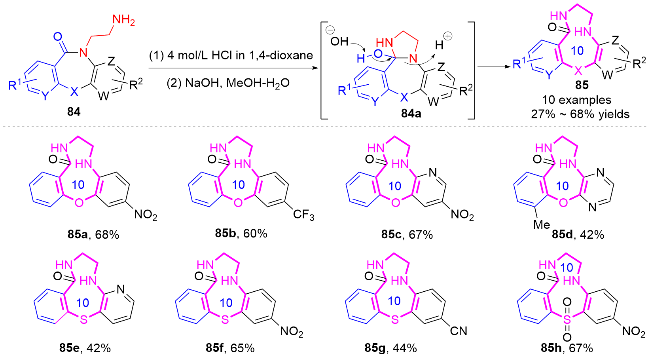
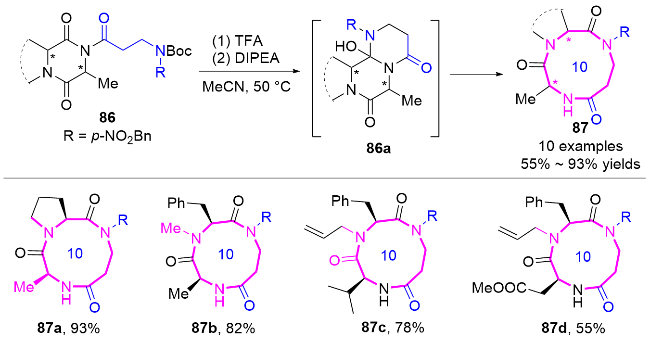
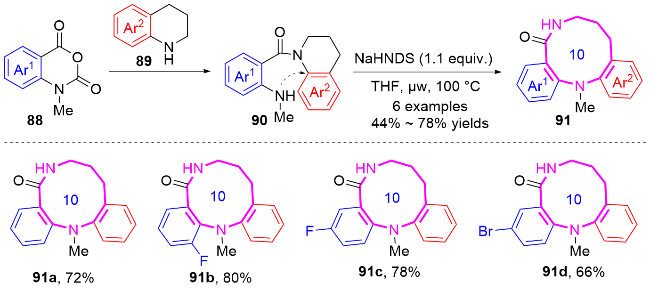
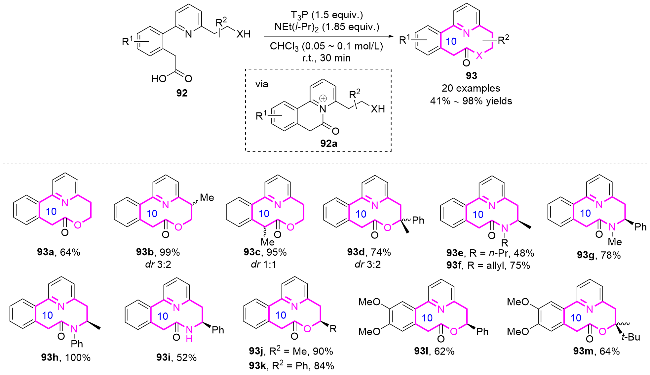
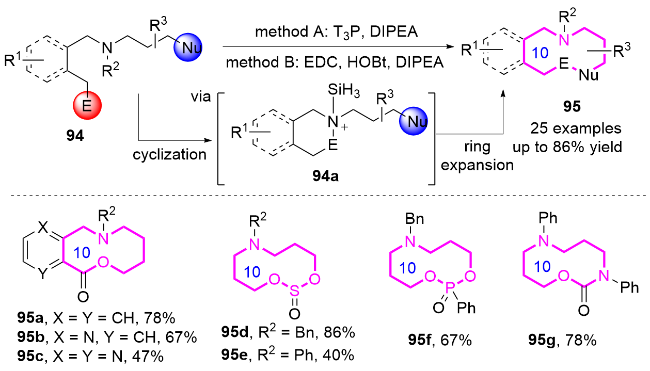
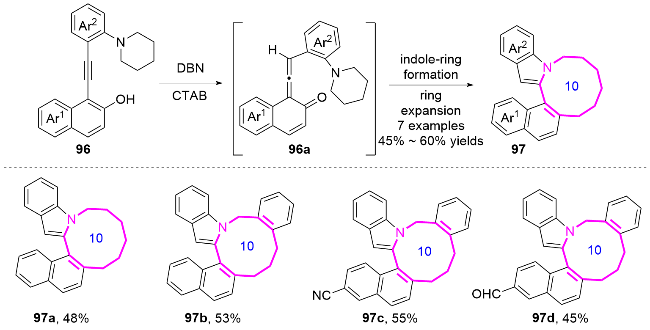
2.2 环化反应构建十元氮杂环化合物
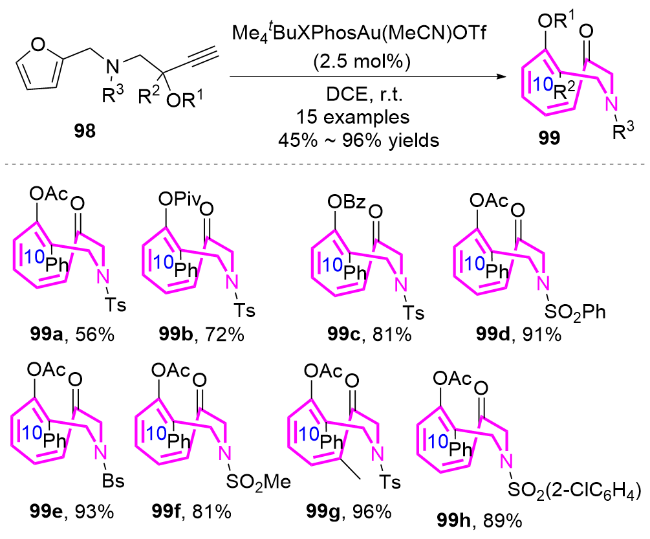
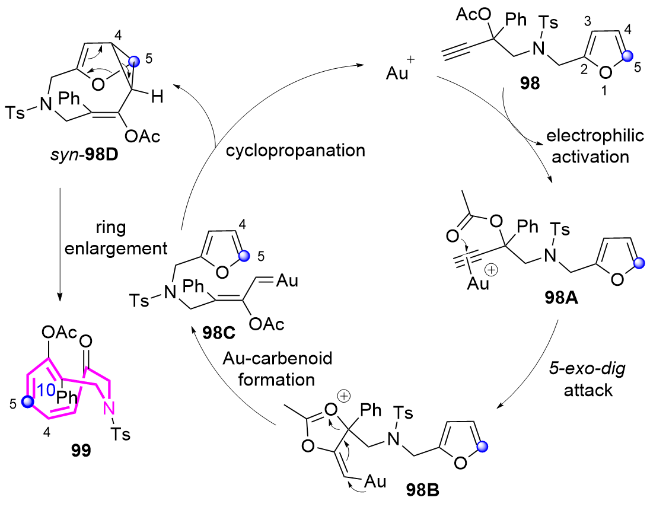
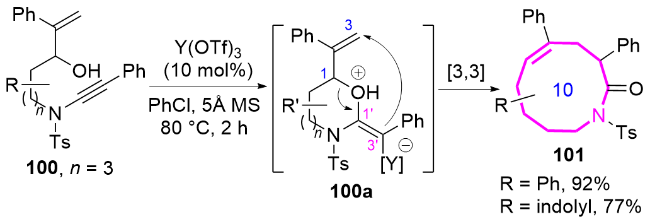
作者推测该反应的机理为: 金催化剂与化合物98中的炔基配位形成金属络合物98A, 该络合物经历分子内5-exo-dig环化产生中间体98B, 随后发生1,2-酰氧基迁移生成高反应活性的金卡宾中间体98C. 该中间体选择性地进攻呋喃环的C5位, 发生分子内环丙烷化, 形成关键的环丙烷中间体syn-98D; 最后通过环丙烷的扩环开环过程, 高效构建含反式双键的十元氮杂环化合物99, 同时金催化剂进入下一催化循环. 这一机理的关键在于金卡宾中间体对呋喃C5位的特异性进攻, 以及后续的扩环过程, 从而克服了传统方法难以合成含反式双键十元环氮杂环化合物的挑战(Scheme 32).
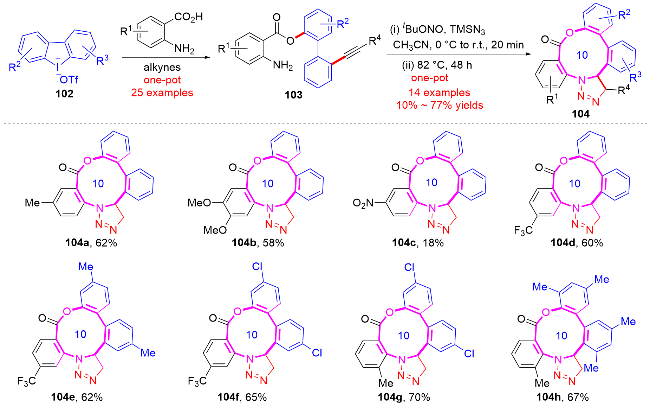
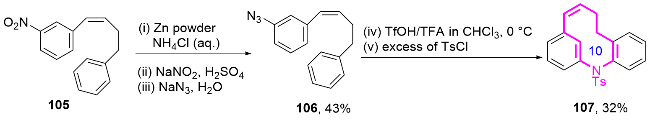
2017年, 张逢质课题组[43]报道了一种基于环状二芳基碘鎓盐102与商业可得邻氨基苯甲酸衍生物制备的目标原料103. 通过分子内环化反应策略, 成功实现了功能化十元氮杂芳基内酯104的高效合成(Scheme 34). 研究发现反应通过铜催化的O-芳基化与Sonogashira偶联的连续转化, 以良好至优异的收率获得具有炔基和胺基双官能团的联芳烃中间体. 该合成策略的创新性在于利用环状二芳基碘鎓盐的双重反应活性, 在无需预活化芳基底物的条件下, 实现了原子经济性和步骤经济性的统一. 值得注意的是, 作者进一步发展了无铜催化的点击化学环化策略, 首次将这类功能化联芳烃成功转化为结构新颖的十元氮杂芳基内酯104.
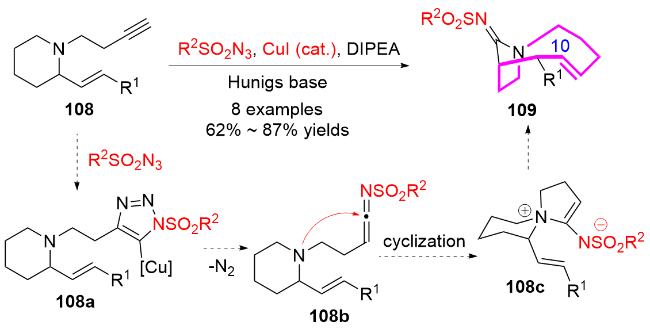
3 含十元氮杂环骨架天然产物和药物分子的合成应用
3.1 在天然产物合成上的应用
十元氮杂环化合物作为中环体系的重要成员之一, 不仅是有机合成中重要的合成砌块, 也是广泛存在于天然产物和药物分子中, 表现出良好的生物活性, 在药物设计和新材料开发中具有不可替代的价值[46].
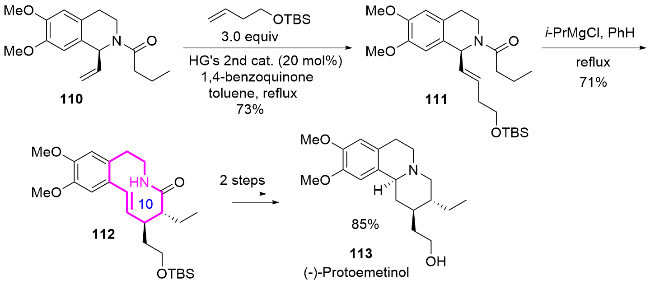
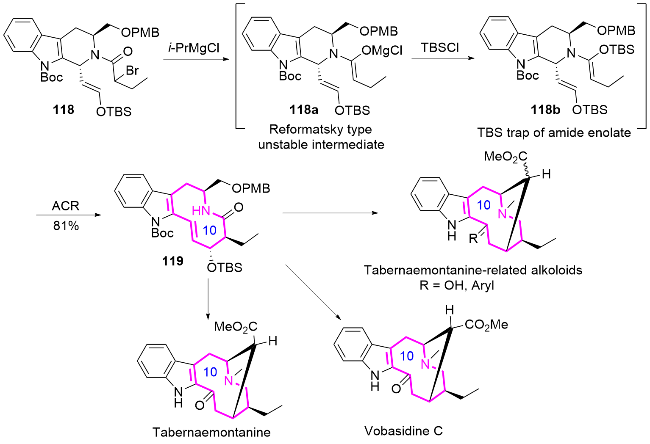
2017年, Paek课题组[50]开发一种高效的串联Reformatsky-aza-Claisen重排反应, 成功构建了taber- naemontanine类吲哚生物碱的关键十元氮杂环核心结构(Scheme 39). 作者通过Pictet-Spengler环化反应合成了四氢-β-咔啉118, 随后通过官能团转化获得关键前体烯醇醚118b, 接着利用iPrMgCl参与的Reformatsky- aza-Claisen重排串联反应, 一步实现含有四个连续手性中心的[5.1.3]双环骨架化合物119的高效构筑, 最后通过进一步环化反应得到含十元环的吲哚生物碱taber- naemontanine及其衍生物.
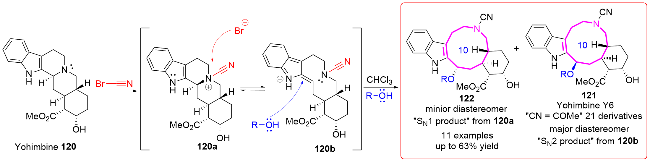
3.2 在药物分子上的应用
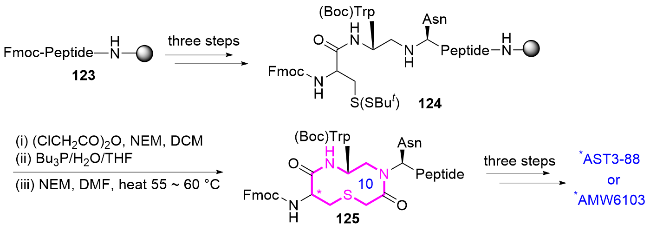
2014年, Carrie课题组[52]报道了一种新型黑皮质素受体激动剂AST3-88的研发, 该化合物通过在Trp7位点引入十元杂环结构对肽骨架进行修饰, 显著提高了对MC4R的选择性(>50倍)和代谢稳定性[53]. 首先, 化合物123在Rink酰胺树脂上逐步组装线性肽链, 经过三步反应成功构建硫醚键前体124, 使用(ClCH2CO)2O/N-乙基马来酰亚胺(NEM)对化合物124进行氯乙酰化, 再通过分子内环化形成关键的十元氮杂化合物125. 最后经三步反应得到AST3-88或AMW610 (Scheme 41). 该工作首次系统阐明了十元环结构在黑皮质素配体设计中的关键作用, 通过精确调控肽骨架的构象约束, 同时实现受体选择性、代谢稳定性和体内活性的多重优化, 为GPCR靶向肽类药物的理性设计提供了重要策略.
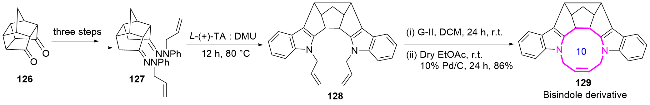
2016年, Jain课题组[54]通过Fischer吲哚化反应和关环复分解反应(RCM)作为关键步骤, 成功设计并合成了一种新型的十元环稠合多环吲哚衍生物129. 作者以易得的Cookson二酮126为起始原料, 经三步反应得到亚胺化合物127. 随后, 化合物127在L-酒石酸与二甲基脲反应条件下实现Fischer吲哚化生成化合物128, 随后通过Grubbs II代催化剂介导的RCM反应, 最终氢化获得十元环双吲哚产物129. 该十环结构的合成仅需六步, 其重要意义在于突破了传统多环吲哚合成的复杂性, 为具有C2对称性的多环吲哚类化合物提供了高效构建策略. 这类结构在生物有机化学中具有特殊价值, 其分子对称性与人类免疫缺陷病毒1型蛋白酶(HIV-1)抑制、抗革兰氏阳性菌活性密切相关[55], 为后续药物开发提供了重要的结构基础(Scheme 42).
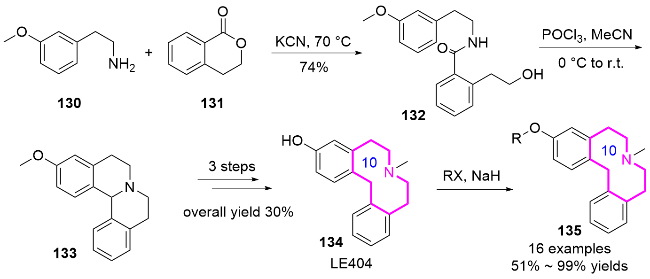
2017年, Andreas课题组[56]报道了抗精神病先导化合物LE404(二苯并氮杂䓬类多巴胺和5-羟色胺受体拮抗剂)的合成优化及其前药衍生物的拓展开发. 该路线以廉价的3-甲氧基苯乙胺130和内酯131为原料, 经氰化钾催化得到苯甲酰胺132, 随后发生Bischler-Napie- ralski-环化得到化合物133, 最后经三步反应合成LE404. 进一步研究表明, LE404与不同氯化物发生亲核反应, 可以转化为具有不同官能团取代的LE404衍生物135. 修饰LE404结构的目的是延长其半衰期, 并增强其作为多巴胺和血清素受体拮抗剂的体内功效. 一些含有丙炔基的LE404衍生物可能会抑制该化合物的肝脏代谢(Scheme 43)[57].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论与展望
十元氮杂环化合物作为中环体系的重要成员之一, 不仅是有机合成中的重要合成砌块, 也广泛存在于天然产物和药物分子中, 因其独特的构象特征和电子性质, 在药物设计和新材料开发中具有重要价值. 当前, 十元氮杂环化合物的合成方法主要包括分子内碳酰化反应、环加成反应、闭环复分解反应(RCM)、光催化氧化策略、金属催化环化反应以及扩环策略等. 尽管这些策略在合成十元氮杂环化合物中取得了不错的进展, 但十元氮杂环的高效合成仍是有机合成化学的重要挑战领域. 当前虽已发展出多种催化策略, 但在原子经济性、步骤效率和立体化学控制等方面仍有明显不足. 未来中环化合物的合成研究可以考虑着力于以下几个方面: (1)开发非贵金属、可持续的催化体系, 特别是将光催化、电催化等绿色化学手段与传统合成方法相结合; (2)结合计算化学和先进表征技术深入理解中环的形成机制; (3)发展模块化、多样性导向的合成策略, 特别是其不对称合成, 解决多手性中心的中环化合物的构建; (4)加强合成方法学与药物化学应用的衔接, 进一步将高效的合成策略应用于药物分子的发现与合成中. 我们相信经过未来的不断努力, 在中环化合物的合成和应用领域将有更多新的突破, 为中环氮杂环的结构创新和功能发现提供新机遇.
(Cheng, F.)